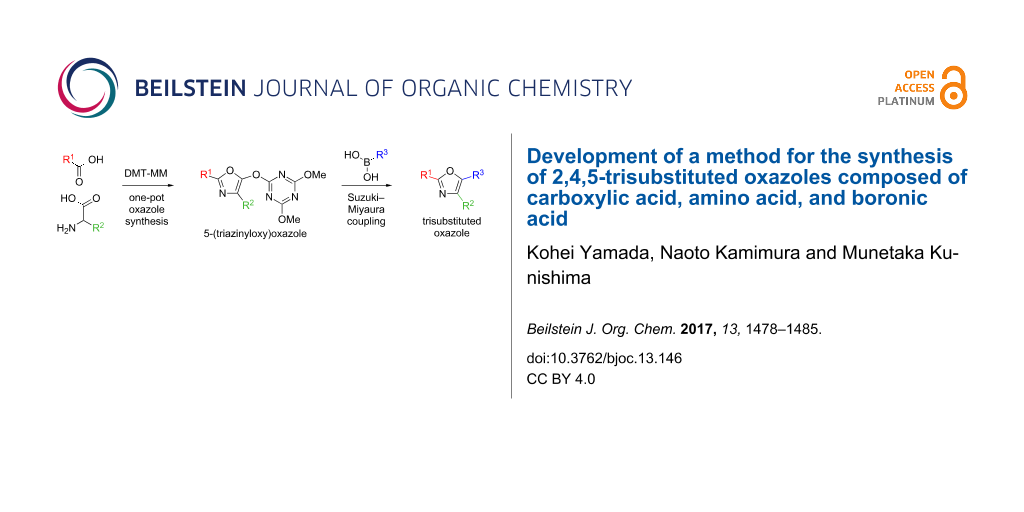
Abstract
A novel method for the synthesis of trisubstituted oxazoles via a one-pot oxazole synthesis/Suzuki–Miyaura coupling sequence has been developed. One-pot formation of 5-(triazinyloxy)oxazoles using carboxylic acids, amino acids and a dehydrative condensing reagent, DMT-MM, followed by Ni-catalyzed Suzuki–Miyaura coupling with boronic acids provided the corresponding 2,4,5-trisubstituted oxazoles in good yields.

Graphical Abstract
Introduction
Oxazoles are found in numerous natural products and are used as a broad range of artificial compounds [1,2]. In particular, 2,4,5-trisubstituted oxazoles attract attention as pharmacologically potent scaffolds because structural diversity can be efficiently generated by the introduction of a variety of substituents. Accordingly, numerous synthetic methods have been developed and can be roughly classified into three synthetic strategies (Scheme 1a).
![[1860-5397-13-146-i1]](/bjoc/content/inline/1860-5397-13-146-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Our strategy for the concise synthesis of 2,4,5-trisubstituted oxazoles.
Scheme 1: Our strategy for the concise synthesis of 2,4,5-trisubstituted oxazoles.
i) The cyclization method: many methods, such as the Robinson–Gabriel oxazole synthesis using α-acylaminoketone [3,4], the Davidson oxazole synthesis with α-acyloxyketone [5], and modifications of these [6,7], have been developed. Moreover, cycloaddition of two starting materials, such as α-haloketones and primary amides [8], alkynes and nitriles [9], amines and α,β-unsaturated carbonyl compounds [10], etc. [11] have been reported. However, these reactions are often conducted under harsh reaction conditions and multistep syntheses of the starting materials are needed. ii) The functionalization method: various regioselective metalations and subsequent functionalizations of the oxazole core skeleton using Cu [12], Pd [13], Mg [14], Zn [14], etc. [15] have been developed. This linear synthetic approach inevitably requires multistep processes and often needs prehalogenation [16]. iii) The multicomponent method: only two strategies have been reported to the best of our knowledge. One is a combination of the Ugi reaction, which uses 2,4-dimethoxybenzylamine, arylglyoxal, carboxylic acid, and isonitrile as components, and a subsequent Robinson–Gabriel reaction [17]. The other is an Au-catalyzed tandem oxazole synthesis using a primary amide, aldehyde, and alkyne [18]. These methods are reasonable for the synthesis of diverse libraries of trisubstituted oxazoles because the combination of three starting materials that are corresponding to the substituents can be readily altered. However, these reactions require strongly acidic conditions or high temperatures. Therefore, a mild method for the synthesis of diverse trisubstituted oxazoles using three commercially available compounds with a wide variety of structures is still desired.
