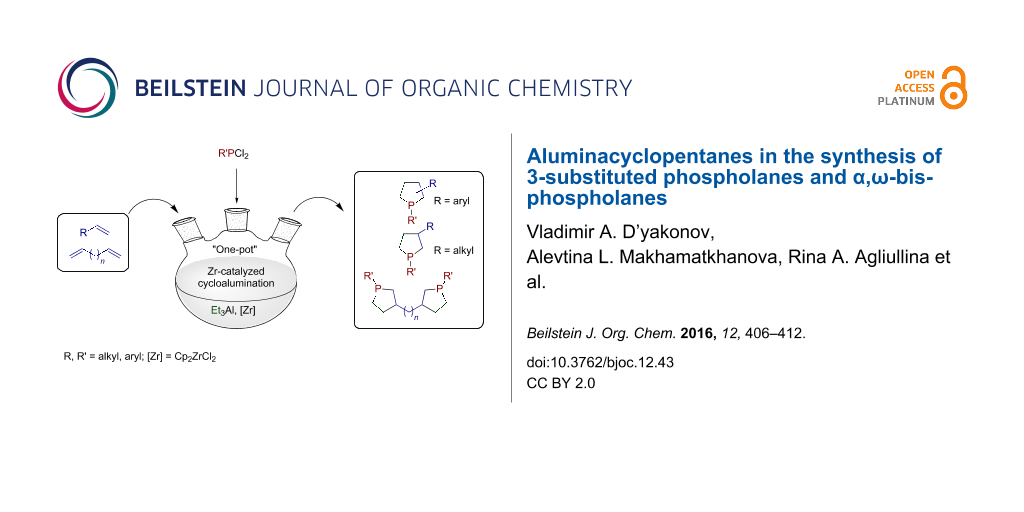
Abstract
An efficient one-pot process for the synthesis of 3-substituted phospholanes and α,ω-bisphospholanes was developed. The method involves the replacement of aluminium in aluminacyclopentanes, prepared in situ by catalytic cycloalumination of α-olefins and α,ω-diolefins, by phosphorus atoms on treatment with dichlorophosphines (R′PCl2). Hydrogen peroxide oxidation and treatment with S8 of the synthesized phospholanes and α,ω-bisphospholanes afforded the corresponding 3-alkyl(aryl)-1-alkyl(phenyl)phospholane 1-oxides, 3-alkyl(aryl)-1-alkyl(phenyl)phospholane 1-sulfides, bisphospholane 1,1'-dioxides, and bisphospholane 1,1'-disulfides in nearly quantitative yields. The complexes LMo(CO)5 (L = 3-hexyl-1-phenylphospholane, 3-benzyl-1-methylphospholane, 1,2-bis(1-phenylphospholan-3-yl)ethane, and 1,6-bis(1-phenylphospholan-3-yl)hexane were prepared by the reaction of 3-substituted phospholanes and α,ω-bisphospholanes with molybdenum hexacarbonyl. The structure of the complexes was proved by multinuclear 1H, 13C, and 31P spectroscopy.

Graphical Abstract
Introduction
A widely used approach for the synthesis of cyclic organophosphorus compounds (OPC) is the direct transformation of five-membered metallacarbocycles based on transition metals to phosphacarbocycles on treatment with phosphorus dihalides. For example, a method for the direct conversion of zirconacyclopentenes [1-4] and zirconacyclopentadienes [5] to substituted phospholenes [6] and phospholes [7] has been reported. This approach was used to obtain materials for light-emitting diodes (LEDs) [8], phosphorus-containing polymers [9,10], bisphospholes [11], bicyclodiphospholanes and spirobicyclodiphospholanes [12,13]. However, this method is faced with some practical complications related to the synthesis of the initial zirconacarbocycles: the reactions proceed at low temperatures (−78 °С) consuming stoichiometric amounts of expensive Cp2ZrCl2. In our opinion, the synthesis of cyclic OPC via catalytic cyclometallation reactions that we developed previously [14-17] would be free from the above-indicated drawbacks.
Furthermore, analysis of world literature demonstrated that the data on direct transformation of aluminacarbocycles [18] to cyclic OPCs are scarce, except for paper [19] in which this approach is implemented for simple olefins. Meanwhile, in our opinion, the development of these reactions would give rise to practically promising one-pot methods for the preparation of a broad range of cyclic and acyclic organophosphorus compounds of specified structure (phospholanes, phospholenes, phospholes and 1,2- and 1,4-diphosphorus compounds) that were difficult to synthesize or unknown before. In order to fill this gap and to extend the scope of applicability of the catalytic cycloalumination of unsaturated compounds, we continued studies along this line, which form the subject of this paper.
Previously [19], it was shown for allylbenzene, hex-1-ene, and oct-1-ene that aluminacyclopentanes, prepared by the reaction of these alkenes with Et3Al in the presence of 5 mol % Cp2ZrCl2 (20 °С, 6–8 h), react in situ with RPCl2 (R = Me, Ph) in toluene for 30 min with replacement of the Al atom by a P atom to give the respective phospholanes in 79–84% yields (Scheme 1).
![[1860-5397-12-43-i1]](/bjoc/content/inline/1860-5397-12-43-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 3-substituted phospholanes according to earlier data [14-17].
Scheme 1: Synthesis of 3-substituted phospholanes according to earlier data [14-17].
The present paper reports the results of experimental studies that further develop the earlier investigation about the substitution of phosphorus atoms for the Al atoms in aluminacyclopentanes with the goal to develop a preparative method for the synthesis of five-membered cyclic organophosphorus compounds.
