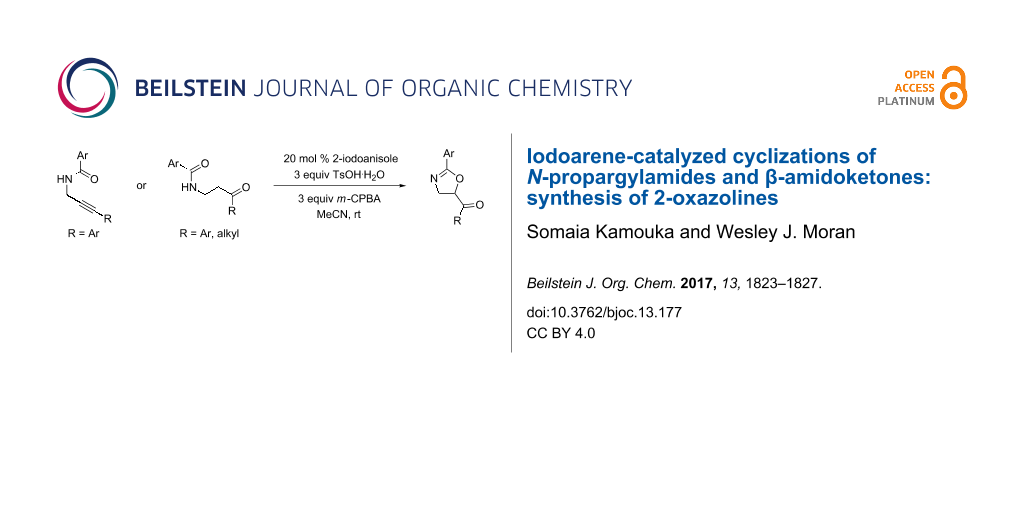
Abstract
Two complementary iodoarene-catalyzed methods for the preparation of 2-oxazolines are presented. The first involves the cyclization of N-propargylamides and the second involves the cyclization of β-amidoketones. These are proposed to proceed through different mechanisms and have different substrate scopes.

Graphical Abstract
Introduction
Hypervalent iodine reagents are of increasing importance in organic synthesis owing to their ease-of-use, low toxicity and relative low cost. Importantly, a wide range of useful reactivity has been uncovered with these compounds and many reviews are available [1-5]. One major advance in recent years is the emergence of conditions to effect catalytic processes with sub-stoichiometric quantities of iodine compound in the presence of an oxidant [6-11].
In this regard, we have reported the use of iodoarenes as precatalysts in the cyclizations of N-alkenylamides 1 [12], δ-alkynyl β-ketoesters 2 [13] and 5-oxo-5-phenylpentanoic acid (3, Scheme 1a–c) [14]. These three cyclizations exemplify three different proposed reaction pathways, i.e., iodine(III) activation of alkenes, alkynes and ketones. These cyclizations can be rendered enantioselective by the generation of non-racemic chiral iodine(III) species from chiral iodoarenes [15-17].
![[1860-5397-13-177-i1]](/bjoc/content/inline/1860-5397-13-177-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Our previous and current iodoarene-catalyzed cyclizations.
Scheme 1: Our previous and current iodoarene-catalyzed cyclizations.
We wished to develop this cyclization methodology further and investigate the cyclization of the amide functional group on to alkynes and methylene groups adjacent to ketones in analogy to our previous work (Scheme 1d). This would provide two complementary routes to substituted 2-oxazolines, which are valuable heterocycles found in ligand scaffolds, natural products such as the leupyrrins [18,19], and potential pharmaceuticals (Figure 1) [20-22]. Traditional routes to this heterocycle include the dehydration of amino alcohols with carboxylic acids, however, this process typically requires forcing conditions such as heating at over 200 °C [23]. Several related processes that operate under milder conditions have been reported in recent years but they suffer from issues such as limited substrate scope or the requirement for expensive reagents or transition metal salts [24-29]. Saito and co-workers have reported the cyclization of propargylamides to form oxazoles rather than oxazolines under stoichiometric and, more recently, catalytic hypervalent iodine conditions [30,31]. Various other methods for the cyclization of unsaturated amides have been reported [32-36]. Gao and co-workers described the I2-catalyzed cyclization of β-acylaminoketones using tert-butyl hydroperoxide as the oxidant; notably, adding DBU led to oxazole formation whereas adding K2CO3 generated oxazolines [37,38]. Our proposed new approaches to oxazoline formation utilise readily available starting materials and operate under mild organocatalytic conditions.
![[1860-5397-13-177-1]](/bjoc/content/figures/1860-5397-13-177-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of biologically-active compounds containing an oxazoline ring.
Figure 1: Examples of biologically-active compounds containing an oxazoline ring.