Previously, we reported a one-pot synthesis of oxazolone from carboxylic acids and amino acids using a dehydrative condensing reagent, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM [19-22])[23]. Formation of 5-(triazinyloxy)oxazole is also reported to occur when an excess of DMT-MM was used. Recently, Jin and co-workers reported that Ni-catalyzed Suzuki–Miyaura coupling between triazinyloxybenzene and arylboronic acids affords the corresponding biaryl compounds [24-33]. In this context, we envisioned application of this Suzuki–Miyaura coupling to a 5-(triazinyloxy)oxazole would provide trisubstituted oxazoles (Scheme 1b). Since many kinds of carboxylic acids, amino acids and boronic acids, which are corresponding to 2-, 4-, and 5-substituents of the oxazole, respectively, are commercially available, this method is suitable for the synthesis of a diverse variety of trisubstituted oxazoles. Herein, we described an efficient method for the synthesis of trisubstituted oxazoles through a one-pot oxazole synthesis and subsequent Suzuki–Miyaura coupling.
Results and Discussion
The study was initiated with the preparation of the key intermediate, 5-(triazinyloxy)oxazole 3, from carboxylic acid 1 and amino acid 2 under conditions improved from [23] (Table 1). A one-pot sequence involving formation of an activated ester from benzoic acid (1a) with DMT-MM, N-benzoylation of alanine (2a), cyclodehydration, and introduction of the triazinyl group was conducted in 1,4-dioxane/H2O to give the desired 5-(triazinyloxy)oxazole 3aa in 78% yield (Table 1, entry 1). A series of carboxylic acids were subjected to the reaction conditions. Aromatic carboxylic acids with both electron-withdrawing and electron-donating groups gave good yields (Table 1, entries 2 and 3). In the case of aliphatic carboxylic acids, 3-phenylpropionic acid (1d) gave a slightly decreased amount of 3da in 54% yield (Table 1, entry 4), whereas the more sterically demanding 1e gave the desired product 3ea in a good yield (Table 1, entry 5). The reaction was carried out with different amino acids, resulting in a varied substitution pattern at the 4-position of the oxazole. The one-pot oxazole synthesis with phenylalanine (2b), valine (2c), leucine (2d), methionine (2e), and phenylglycine (2f) proceeded to give the corresponding intermediates in good yields (Table 1, entries 6–10). Despite the existence of highly polar starting materials and relatively more lipophilic activated esters and oxazolone intermediates during the course of the reaction, various 5-(triazinyloxy)oxazoles were uneventfully synthesized under these improved conditions.
Table 1: One-pot synthesis of 5-(triazinyloxy)oxazole under improved conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-146-i3.svg?max-width=637&scale=1.0)
|
|||
| entry | carboxylic acid 1 | amino acid 2 | yield of 3 (%)a |
|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-146-i4.svg?max-width=637&scale=1.0)
1a |
alanine (2a) | 3aa, 78 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-146-i5.svg?max-width=637&scale=1.0)
1b |
2a | 3ba, 69 |
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-146-i6.svg?max-width=637&scale=1.0)
1c |
2a | 3ca, 60 |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-146-i7.svg?max-width=637&scale=1.0)
1d |
2a | 3da, 54 |
| 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-146-i8.svg?max-width=637&scale=1.0)
1e |
2a | 3ea, 71 |
| 6 | 1a | phenylalanine (2b) | 3ab, 78 |
| 7 | 1a | valine (2c) | 3ac, 83 |
| 8 | 1a | leucine (2d) | 3ad, 70 |
| 9 | 1a | methionine (2e) | 3ae, 70 |
| 10 | 1a | phenylglycine (2f) | 3af, 78 |
aIsolated yield.