Results and Discussion
First, we studied the effect of the structure of substituent in position 3 of the initial aluminacyclopentanes on the yield of target phospholanes. Under the selected conditions (toluene, 20–22 °С, 30 min), 3-alkyl-substituted aluminacyclopentanes 1a–c react with phenyldichlorophosphine to give 3-butyl-1-phenylphospholane (2a), 3-hexyl-1-phenylphospholane (2b), or 3-octyl-1-phenylphospholane (2c) in ca. 92, 91, and 87% yields, respectively (Scheme 2, Table 1). The isolated organophosphorus compounds 2a–с are 3:2 mixtures of diastereomers formed due to the presence of two asymmetric centres in the molecule at С-3 and P-1. The latter exists due to the high barrier for inversion of configuration at the phosphorus atom [20].
![[1860-5397-12-43-i2]](/bjoc/content/inline/1860-5397-12-43-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 3-substituted phospholanes.
Scheme 2: Synthesis of 3-substituted phospholanes.
Table 1: Results of 3-substituted phospholanes a.
| Entry | R | R' | Product | Yield (%) | trans/cis |
|---|---|---|---|---|---|
| 1 | Bu | Ph | 2ab | 92 | 3/2 |
| 2 | Hex | Ph | 2b | 91 | 3/2 |
| 3 | Oct | Ph | 2c | 87 | 3/2 |
| 4 | cyclohexyl | Ph | 2d | 93 | 3/2 |
| 5 | cyclohexen-3-yl | Ph | 2e | 90 | 3/2 |
| 6 | Bn | Ph | 2fb | 82 | 3/2 |
| 7 | Bn | Me | 2gb | 84 | 2/1 |
| 8 | Bn | Bu | 2h | 86 | 2/1 |
aReaction conditions: (i) 10 mmol Et3Al, 5 mol % Cp2ZrCl2, toluene (25 mL), room temperature, 12 hours; (ii) 10 mmol R’PCl2, −5 °C to room temperature, 30 minutes. bThe cyclic OPCs have been previously synthesized and described in [19].
The diastereomeric mixture was identified by multinuclear 1H, 13C, and 31P NMR spectroscopy and conventionally designated as the syn and anti isomers in which, for example, the high-priority substituents at Р-1 and С-3 are proximate in the syn isomer. Using the homo- and heteronuclear 2D NMR spectroscopy (H,H-COSY, HSQC, HMBC), parameters of the NMR spectra of some compounds 2 were determined and proved to agree with published data [19]. Specifically, 1-phenyl-3-substituted phospholanes typically show 31Р NMR signals in area of −14 to −13 ppm, except for the 1-alkyl-3-benzylphospholane 2g and 2h isomers, for which the signals occur at δP = −34 ppm and −35 ppm, and −22 to −23 ppm, respectively. The presence of a magnetically active phosphorus atom in the five-membered heterocycle induces the doublet splitting of the carbon signals in the 13С NMR spectra of 2a–h for both the P-substituent and the ring. The highest heteronuclear constants J(31P13C) for the ring, 1JPC ≈ 10–20 Hz, are found for the α-carbon atoms (С-2 and С-5) of phospholanes 2a–h.
Allylbenzene reacts with AlEt3 (5 mol % Cp2ZrCl2, 20 °С, 12 h) to give 3-benzyl-1-ethylaluminacyclopentane 1f, which then reacts with phenyldichlorophosphine giving rise to 3-benzyl-1-phenylphospholane 2f in a yield of 82% (Scheme 2).
When PhPCl2 is replaced by MePCl2 or BuPCl2, the reaction with 3-benzyl-1-ethylaluminacyclopentane 1f affords the corresponding phospholanes 2g,h with one of the isomers predominating (Table 1).
In the case of 3-cyclohexyl- and 3-(сyclohex-3-en-1-yl)-1-phenylphospholanes (2d and 2e), the number of stereoisomers increases owing to the additional asymmetric centre С(1′) in the substituent, and the total yield of these compounds is 90–93%.
Phospholanes 2а–h readily react with H2O2 in chloroform, owing to the presence of a lone electron pair at phosphorus to give phospholane 1-oxides 3а–h in quantitative yields. The reaction of 2а–h with S8 affords phospholane 1-sulfides 4а–h also in quantitative yields (Scheme 3).
![[1860-5397-12-43-i3]](/bjoc/content/inline/1860-5397-12-43-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 3-substituted phospholane oxides and sulfides.
Scheme 3: Synthesis of 3-substituted phospholane oxides and sulfides.
In the 31Р NMR spectra of phospholane oxides 3a–h and phospholane sulfides 4a–h, the phosphorus signal shifts downfield to ca. 57–70 ppm relative to the initial phospholanes, and the heteronuclear constants 1J(31P13C2) and 1J(31P13C5) observed in the 13С NMR spectra increase to ca. 53–66 Hz.
When styrene or 2-vinylnaphthalene reacts with AlEt3 in the presence of Cp2ZrCl2, apart from 3-phenyl(naphthyl)-1-ethylaluminacyclopentanes 5a–f, the reaction mixture contains 2-phenyl(naphthyl)-1-ethylaluminacyclopentane 6a–f [21]. Both regioisomers react in situ with phosphorus dihalides and hydrogen peroxide to afford 1-phenyl(alkyl)-2-arylphospholane oxides 7a–f and 1-phenyl(alkyl)-3-arylphospholane oxides 8a–f in 2:1 ratio in a 69–87% total yield (Table 2). The regioisomers were isolated by column chromatography (hexane/ethyl acetate/methanol = 5:3:1) and characterized in a separate fraction (Scheme 4).
![[1860-5397-12-43-i4]](/bjoc/content/inline/1860-5397-12-43-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 3-substituted 7a–f and 2-substituted 8a–f phospholanes.
Scheme 4: Synthesis of 3-substituted 7a–f and 2-substituted 8a–f phospholanes.
It should be noted that the phosphorus signals of major intensity of 2-aryl phospholane oxides 8a, 8b, 8d, 8e, 8f, corresponding to one of the stereoisomers, are shifted upfield by ca. 5–7 ppm with respect to those of 3-aryl-substituted phospholane oxides 7. In contrast, the 31P NMR signal of the second isomer of 8 is shifted to lower field.
For further development of this study, it appeared pertinent to apply our method in the preparation of α,ω-bisphospholane compounds by the reaction of phosphorus dihalides with bisaluminacyclopentanes (Scheme 5).
![[1860-5397-12-43-i5]](/bjoc/content/inline/1860-5397-12-43-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Thus bisaluminacyclopentane 9a, synthesized by catalytic cycloalumination of 1,7-octadiene, was subjected without isolation to the substitution reaction with aryl(alkyl)dichlorophosphine with replacement of Al by Р (ca. 20 °С, 30 min) to give bisphospholane 10a as a mixture of isomers in a total yield of 84–85%.
Under the selected conditions, bisaluminacyclopentanes 9b–e, prepared by catalytic cycloalumination of 1,5-hexadiene and 1,9-decadiene, react with phenyl(methyl, butyl)dichlorophosphine to give 1,2-bis(1-phenylphospholan-3-yl)ethane (10b), 1,2-bis(1-butylphospholan-3-yl)ethane (10d), 1,4-bis(1-methylphospholan-3-yl)butane (10e), and 1,6-bis(1-phenylphospholan-3-yl)hexane (10с) in 85 % yields.
Similarly to phospholanes 2a–h, the resulting bisphospholanes 10a–c readily react with H2O2 in chloroform or with elemental sulfur to furnish the corresponding bisphospholane 1,1'-dioxides 11a–c and bisphospholane 1,1'-disulfides 12a–c (Scheme 6).
![[1860-5397-12-43-i6]](/bjoc/content/inline/1860-5397-12-43-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of bisphospholane-1,1'-oxides and bisphospholane-1,1'-sulfides.
Scheme 6: Synthesis of bisphospholane-1,1'-oxides and bisphospholane-1,1'-sulfides.
Diastereomeric cleavage of signals in the 13С NMR spectra of bisphospholanes observed only for compounds 10а, 10d, 11а, 12а in which the phospholane moieties are linked by two methylene groups, obviously as a result of mutual influence of the proximate asymmetric centres at C-3 and С-3′. Ratios of stereoisomers were determined by HPLC method as 2:1 (see Supporting Information File 2, Figure S1).
Organophosphorus compounds, including cyclic ones, are known to readily form complexes with transition metals, which are extensively studied and used in homogeneous catalytic reactions. Therefore, in order to study the properties of the cyclic OPC that we prepared and to demonstrate how they could be used as ligands, we investigated the reaction of 3-substituted phospholanes and bisphospholanes with Mo(CO)6, resulting in the preparation of molybdenum complexes. For example, 3-hexyl-1-phenylphospholane and 3-benzyl-1-methylphospholane react with Mo(CO)6 furnishing molybdenum complexes 13b and 14g (Scheme 7).
![[1860-5397-12-43-i7]](/bjoc/content/inline/1860-5397-12-43-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of the molybdenum complex (3-hexyl(benzyl)-1-phenyl(methyl)phospholane)Mo(CO)5.
Scheme 7: Synthesis of the molybdenum complex (3-hexyl(benzyl)-1-phenyl(methyl)phospholane)Mo(CO)5.
The complex formation is evidenced by changes in the NMR spectral parameters of compounds 13b and 14g in relation to the initial phospholanes. A typical feature is the downfield shift of the 31Р NMR signals. As a result, both compounds were found to exhibit close chemical shifts δP ≈ 25 ppm and δP ≈ 26 ppm for the diastereomers formed, irrespective of the nature of substituent R', which is indicative of equal pyramidality of the bonds of the phosphorus atom incorporated in the molybdenum complex. In addition, the С-5 signal in the 13С NMR spectra is shifted downfield by ca. 8 ppm and the constant JPC5 = 24.1 Hz (for example, for 13b) is much higher not only compared to the corresponding atom in the initial phospholane 2b but also than JPC2 = 3.0 Hz in compound 13b. In view of the fact that the phosphorus–carbon constants obey the Karplus equation [22], this result suggests that the conformation of the five-membered heterocycle changes upon the formation of the complex with molybdenum hexacarbonyl. Similarly, bisphospholanes 10a and 10c react with Mo(CO)6 to form molybdenum complexes 15a and 16c (Scheme 8). The resulting molybdenum complexes are oily liquids. The structure of the complexes was also proved by spectral data.
![[1860-5397-12-43-i8]](/bjoc/content/inline/1860-5397-12-43-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of molybdenum complexes (1,2(1,6)-bis(1-phenylphospholan-3-yl)ethane(hexane))Mo(CO)5.