According to the procedure previously reported [24], we examined Ni-catalyzed Suzuki–Miyaura coupling between 5-(triazinyloxy)oxazole 3aa and phenylboronic acid (4a, Table 2). As expected, the desired trisubstituted oxazole 5aaa was obtained in 21% yield (Table 2, entry 1). The use of different bidentate (dppp) or monodentate (PCy3) phosphines as ligands for the Ni catalyst resulted in poor yields (Table 2, entries 2 and 3). Notably, we found that 3 equiv of LiCl was an effective additive for shortening the reaction time (3 h) and improving the yield (73%, Table 2, entry 4) [34]. Other lithium halides, except for LiF, were also effective (Table 2, entries 5–7). However, utilization of Na+ and K+ as counter cations for the additive provided inferior results (Table 2, entries 8 and 9). Interestingly, switching the counter cation of the base to Li+ did not afford any product (Table 2, entry 10). The reaction with PdCl2(dppf) instead of NiCl2(dppf) reduced the outcome of the reaction (Table 2, entry 11). No product was obtained when boronic acid pinacol ester 6 and borate salt 7 were used as a coupling partner (Table 2, entries 12 and 13) [35]. Dimethoxyethane (DME) and 1,4-dioxane as ethereal solvents did not improve the yields (Table 2, entries 14 and 15). Decreasing the temperature to 80 °C or increasing to 160 °C by microwave irradiation were not effective for improving the reaction (Table 2, entries 16 and 17). Consequently, we found that the reaction shown in entry 4 afforded the optimal result (see Table S1 in Supporting Information File 1 for further manipulation of the reaction conditions).
Table 2: Screening of reaction conditions of Suzuki–Miyaura coupling with 3aa.
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-146-i9.svg?max-width=637&scale=1.0)
|
||||||
| entry | metal cat. | base | additive (3 equiv) | solvent | time (h) | yield of 5aaa (%)a |
|---|---|---|---|---|---|---|
| 1 | NiCl2(dppf) | K3PO4 | – | toluene | 19 | 21 |
| 2 | NiCl2(dppp) | K3PO4 | – | toluene | 12 | 9 |
| 3 | NiCl2(PCy3)2 | K3PO4 | – | toluene | 20 | 12 |
| 4 | NiCl2(dppf) | K3PO4 | LiCl | toluene | 3 | 73 (68%)b |
| 5 | NiCl2(dppf) | K3PO4 | LiF | toluene | 21 | 35 |
| 6 | NiCl2(dppf) | K3PO4 | LiBr | toluene | 3 | 64 |
| 7 | NiCl2(dppf) | K3PO4 | LiI | toluene | 3 | 70 |
| 8 | NiCl2(dppf) | K3PO4 | NaI | toluene | 24 | 0 |
| 9 | NiCl2(dppf) | K3PO4 | KI | toluene | 17 | 51 |
| 10 | NiCl2(dppf) | Li3PO4 | LiCl | toluene | 26 | 0 |
| 11 | PdCl2(dppf) ·CH2Cl2 | K3PO4 | LiCl | toluene | 22 | 0 |
| 12c | NiCl2(dppf) | K3PO4 | LiCl | toluene | 20 | 0 |
| 13d | NiCl2(dppf) | – | LiCl | toluene | 12 | 0 |
| 14 | NiCl2(dppf) | K3PO4 | LiCl | DME | 10 | 0 |
| 15 | NiCl2(dppf) | K3PO4 | LiCl | 1,4-dioxane | 21 | 0 |
| 16 | NiCl2(dppf) | K3PO4 | LiCl | toluene 80 °C | 23 | 36 |
| 17 | NiCl2(dppf) | K3PO4 | LiCl | toluene 160 °C (MW) | 20 min | 30 |
aNMR yield. bIsolated yield. cBoronic acid pinacol ester 6 was used instead of boronic acid 4a. dBorate 7 was used instead of boronic acid 4a.
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-146-i10.svg?max-width=637&scale=1.18182)
A number of trisubstituted oxazoles were synthesized using 5-(triazinyloxy)oxazoles 3 and various boronic acids 4 (Table 3). To our disappointment, the reaction of 3aa with the arylboronic acid possessing an electron-withdrawing group 4b decreased the yield of 5aab (25%, Table 3, entry 1). Further investigation of the reaction conditions revealed that the reaction with additional dppf (5 mol %) in a sealed tube increased the yield to 64% (Table 3, entry 2) [36,37]. These reaction conditions were defined as conditions B, whereas the conditions in Table 2, entry 4 are defined as conditions A. The arylboronic acid with an electron-donating group 4c also provided a better yield under conditions B rather than conditions A (Table 3, entries 3 and 4). The desired naphthyloxazole 5aad was obtained in a high yield (77%, Table 3, entry 5). Introduction of a p-tolyl group afforded a good yield of 71% (Table 3, entry 6), whereas reactions of the o-tolyl group resulted in moderate yields under both conditions owing to the steric effect (Table 3, entries 7 and 8). The reaction with 3-thienylboronic acid (4g) under conditions B proceeded to give 5aag in 68% yield (Table 3, entries 9 and 10). No product was obtained when n-butylboronic acid (4h) was used as an aliphatic boronic acid (Table 3, entry 11). Subsequently, the effect of substituents at the 2-position, which was derived from the carboxylic acids, was tested. Aryl substituents possessing both electron-withdrawing and electron-donating groups proceeded to give the corresponding oxazoles in good yields (Table 3, entries 12 and 13). Aliphatic substituents are innocent in the reaction outcome (Table 3, entries 14 and 15). Suzuki–Miyaura coupling of several intermediates 3ab–3af, which have different substituents at the 4-position, were examined (Table 3, entries 16–20). Compared with 3aa, the yields using these compounds were slightly lower, especially in the case of sterically more hindered 3ac and 3af (Table 3, entries 17 and 20). Thus, Suzuki–Miyaura coupling is affected by steric hindrance from the 4-substituent of oxazoles.