Scheme 8: Synthesis of molybdenum complexes (1,2(1,6)-bis(1-phenylphospholan-3-yl)ethane(hexane))Mo(CO)5.
Conclusion
Thus, as a continuation of investigations dealing with the search for effective methods for the synthesis of five-membered cyclic organophosphorus compounds, we developed a preparative process for the one-pot conversion of aluminacyclopentanes, obtained in situ by catalytic cycloalumination of olefins or diolefins with AlEt3, to phospholanes, phospholane oxides or phospholane sulfides, including bisphospholanes and their derivatives in high yields.
The developed methods are distinguished by easy implementation of the reaction with the use of accessible reagents and monomers, which makes them quite promising for the use in both the laboratory practice and industry.
Experimental
Preparation of 3-alkyl(aryl)phospholanes (general procedure)
A glass reactor maintained under dry argon at 0 °C was successively charged, with stirring, with toluene (25 mL), Cp2ZrCl2 (0.298 g, 1 mmol), olefin (10 mmol), and AlEt3 (1.8 mL, 10 mmol) The mixture was warmed up to room temperature (ca. 20 °C) and stirred for 12 h. Then the reaction mixture was cooled down to −5 to −10 °С, alkyl(phenyl)dichlorophosphine (10 mmol) was added dropwise, and the mixture was stirred at room temperature for additional 30 min. Then the reaction mixture was treated with a saturated aqueous solution of NH4Cl and the reaction products were extracted with diethyl ether and dried with MgSO4. The solvent was evaporated and the target phospholanes were isolated by vacuum distillation. All operations were carried out in an argon flow.
The general procedure and analytical data for compounds 2a, 2b, 2f. 2g and 3a, 3b, 3f, 3g were previously described in [19].
3-Octyl-1-phenylphospholane (2c) in the mixture in a ratio of 3:2: Colorless oil (87%); bp 215–218 °C (9 torr); calcd for C18H29P: C, 78.22%; H, 10.58%; found: C, 78.3%; H, 10.6%; 1H NMR (400.13 MHz, CDCl3) δ 0.95 (t, 3J = 6.8 Hz, 6H, C(8')H), 1.27–1.63 (m, 32H, C(1')H, C(2')H, C(3')H, C(4')H, C(5')H, C(6′)H, C(7′))H, C(4)Ha, C(2)Ha), 1.94–2.03 (m, 4H, C(3)H, C(5)Ha), 2.10–2.20 (m, 4H, C(2)Hb, C(4)Hb), 2.26 (m, 2H, C(5)Hb), 7.24–7.30, 7.32–7.38, 7.42–7.48 (m, 10H, Ph); 13C NMR (100.62 MHz, CDCl3) δ 14.15 (C(8')), 22.70 (C(7')), 25.79 (JPC = 12.1 Hz, C(5)), 26.60 (JPC = 10.1 Hz, C(5)), 28.49, 28.61 (C(2')), 29.04, 29.05 (C(4')), 29.32 (C(5')), 29.57, 29.64 (C(3')), 31.63 (C(6')), 32.92 (JPC = 13.1 Hz, C(2)), 33.16 (JPC = 11.1 Hz, C(2)), 33.95 (JPC = 3.0 Hz, C(4)), 34.24 (JPC = 4.0 Hz, C(4)), 35.60 (JPC = 3.0 Hz, C(1')), 35.86 (JPC = 5.0 Hz, C(1')), 41.82 (JPC = 4.0 Hz, C(3)), 43.02 (JPC = 1.0 Hz, C(3)), 126.87 (C(9)), 127.90, 127.91 (JPC = 5.0 Hz, C(8), C(10)), 130.07 (JPC = 16.1 Hz, C(7), C(11)), 130.13 (JPC = 15.1 Hz, C(7), C(11)), 142.47, 142.84 (JPC = 23.1 Hz, C(6)) (P-Ph); 31P NMR (161.97 MHz, CDCl3) δ −13.5, −13.9; MALDI–TOF: m/z сalcd for C18H30P ([M + H]+): 277.4046; found: 277.4.
Preparation of 3-alkyl(aryl)phospholane-1-oxides (general procedure)
A 30% solution of hydrogen peroxide (0.7 mL, 6 mmol) was slowly added dropwise with vigorous stirring to a solution of 3-alkyl(benzyl)-1-alkyl(phenyl)phospholane (5 mmol), synthesized as described above, in chloroform (10 mL) and the mixture was stirred for 1 h. Then the reaction mixture was washed with water (3 × 5 mL) and the organic layer was dried with MgSO4. The solvent was evaporated and the residue was chromatographed on silica gel (hexane/ethyl acetate/methanol 5:3:1). 3-Octyl-1-phenylphospholane-1-oxide (3c) in the mixture in a ratio of 3:2: Calcd for C18H29OP: C, 73.94; H, 10.00%; found: C, 73.7%; H, 9.8%; 1H NMR (400.13 MHz, CDCl3) δ 0.81 (t, 3J = 7.2 Hz, 6H, C(8')H), 1.17–1.60 (m, 30H, C(2)Ha, C(4)Ha, C(1')H, C(2')H, C(3')H, C(4')H, C(5')H, C(6')H, C(7')H), 1.70 (m, 1H, C(2)Ha), 1.80 (m, 1H, C(4)Ha), 1.87 (m, 1H, C(5)Ha), 1.94–2.12 (m, 2H, C(3)H, C(5)Ha), 2.14–2.35 (m, 7H, C(2)Hb, C(3)H, C(4)Hb, C(5)Hb), 7.36–7.56, 7.60–7.78 (m, 10H, Ph); 13C NMR (100.62 MHz, CDCl3) δ 13.82 (C(8')), 22.35 (C(7')), 27.51, 27.60 (C(2')), 28.94, 28.96 (C(4')), 29.19 (JPC = 66.4 Hz, C(5)), 29.21 (C(5')), 29.32, 29.36 (C(3')), 30.18 (JPC = 66.4 Hz, C(5)), 30.99 (JPC = 6.0 Hz, C(4)), 31.55 (C(6')), 31.93 (JPC = 7.0 Hz, C(4)), 35.87 (JPC = 55.3 Hz, C(2)), 35.99 (JPC = 12.1 Hz, C(1')), 36.04 (JPC = 14.1 Hz, C(1')), 36.16 (JPC = 53.3 Hz, C(2)), 38.60, 40.21 (JPC = 8.0 Hz, C(3)), 128.39 (JPC = 12.1 Hz, C(8), C(10)), 129.55 (JPC = 10.1 Hz, C(7), C(11)), 131.40 (JPC = 3.0 Hz, C(9)), 133.90 (JPC = 89.5 Hz, C(6)), 134.00 (JPC = 90.5 Hz, C(6)) (P-Ph); 31P NMR (161.97 MHz, CDCl3) δ 59.7, 59.4; MALDI TOF: m/z calculated for C18H30OP ([M+ H]+): 293.4040; found: 293.5.
General procedure for the preparation of 3-alkyl(aryl)phospholane-1-sulfides
Reactions were performed under argon. Sulfur (0.13 g, 4 mmol) was added with cooling to a solution of 3-alkyl(aryl)phospholane (4 mmol) (prepared as described above) in 10 mL toluene, and the mixture was stirred for 4 h. After filtration through a thin layer of silica gel the solvent was evaporated to give a colorless oil. 3-Hexyl-1-phenylphospholane-1-sulfide (4b) in the mixture in a ratio of 3:2: Calculated for C16H25PS: C, 68.53%; H, 8.99%; found: C, 68.4%; H, 8.0%; 1H NMR (400.13 MHz, CDCl3) δ 0.91 (t, 3J = 6.8 Hz, 6H, C(6')H), 1.25–1.43 (m, 16H, C(2')H, C(3')H, C(4')H, C(5')H), 1.43–1.61 (m, 5H, C(1')H, C(4)Ha), 1.83–1.95 (m, 2H, C(2)Ha, C(4)Hа), 1.99 (m, 1H, C(2)Ha), 2.11–2.52 (m, 8H, C(2)Hb, C(3)H, C(4)Hb, C(5)Ha, C(5)Hb), 2.60 (m, 1H, C(5)Hb), 2.66 (m, 1H, C(2)Hb), 7.42–7.53, 7.82–7.94 (m, 10H, Ph); 13C NMR (100.62 MHz, CDCl3) δ 13.77 (C(6')), 22.27, 22.29 (C(5')), 27.62, 27.68 (C(2')), 29.00 (C(3')), 31.41, 31.43 (C(4')), 31.86 (JPC = 6.0 Hz, C(4)), 33.62 (JPC = 4.0 Hz, C(4)), 35.44 (JPC = 14.1 Hz, C(1')), 35.64 (JPC = 53.3 Hz, C(5)), 35.71 (JPC = 12.1 Hz, C(1')), 36.49 (JPC = 53.3 Hz, C(5)), 39.74 (JPC = 8.0 Hz, C(3)), 41.78 (JPC = 6.0 Hz, C(3)), 41.94 (JPC = 54.3 Hz, С(2)), 42.38 (JPC = 53.3 Hz, C(2)), 128.30 (JPC = 12.1 Hz, C(8), C(10)), 130.00 (JPC = 11.1 Hz, C(7), C(11)), 131.07 (JPC = 2.0 Hz, C(9)), 133.76, 133.84 (JPC = 70.4 Hz, C(6)) (P-Ph); 31P NMR (161.97 MHz, CDCl3) δ 57.72; MALDI TOF: m/z сalculated for C16H26PS ([M + H]+): 281.4174; found: 281.3.