Table 3: Synthesis of various trisubstituted oxazoles.
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-146-i11.svg?max-width=637&scale=1.0)
|
|||||
| entry | 3 | 4 | product 5 | conditions | yield of 5 (%)a |
|---|---|---|---|---|---|
| 1 | 3aa | 4b |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-146-i12.svg?max-width=637&scale=1.0)
5aab |
A | 25 |
| 2 | 3aa | 4b | 5aab | B | 64 |
| 3 | 3aa | 4c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-146-i13.svg?max-width=637&scale=1.0)
5aac |
A | 54 |
| 4 | 3aa | 4c | 5aac | B | 67 |
| 5 | 3aa | 4d |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-146-i14.svg?max-width=637&scale=1.0)
5aad |
A | 77 |
| 6 | 3aa | 4e |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-146-i15.svg?max-width=637&scale=1.0)
5aae |
A | 71 |
| 7 | 3aa | 4f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-146-i16.svg?max-width=637&scale=1.0)
5aaf |
A | 42 |
| 8 | 3aa | 4f | 5aaf | B | 46 |
| 9 | 3aa | 4g |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-146-i17.svg?max-width=637&scale=1.0)
5aag |
A | 30 |
| 10 | 3aa | 4g | 5aag | B | 68 |
| 11 | 3aa | 4h |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-146-i18.svg?max-width=637&scale=1.0)
5aah |
B | 0 |
| 12 | 3ba | 4a |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-146-i19.svg?max-width=637&scale=1.0)
5baa |
A | 69 |
| 13 | 3ca | 4a |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-146-i20.svg?max-width=637&scale=1.0)
5caa |
A | 64 |
| 14 | 3da | 4a |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-146-i21.svg?max-width=637&scale=1.0)
5daa |
A | 68 |
| 15 | 3ea | 4a |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-146-i22.svg?max-width=637&scale=1.0)
5eaa |
A | 73 |
| 16 | 3ab | 4a |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-146-i23.svg?max-width=637&scale=1.0)
5aba |
A | 60 |
| 17 | 3ac | 4a |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-146-i24.svg?max-width=637&scale=1.0)
5aca |
B | 54 |
| 18 | 3ad | 4a |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-146-i25.svg?max-width=637&scale=1.0)
5ada |
B | 62 |
| 19 | 3ae | 4a |
![[Graphic 24]](/bjoc/content/inline/1860-5397-13-146-i26.svg?max-width=637&scale=1.0)
5aea |
A | 62 |
| 20 | 3af | 4a |
![[Graphic 25]](/bjoc/content/inline/1860-5397-13-146-i27.svg?max-width=637&scale=1.0)
5afa |
B | 47 |
aIsolated yield.
It is noteworthy that the synthesis of bis-oxazole intermediate 3fa with highly polar terephthalic acid (1f) and subsequent double coupling reaction with 4a successfully proceeded to give DMPOPOP (5faa), which is used as a liquid scintillator [38], in a good yield (Scheme 2).
![[1860-5397-13-146-i2]](/bjoc/content/inline/1860-5397-13-146-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
We have successfully developed a new synthetic method for 2,4,5-trisubstituted oxazoles comprising of carboxylic acids, amino acids, and boronic acids in a one-pot oxazole synthesis with following Ni-catalyzed Suzuki–Miyaura coupling. The combination of various starting materials, which are commercially available, provided the corresponding 2,4,5-trisubstituted oxazoles in good yields. Furthermore, several functionalities, such as ethoxycarbonyl, formyl, and methylsulfanyl groups, which are sensitive to acids, bases, nucleophiles, electrophiles and oxidants, were able to tolerate these reaction conditions (Table 1, entries 3, 9 and Table 3, entries 2, 13 and 19). Therefore, this method is suitable for the synthesis of numerous oxazoles with diverse functionalities.