General procedure for the preparation of Mo complexes 13b, 14g
Reactions was performed under argon using standard Schlenk techniques. A mixture of Mo(CO)6 (0.79 g, 3 mmol) and 3-hexyl(benzyl)-1-phenyl(methyl)phospholane 2b, 2g (3 mmol) was stirred under reflux in 15 mL THF for 24 h. The colorless solution became dark-brown during this time. The solvent was removed under vacuum and the residue was chromatographed on silica gel (hexane). The product was obtained as dark green thick liquid. (3-Hexyl-1-phenylphospholane)pentacarbonylmolybdenum (13b) in the mixture in a ratio of 1:1: Calcd for C21H25MoO5P: C, 52.08%; H, 5.20%; found: C, 52.2%; H, 5.3%; 1H NMR (400.13 MHz, CDCl3) δ 0.20–1.01 (s, 6H, C(6′)H), 1.30–1.56 (s, 20H, C(5′)H, C(3′)H, C(2′)H, C(4′)H, C(1′)H), 1.58–1.64 (m, 2H, C(4)H), 1.72–1.82 (m, 1H, C(2)Ha), 1.93–2.02 (m, 1H, C(3)H), 2.06–2.27 (m, 4H, C(4)Hb, C(2)Hb, C(3)H), 2.41–2.57 (m, 3H, C(5)Ha, C(5)Hb), 2.58–2.80 (m, 3Н, С(5)Hb, С(2)Ha, С(2)Hb), 7.23–7.26, 7.31–7.33, 7.37–7.44, 7.45–7.56 (m, 10Н, Ph); 13C NMR (100.62 MHz, CDCl3) δ 14.10 (C(6')), 22.66, 22.72 (C(5')), 28.39, 28.42 (C(3')), 29.37, 29.44 (C(2')), 31.45 (JPC = 22.1 Hz, C(5)), 31.81 (C(4')), 32.16 (JPC = 24.1 Hz, C(5)), 33.42, 34.13 (C(4)), 35.70 (JPC = 8.0 Hz, C(1')), 35.75 (JPC = 9.1 Hz, C(1')), 38.19 (C(2)), 38.41 (JPC = 3.0 Hz, C(2)), 41.78 (JPC = 1.0 Hz, C(3)), 42.30 (C(3)), 128.81, 128.88 (JPC = 10.1 Hz), 128.95 (JPC = 11.1 Hz), 129.08, 139.67 (JPC = 27.2 Hz, C(6)), 140.24 (JPC = 28.2 Hz, C(6)) (P-Ph), 205.75 (JPC = 9.1 Hz, COcis), 205.77 (JPC = 9.1 Hz, COcis), 210.51 (JPC = 22.1 Hz, COtrans), 210.57 (JPC = 21.1 Hz, COtrans); 31P NMR (161.97 MHz, CDCl3): δ 26.00, 25.20.
General procedure for the preparation of Mo complexes 15a, 16c
Reactions were performed under argon using standard Schlenk techniques. A mixture of Mo(CO)6 (0.79 g, 3 mmol) and 1,2(1,6)-bis(1-phenylphospholan-3-yl)ethane(hexane) 10a, 10c (3 mmol) was stirred under reflux in 15 mL THF for 24 h. The colorless solution became brown during this time. The solvent was removed under vacuum and the crude product was purified by silica gel column chromatography (hexane) giving a dark brown, thick liquid. 1,2-Bis(1-pentacarbonylmolybdenum-1-phenylphospholan-3-yl)ethane (15a) as a mixture of isomers. Calcd for C32H28Mo2O10P2: C, 46.51%; H, 3.42%; found: C, 46.5%; H, 3.5%; 1H NMR (400.13 MHz, CDCl3) δ 1.28–1.39 (m, 2H, C(4)Ha, C(4)Hb), 1.40–1.60 (m, 2H, C(4)Ha, C(4)Hb), 1.64–1.73 (m, 1H, C(2)Ha), 1.84–1.94 (m, 1H, C(3)H), 1.98–2.22 (m, 6H, C(6′), C(2)Hb, C(3)H), 2.35–2.58 (m, 3H, C(5)Ha, C(5)Hb, C(2)Ha), 2.60–2.70 (m, 3H, C(5)Ha, C(5)Hb, С(2)Hb), 7.34–7.56 (m, 10H, P-Ph); 13C NMR (100.62 MHz, CDCl3) δ 31.29, 31.35 (JPC = 23.1 Hz, C(5)), 32.01 (JPC = 23.1 Hz, C(5)), 32.05 (JPC = 24.1 Hz, C(5)), 33.35, 33.40, 34.13, 34.17, 34.22 (С(4)), 34.37, 34.44, 34.52, 34.59 (C(6')), 38.31, 38.35, 38.37 (JPC = 23.1 Hz, C(2)), 38.50, 38.54 (C(2)), 41.74, 41.82, 41.91, 42.36, 42.42 (C(3)), 128.84, 128.94, 129.03, 129.19, 139.44, 140.11 (JPC = 28.2 Hz, C(6)) (Р-Ph), 205.74, 205.83 (COcis), 210.49, 210.55 (JPC = 22.1 Hz, COtrans); 31P NMR (161.97 MHz, CDCl3) δ 25.3, 26.3.
Supporting Information
Detailed synthesis and characterization procedures are provided for all compounds synthesized and characterized. NMR spectra are provided for all compounds for which NMR data are reported.