Supporting Information
| Supporting Information File 1: General information, Table S1, experimental procedure and characterization data for products, and 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Turchi, I. J.; Dewar, M. J. S. Chem. Rev. 1975, 75, 389–437. doi:10.1021/cr60296a002
Return to citation in text: [1] -
Wipf, P. Chem. Rev. 1995, 95, 2115–2134. doi:10.1021/cr00038a013
Return to citation in text: [1] -
Robinson, R. J. Chem. Soc. 1909, 95, 2167–2174. doi:10.1039/CT9099502167
Return to citation in text: [1] -
Gabriel, S. Ber. Dtsch. Chem. Ges. 1910, 43, 134–138. doi:10.1002/cber.19100430117
Return to citation in text: [1] -
Davidson, D.; Weiss, M.; Jelling, M. J. Org. Chem. 1937, 2, 328–334. doi:10.1021/jo01227a005
Return to citation in text: [1] -
Wipf, P.; Aoyama, Y.; Benedum, T. E. Org. Lett. 2004, 6, 3593–3595. doi:10.1021/ol0485058
Return to citation in text: [1] -
Patil, P. C.; Luzzio, F. A.; Demuth, D. R. Tetrahedron Lett. 2015, 56, 3039–3041. doi:10.1016/j.tetlet.2014.11.014
Return to citation in text: [1] -
Bailey, J. L.; Sudini, R. R. Tetrahedron Lett. 2014, 55, 3674–3677. doi:10.1016/j.tetlet.2014.05.002
Return to citation in text: [1] -
Saito, A.; Taniguchi, A.; Kambara, Y.; Hanzawa, Y. Org. Lett. 2013, 15, 2672–2675. doi:10.1021/ol4009816
Return to citation in text: [1] -
Liu, D.; Yu, J.; Cheng, J. Tetrahedron 2014, 70, 1149–1153. doi:10.1016/j.tet.2013.12.077
Return to citation in text: [1] -
Zhang, L.; Zhao, X. Org. Lett. 2015, 17, 184–186. doi:10.1021/ol5030986
Return to citation in text: [1] -
Yoshizumi, T.; Satoh, T.; Hirano, K.; Matsuo, D.; Orita, A.; Otera, J.; Miura, M. Tetrahedron Lett. 2009, 50, 3273–3276. doi:10.1016/j.tetlet.2009.02.039
Return to citation in text: [1] -
Théveau, L.; Verrier, C.; Lassalas, P.; Martin, T.; Dupas, G.; Querolle, O.; Van Hijfte, L.; Marsais, F.; Hoarau, C. Chem. – Eur. J. 2011, 17, 14450–14463. doi:10.1002/chem.201101615
Return to citation in text: [1] -
Haas, D.; Mosrin, M.; Knochel, P. Org. Lett. 2013, 15, 6162–6165. doi:10.1021/ol403019c
Return to citation in text: [1] [2] -
Amaike, K.; Muto, K.; Yamaguchi, J.; Itami, K. J. Am. Chem. Soc. 2012, 134, 13573–13576. doi:10.1021/ja306062c
Return to citation in text: [1] -
Hodgetts, K. J.; Kershaw, M. T. Org. Lett. 2002, 4, 2905–2907. doi:10.1021/ol0262800
Return to citation in text: [1] -
Shaw, A. Y.; Xu, Z.; Hulme, C. Tetrahedron Lett. 2012, 53, 1998–2000. doi:10.1016/j.tetlet.2012.02.030
Return to citation in text: [1] -
Querard, P.; Girard, S. A.; Uhlig, N.; Li, C.-J. Chem. Sci. 2015, 6, 7332–7335. doi:10.1039/C5SC02933C
Return to citation in text: [1] -
Kunishima, M.; Kawachi, C.; Iwasaki, F.; Terao, K.; Tani, S. Tetrahedron Lett. 1999, 40, 5327–5330. doi:10.1016/S0040-4039(99)00968-5
Return to citation in text: [1] -
Kunishima, M.; Kawachi, C.; Monta, J.; Terao, K.; Iwasaki, F.; Tani, S. Tetrahedron 1999, 55, 13159–13170. doi:10.1016/S0040-4020(99)00809-1
Return to citation in text: [1] -
Kunishima, M.; Kawachi, C.; Hioki, K.; Terao, K.; Tani, S. Tetrahedron 2001, 57, 1551–1558. doi:10.1016/S0040-4020(00)01137-6
Return to citation in text: [1] -
Kitamura, M.; Kunishima, M. 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. In e-EROS (Encyclopedia of Reagents for Organic Synthesis) [Online]; Crich, D.; Charette, A. B.; Fuchs, P. L., Eds.; John Wiley & Sons, Ltd: West Sussex, 2013.