| Supporting Information File 1: Experimental details, characterization data of all products. | ||
| Format: PDF | Size: 395.1 KB | Download |
| Supporting Information File 2: NMR spectra. | ||
| Format: PDF | Size: 5.4 MB | Download |
References
-
Fagan, P. J.; Nugent, W. A. J. Am. Chem. Soc. 1988, 110, 2310–2312. doi:10.1021/ja00215a057
Return to citation in text: [1] -
Fagan, P. J.; Nugent, W. A.; Calabrese, J. C. J. Am. Chem. Soc. 1994, 116, 1880–1889. doi:10.1021/ja00084a031
Return to citation in text: [1] -
Hydrio, J.; Gouygou, M.; Dallemer, F.; Daran, J.-C.; Balavoine, G. G. A. J. Organomet. Chem. 2000, 595, 261–267. doi:10.1016/S0022-328X(99)00635-X
Return to citation in text: [1] -
Zhou, Y.; Wang, S.; Chen, C.; Xi, C. RSC Adv. 2015, 5, 71724–71727. doi:10.1039/C5RA13818C
Return to citation in text: [1] -
Crassous, J.; Réau, R. Dalton Trans. 2008, 6865–6876. doi:10.1039/b810976a
Return to citation in text: [1] -
Zhou, Y.; Yan, X.; Xi, C. Tetrahedron Lett. 2010, 51, 6136–6138. doi:10.1016/j.tetlet.2010.09.061
Return to citation in text: [1] -
Hu, G.; Zhang, Y.; Su, J.; Li, Z.; Gao, Y.; Zhao, Y. Org. Biomol. Chem. 2015, 13, 8221–8231. doi:10.1039/C5OB00959F
Return to citation in text: [1] -
Fave, C.; Hissler, M.; Kárpáti, T.; Rault-Berthelot, J.; Deborde, V.; Toupet, L.; Nyulászi, L.; Réau, R. J. Am. Chem. Soc. 2004, 126, 6058–6063. doi:10.1021/ja0317067
Return to citation in text: [1] -
Mao, S. S. H.; Tilley, T. D. Macromolecules 1997, 30, 5566–5569. doi:10.1021/ma9701402
Return to citation in text: [1] -
Makhamatkhanova, A. L.; Dil’mukhametova, L. K.; Tyumkina, T. V.; D’yakonov, V. A.; Dzhemilev, U. M. Russ. Chem. Bull. 2013, 62, 2467–2471. doi:10.1007/s11172-013-0357-x
Return to citation in text: [1] -
Doherty, S.; Eastham, G. R.; Tooze, R. P.; Scanlan, T. H.; Williams, D.; Elsegood, M. R. J.; Clegg, W. Organometallics 1999, 18, 3558–3560. doi:10.1021/om990346m
Return to citation in text: [1] -
Mirza-Aghayan, M.; Boukherroub, R.; Etemad-Moghadam, G.; Manuel, G.; Koenig, M. Tetrahedron Lett. 1996, 37, 3109–3112. doi:10.1016/0040-4039(96)00503-5
Return to citation in text: [1] -
Oba, G.; Phok, S.; Manuel, G.; Koenig, M. Tetrahedron 2000, 56, 121–127. doi:10.1016/S0040-4020(99)00780-2
Return to citation in text: [1] -
Dzhemilev, U. M.; Ibragimov, A. G. J. Organomet. Chem. 2010, 695, 1085–1110. doi:10.1016/j.jorganchem.2010.01.002
Return to citation in text: [1] [2] -
Dzhemilev, U. M.; D'yakonov, V. A. Hydro-, Carbo- and Cycloalumination of Unsaturated Compounds. In Modern Organoaluminum Reagents: Preparation, Structure, Reactivity and Use; Woodward, S.; Dagorne, S., Eds.; Springer: Berlin, Heidelberg, 2013; Vol. 41, pp 312 ff.
Return to citation in text: [1] [2] -
D'yakonov, V. A.; Trapeznikova, O. A.; de Meijere, A.; Dzhemilev, U. M. Chem. Rev. 2014, 114, 5775–5814. doi:10.1021/cr400291c
Return to citation in text: [1] [2] -
D'yakonov, V. A.; Dzhemilev, U. M. Reactions in Organic and Organometallic Synthesis; NOVA Sci. Publ.: New York, 2010; p 96.
Return to citation in text: [1] [2] -
Tyumkina, T. V.; Islamov, D. N.; Parfenova, L. V.; Khalilov, L. M.; Dzhemilev, U. M. Magn. Reson. Chem. 2016, 54, 62–74. doi:10.1002/mrc.4311
Return to citation in text: [1] -
D’yakonov, V. A.; Makhamatkhanova, A. L.; Tyumkina, T. V.; Dzhemilev, U. M. Russ. Chem. Bull. 2012, 61, 1556–1559. doi:10.1007/s11172-012-0205-4
Return to citation in text: [1] [2] [3] [4] [5] -
Potapov, V. M. Stereochemistry; Chemistry, R. F.: Moscow, 1988.
Return to citation in text: [1] -
Ibragimov, A. G.; Khafizova, L. O.; Satenov, K. G.; Khalilov, L. M.; Yakovleva, L. G.; Rusakov, S. V.; Dzhemilev, U. M. Russ. Chem. Bull. 1999, 48, 1574–1580. doi:10.1007/BF02496415
Return to citation in text: [1] -
Kühl, O. Phosphorus-31 NMR spectroscopy: a concise introduction for the synthetic organic and organometallic chemist; Springer: Berlin, 2008.