Return to citation in text: [1] -
Fujita, H.; Kunishima, M. Chem. Pharm. Bull. 2012, 60, 907–912. doi:10.1248/cpb.c12-00291
Return to citation in text: [1] [2] -
Li, X.-J.; Zhang, J.-L.; Geng, Y.; Jin, Z. J. Org. Chem. 2013, 78, 5078–5084. doi:10.1021/jo4005537
Return to citation in text: [1] [2] -
Iranpoor, N.; Panahi, F. Adv. Synth. Catal. 2014, 356, 3067–3073. doi:10.1002/adsc.201400460
See for amination.
Return to citation in text: [1] -
Iranpoor, N.; Panahi, F. Org. Lett. 2015, 17, 214–217. doi:10.1021/ol503560e
See for reduction.
Return to citation in text: [1] -
Yu, B.; Sun, H.; Xie, Z.; Zhang, G.; Xu, L.-W.; Zhang, W.; Gao, Z. Org. Lett. 2015, 17, 3298–3301. doi:10.1021/acs.orglett.5b01466
See for Sonogashira coupling.
Return to citation in text: [1] -
Yamada, K.; Fujita, H.; Kunishima, M. Org. Lett. 2012, 14, 5026–5029. doi:10.1021/ol302222p
We have developed acid-catalyzed triazine-type alkylating reagents, in which triazinyloxy group works as a leaving group. See also refs. [29-33].
Return to citation in text: [1] -
Yamada, K.; Yoshida, S.; Fujita, H.; Kitamura, M.; Kunishima, M. Eur. J. Org. Chem. 2015, 36, 7997–8002. doi:10.1002/ejoc.201501172
Return to citation in text: [1] [2] -
Fujita, H.; Hayakawa, N.; Kunishima, M. J. Org. Chem. 2015, 80, 11200–11205. doi:10.1021/acs.joc.5b02059
Return to citation in text: [1] [2] -
Yamada, K.; Fujita, H.; Kitamura, M.; Kunishima, M. Synthesis 2013, 45, 2989–2997. doi:10.1055/s-0033-1339713
Return to citation in text: [1] [2] -
Yamada, K.; Hayakawa, N.; Fujita, H.; Kitamura, M.; Kunishima, M. Eur. J. Org. Chem. 2016, 4093–4098. doi:10.1002/ejoc.201600663
Return to citation in text: [1] [2] -
Yamada, K.; Hayakawa, N.; Fujita, H.; Kitamura, M.; Kunishima, M. Chem. Pharm. Bull. 2017, 65, 112–115. doi:10.1248/cpb.c16-00744
Return to citation in text: [1] [2] -
Scott, W. J.; Crisp, G. T.; Stille, J. K. J. Am. Chem. Soc. 1984, 106, 4630–4632. doi:10.1021/ja00328a063
Return to citation in text: [1] -
Yamamoto, Y.; Takizawa, M.; Yu, X.-Q.; Miyaura, N. Angew. Chem., Int. Ed. 2008, 47, 928–931. doi:10.1002/anie.200704162
Return to citation in text: [1] -
Saito, S.; Oh-tani, S.; Miyaura, N. J. Org. Chem. 1997, 62, 8024–8030. doi:10.1021/jo9707848
Return to citation in text: [1] -
The reaction of 3aa and 4a under conditions B afforded a comparable yield (65%, Table S1, entry 16).
Return to citation in text: [1] -
Nemchenok, I. B.; Babin, V. I.; Brudanin, V. B.; Kochetov, O. I.; Timkin, V. V. Phys. Part. Nucl. Lett. 2011, 8, 129–135. doi:10.1134/S1547477111020099
Return to citation in text: [1]
| 1. | Turchi, I. J.; Dewar, M. J. S. Chem. Rev. 1975, 75, 389–437. doi:10.1021/cr60296a002 |
| 2. | Wipf, P. Chem. Rev. 1995, 95, 2115–2134. doi:10.1021/cr00038a013 |
| 8. | Bailey, J. L.; Sudini, R. R. Tetrahedron Lett. 2014, 55, 3674–3677. doi:10.1016/j.tetlet.2014.05.002 |
| 17. | Shaw, A. Y.; Xu, Z.; Hulme, C. Tetrahedron Lett. 2012, 53, 1998–2000. doi:10.1016/j.tetlet.2012.02.030 |
| 6. | Wipf, P.; Aoyama, Y.; Benedum, T. E. Org. Lett. 2004, 6, 3593–3595. doi:10.1021/ol0485058 |
| 7. | Patil, P. C.; Luzzio, F. A.; Demuth, D. R. Tetrahedron Lett. 2015, 56, 3039–3041. doi:10.1016/j.tetlet.2014.11.014 |
| 18. | Querard, P.; Girard, S. A.; Uhlig, N.; Li, C.-J. Chem. Sci. 2015, 6, 7332–7335. doi:10.1039/C5SC02933C |
| 5. | Davidson, D.; Weiss, M.; Jelling, M. J. Org. Chem. 1937, 2, 328–334. doi:10.1021/jo01227a005 |
| 15. | Amaike, K.; Muto, K.; Yamaguchi, J.; Itami, K. J. Am. Chem. Soc. 2012, 134, 13573–13576. doi:10.1021/ja306062c |
| 3. | Robinson, R. J. Chem. Soc. 1909, 95, 2167–2174. doi:10.1039/CT9099502167 |
| 4. | Gabriel, S. Ber. Dtsch. Chem. Ges. 1910, 43, 134–138. doi:10.1002/cber.19100430117 |
| 16. | Hodgetts, K. J.; Kershaw, M. T. Org. Lett. 2002, 4, 2905–2907. doi:10.1021/ol0262800 |
| 12. | Yoshizumi, T.; Satoh, T.; Hirano, K.; Matsuo, D.; Orita, A.; Otera, J.; Miura, M. Tetrahedron Lett. 2009, 50, 3273–3276. doi:10.1016/j.tetlet.2009.02.039 |
| 14. | Haas, D.; Mosrin, M.; Knochel, P. Org. Lett. 2013, 15, 6162–6165. doi:10.1021/ol403019c |
| 14. | Haas, D.; Mosrin, M.; Knochel, P. Org. Lett. 2013, 15, 6162–6165. doi:10.1021/ol403019c |
| 10. | Liu, D.; Yu, J.; Cheng, J. Tetrahedron 2014, 70, 1149–1153. doi:10.1016/j.tet.2013.12.077 |
| 9. | Saito, A.; Taniguchi, A.; Kambara, Y.; Hanzawa, Y. Org. Lett. 2013, 15, 2672–2675. doi:10.1021/ol4009816 |
| 13. | Théveau, L.; Verrier, C.; Lassalas, P.; Martin, T.; Dupas, G.; Querolle, O.; Van Hijfte, L.; Marsais, F.; Hoarau, C. Chem. – Eur. J. 2011, 17, 14450–14463. doi:10.1002/chem.201101615 |
| 24. | Li, X.-J.; Zhang, J.-L.; Geng, Y.; Jin, Z. J. Org. Chem. 2013, 78, 5078–5084. doi:10.1021/jo4005537 |
| 25. |
Iranpoor, N.; Panahi, F. Adv. Synth. Catal. 2014, 356, 3067–3073. doi:10.1002/adsc.201400460
See for amination. |
| 26. |
Iranpoor, N.; Panahi, F. Org. Lett. 2015, 17, 214–217. doi:10.1021/ol503560e
See for reduction. |
| 27. |
Yu, B.; Sun, H.; Xie, Z.; Zhang, G.; Xu, L.-W.; Zhang, W.; Gao, Z. Org. Lett. 2015, 17, 3298–3301. doi:10.1021/acs.orglett.5b01466
See for Sonogashira coupling. |
| 28. |
Yamada, K.; Fujita, H.; Kunishima, M. Org. Lett. 2012, 14, 5026–5029. doi:10.1021/ol302222p
We have developed acid-catalyzed triazine-type alkylating reagents, in which triazinyloxy group works as a leaving group. See also refs. [29-33]. |