Return to citation in text: [1]
| 19. | D’yakonov, V. A.; Makhamatkhanova, A. L.; Tyumkina, T. V.; Dzhemilev, U. M. Russ. Chem. Bull. 2012, 61, 1556–1559. doi:10.1007/s11172-012-0205-4 |
| 21. | Ibragimov, A. G.; Khafizova, L. O.; Satenov, K. G.; Khalilov, L. M.; Yakovleva, L. G.; Rusakov, S. V.; Dzhemilev, U. M. Russ. Chem. Bull. 1999, 48, 1574–1580. doi:10.1007/BF02496415 |
| 22. | Kühl, O. Phosphorus-31 NMR spectroscopy: a concise introduction for the synthetic organic and organometallic chemist; Springer: Berlin, 2008. |
| 1. | Fagan, P. J.; Nugent, W. A. J. Am. Chem. Soc. 1988, 110, 2310–2312. doi:10.1021/ja00215a057 |
| 2. | Fagan, P. J.; Nugent, W. A.; Calabrese, J. C. J. Am. Chem. Soc. 1994, 116, 1880–1889. doi:10.1021/ja00084a031 |
| 3. | Hydrio, J.; Gouygou, M.; Dallemer, F.; Daran, J.-C.; Balavoine, G. G. A. J. Organomet. Chem. 2000, 595, 261–267. doi:10.1016/S0022-328X(99)00635-X |
| 4. | Zhou, Y.; Wang, S.; Chen, C.; Xi, C. RSC Adv. 2015, 5, 71724–71727. doi:10.1039/C5RA13818C |
| 8. | Fave, C.; Hissler, M.; Kárpáti, T.; Rault-Berthelot, J.; Deborde, V.; Toupet, L.; Nyulászi, L.; Réau, R. J. Am. Chem. Soc. 2004, 126, 6058–6063. doi:10.1021/ja0317067 |
| 19. | D’yakonov, V. A.; Makhamatkhanova, A. L.; Tyumkina, T. V.; Dzhemilev, U. M. Russ. Chem. Bull. 2012, 61, 1556–1559. doi:10.1007/s11172-012-0205-4 |
| 7. | Hu, G.; Zhang, Y.; Su, J.; Li, Z.; Gao, Y.; Zhao, Y. Org. Biomol. Chem. 2015, 13, 8221–8231. doi:10.1039/C5OB00959F |
| 19. | D’yakonov, V. A.; Makhamatkhanova, A. L.; Tyumkina, T. V.; Dzhemilev, U. M. Russ. Chem. Bull. 2012, 61, 1556–1559. doi:10.1007/s11172-012-0205-4 |
| 6. | Zhou, Y.; Yan, X.; Xi, C. Tetrahedron Lett. 2010, 51, 6136–6138. doi:10.1016/j.tetlet.2010.09.061 |
| 14. | Dzhemilev, U. M.; Ibragimov, A. G. J. Organomet. Chem. 2010, 695, 1085–1110. doi:10.1016/j.jorganchem.2010.01.002 |
| 15. | Dzhemilev, U. M.; D'yakonov, V. A. Hydro-, Carbo- and Cycloalumination of Unsaturated Compounds. In Modern Organoaluminum Reagents: Preparation, Structure, Reactivity and Use; Woodward, S.; Dagorne, S., Eds.; Springer: Berlin, Heidelberg, 2013; Vol. 41, pp 312 ff. |
| 16. | D'yakonov, V. A.; Trapeznikova, O. A.; de Meijere, A.; Dzhemilev, U. M. Chem. Rev. 2014, 114, 5775–5814. doi:10.1021/cr400291c |
| 17. | D'yakonov, V. A.; Dzhemilev, U. M. Reactions in Organic and Organometallic Synthesis; NOVA Sci. Publ.: New York, 2010; p 96. |
| 14. | Dzhemilev, U. M.; Ibragimov, A. G. J. Organomet. Chem. 2010, 695, 1085–1110. doi:10.1016/j.jorganchem.2010.01.002 |
| 15. | Dzhemilev, U. M.; D'yakonov, V. A. Hydro-, Carbo- and Cycloalumination of Unsaturated Compounds. In Modern Organoaluminum Reagents: Preparation, Structure, Reactivity and Use; Woodward, S.; Dagorne, S., Eds.; Springer: Berlin, Heidelberg, 2013; Vol. 41, pp 312 ff. |
| 16. | D'yakonov, V. A.; Trapeznikova, O. A.; de Meijere, A.; Dzhemilev, U. M. Chem. Rev. 2014, 114, 5775–5814. doi:10.1021/cr400291c |
| 17. | D'yakonov, V. A.; Dzhemilev, U. M. Reactions in Organic and Organometallic Synthesis; NOVA Sci. Publ.: New York, 2010; p 96. |
| 19. | D’yakonov, V. A.; Makhamatkhanova, A. L.; Tyumkina, T. V.; Dzhemilev, U. M. Russ. Chem. Bull. 2012, 61, 1556–1559. doi:10.1007/s11172-012-0205-4 |
| 12. | Mirza-Aghayan, M.; Boukherroub, R.; Etemad-Moghadam, G.; Manuel, G.; Koenig, M. Tetrahedron Lett. 1996, 37, 3109–3112. doi:10.1016/0040-4039(96)00503-5 |
| 13. | Oba, G.; Phok, S.; Manuel, G.; Koenig, M. Tetrahedron 2000, 56, 121–127. doi:10.1016/S0040-4020(99)00780-2 |
| 19. | D’yakonov, V. A.; Makhamatkhanova, A. L.; Tyumkina, T. V.; Dzhemilev, U. M. Russ. Chem. Bull. 2012, 61, 1556–1559. doi:10.1007/s11172-012-0205-4 |
| 11. | Doherty, S.; Eastham, G. R.; Tooze, R. P.; Scanlan, T. H.; Williams, D.; Elsegood, M. R. J.; Clegg, W. Organometallics 1999, 18, 3558–3560. doi:10.1021/om990346m |
| 9. | Mao, S. S. H.; Tilley, T. D. Macromolecules 1997, 30, 5566–5569. doi:10.1021/ma9701402 |
| 10. | Makhamatkhanova, A. L.; Dil’mukhametova, L. K.; Tyumkina, T. V.; D’yakonov, V. A.; Dzhemilev, U. M. Russ. Chem. Bull. 2013, 62, 2467–2471. doi:10.1007/s11172-013-0357-x |
| 18. | Tyumkina, T. V.; Islamov, D. N.; Parfenova, L. V.; Khalilov, L. M.; Dzhemilev, U. M. Magn. Reson. Chem. 2016, 54, 62–74. doi:10.1002/mrc.4311 |
© 2016 D’yakonov et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)