| 29. | Yamada, K.; Yoshida, S.; Fujita, H.; Kitamura, M.; Kunishima, M. Eur. J. Org. Chem. 2015, 36, 7997–8002. doi:10.1002/ejoc.201501172 |
| 30. | Fujita, H.; Hayakawa, N.; Kunishima, M. J. Org. Chem. 2015, 80, 11200–11205. doi:10.1021/acs.joc.5b02059 |
| 31. | Yamada, K.; Fujita, H.; Kitamura, M.; Kunishima, M. Synthesis 2013, 45, 2989–2997. doi:10.1055/s-0033-1339713 |
| 32. | Yamada, K.; Hayakawa, N.; Fujita, H.; Kitamura, M.; Kunishima, M. Eur. J. Org. Chem. 2016, 4093–4098. doi:10.1002/ejoc.201600663 |
| 33. | Yamada, K.; Hayakawa, N.; Fujita, H.; Kitamura, M.; Kunishima, M. Chem. Pharm. Bull. 2017, 65, 112–115. doi:10.1248/cpb.c16-00744 |
| 19. | Kunishima, M.; Kawachi, C.; Iwasaki, F.; Terao, K.; Tani, S. Tetrahedron Lett. 1999, 40, 5327–5330. doi:10.1016/S0040-4039(99)00968-5 |
| 20. | Kunishima, M.; Kawachi, C.; Monta, J.; Terao, K.; Iwasaki, F.; Tani, S. Tetrahedron 1999, 55, 13159–13170. doi:10.1016/S0040-4020(99)00809-1 |
| 21. | Kunishima, M.; Kawachi, C.; Hioki, K.; Terao, K.; Tani, S. Tetrahedron 2001, 57, 1551–1558. doi:10.1016/S0040-4020(00)01137-6 |
| 22. | Kitamura, M.; Kunishima, M. 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. In e-EROS (Encyclopedia of Reagents for Organic Synthesis) [Online]; Crich, D.; Charette, A. B.; Fuchs, P. L., Eds.; John Wiley & Sons, Ltd: West Sussex, 2013. |
| 23. | Fujita, H.; Kunishima, M. Chem. Pharm. Bull. 2012, 60, 907–912. doi:10.1248/cpb.c12-00291 |
| 29. | Yamada, K.; Yoshida, S.; Fujita, H.; Kitamura, M.; Kunishima, M. Eur. J. Org. Chem. 2015, 36, 7997–8002. doi:10.1002/ejoc.201501172 |
| 30. | Fujita, H.; Hayakawa, N.; Kunishima, M. J. Org. Chem. 2015, 80, 11200–11205. doi:10.1021/acs.joc.5b02059 |
| 31. | Yamada, K.; Fujita, H.; Kitamura, M.; Kunishima, M. Synthesis 2013, 45, 2989–2997. doi:10.1055/s-0033-1339713 |
| 32. | Yamada, K.; Hayakawa, N.; Fujita, H.; Kitamura, M.; Kunishima, M. Eur. J. Org. Chem. 2016, 4093–4098. doi:10.1002/ejoc.201600663 |
| 33. | Yamada, K.; Hayakawa, N.; Fujita, H.; Kitamura, M.; Kunishima, M. Chem. Pharm. Bull. 2017, 65, 112–115. doi:10.1248/cpb.c16-00744 |
| 36. | Saito, S.; Oh-tani, S.; Miyaura, N. J. Org. Chem. 1997, 62, 8024–8030. doi:10.1021/jo9707848 |
| 37. | The reaction of 3aa and 4a under conditions B afforded a comparable yield (65%, Table S1, entry 16). |
| 38. | Nemchenok, I. B.; Babin, V. I.; Brudanin, V. B.; Kochetov, O. I.; Timkin, V. V. Phys. Part. Nucl. Lett. 2011, 8, 129–135. doi:10.1134/S1547477111020099 |
| 34. | Scott, W. J.; Crisp, G. T.; Stille, J. K. J. Am. Chem. Soc. 1984, 106, 4630–4632. doi:10.1021/ja00328a063 |
| 35. | Yamamoto, Y.; Takizawa, M.; Yu, X.-Q.; Miyaura, N. Angew. Chem., Int. Ed. 2008, 47, 928–931. doi:10.1002/anie.200704162 |
| 23. | Fujita, H.; Kunishima, M. Chem. Pharm. Bull. 2012, 60, 907–912. doi:10.1248/cpb.c12-00291 |
| 24. | Li, X.-J.; Zhang, J.-L.; Geng, Y.; Jin, Z. J. Org. Chem. 2013, 78, 5078–5084. doi:10.1021/jo4005537 |
© 2017 Yamada et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)