Results and Discussion
We initiated our study with readily available alkyne 4a and subjected it to reaction conditions similar to those we have previously reported (Table 1). By using 2-iodoanisole as precatalyst in the presence of p-toluenesulfonic acid and m-chloroperbenzoic acid in acetonitrile, 4a cyclized to 6a in 92% yield as determined by 1H NMR analysis of the crude reaction mixture (Table 1, entry 1). In line with our previous findings with N-alkenylamides, the use of iodobenzene in place of 2-iodoanisole provided a diminished yield of 6a (Table 1, entry 2) [12]. The iodoarene was found to be essential for the conversion of the starting material, as its absence led to complete return of 4a (Table 1, entry 3). Using Oxone as oxidant led to essentially no conversion of 4a (Table 1, entry 4). Changing the acid to TFA or changing the solvent to CH2Cl2 led to significantly lower yields of 6a (Table 1, entries 5 and 6). Reducing the number of equivalents of oxidant and/or acid also led to lower yields (Table 1, entries 7–9). Importantly, formation of the six-membered ring was not observed under any conditions studied.
Table 1: Investigation of reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-177-i8.svg?max-width=637&scale=1.0)
|
||
| Entry | Deviations from the standard conditions | Yield [%]a |
| 1 | none | 92 (73)b |
| 2 | iodobenzene instead of 2-iodoanisole | 60 |
| 3 | no 2-iodoanisole | 0 |
| 4 | Oxone instead of m-CPBA | <5 |
| 5 | TFA instead of TsOH·H2O | 19 |
| 6 | CH2Cl2 instead of MeCN | 37 |
| 7 | 2 equiv m-CPBA and 2 equiv TsOH·H2O | 54 |
| 8 | 1 equiv m-CPBA and 1 equiv TsOH·H2O | 44 |
| 9 | 3 equiv m-CPBA and 1 equiv TsOH·H2O | 41 |
aDetermined by NMR analysis by comparison to a known quantity of 1,3,5-trimethoxybenzene. bYield of isolated compound.
With the optimal cyclization conditions in hand, the scope of this cyclization process was investigated for a range of propargylamides 4 which are readily accessible from propargylamine by amidation and Sonogoshira coupling (Scheme 2) [39]. The cyclization was successful in all cases studied with various arylamide and alkyne substituents. All functional groups were well tolerated apart from an alkyne terminated with an alkyl substituted arene which led to a diminished yield of product, i.e., 6g.
![[1860-5397-13-177-i2]](/bjoc/content/inline/1860-5397-13-177-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: 2-Iodoanisole-catalyzed cyclization of N-propargylamides.
Scheme 2: 2-Iodoanisole-catalyzed cyclization of N-propargylamides.
The mechanism of this cyclization is proposed to proceed though activation of the alkyne by an in situ generated iodine(III) species followed by intramolecular attack by the amide (Scheme 3). Subsequent addition of water leads to the loss of the iodoarene and tautomerization of the resulting enol generates the ketone 6.
![[1860-5397-13-177-i3]](/bjoc/content/inline/1860-5397-13-177-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Postulated mechanism for N-propargylamide cyclization.
Scheme 3: Postulated mechanism for N-propargylamide cyclization.
With these results in hand, we envisaged an alternative approach to 2-oxazoline formation through the iodoarene-catalyzed cyclization of β-amidoketones 5. These are readily prepared by alkylation of the corresponding β-ketoester followed by decarboxylation (Scheme 4) [40,41].
![[1860-5397-13-177-i4]](/bjoc/content/inline/1860-5397-13-177-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The cyclization of β-amidoketones 5 was successful with the same conditions as propargylamides 4 (Scheme 5). In line with the results for the propargylamides, iodobenzene was an inferior pre-catalyst to 2-iodoanisole and other oxidants, acids and solvents led to lower yields of 6.
![[1860-5397-13-177-i5]](/bjoc/content/inline/1860-5397-13-177-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: 2-Iodoanisole-catalyzed cyclization of β-amidoketones 5.
Scheme 5: 2-Iodoanisole-catalyzed cyclization of β-amidoketones 5.
The scope of this cyclization process was explored and different aromatic amide and ketone groups were well tolerated. Alkylketone substrates were also successfully converted to 2-oxazolines. Installation of substituents on the tether led to facile formation of product 6m containing a quaternary carbon and compounds 6n and 6o but without any observed diastereoselectivity. Interestingly, the selectivity for 6o could be improved to 5:1 by substituting p-toluenesulfonic acid with trifluoroacetic acid albeit with a loss of yield.
When the p-nitrophenylamide 5p was subjected to the reaction conditions, the expected oxazoline 6p was not observed (Scheme 6). Instead, alcohol 8 was isolated in 66% yield. Presumably, 6p is formed under the reaction conditions but the oxazoline ring is readily hydrolysed due to the influence of the electron-withdrawing nitro group on the aromatic ring.
![[1860-5397-13-177-i6]](/bjoc/content/inline/1860-5397-13-177-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: In situ oxazoline ring hydrolysis.
Scheme 6: In situ oxazoline ring hydrolysis.
The mechanism of this cyclization is proposed to proceed through the formation of iodine(III)-enolate 9 followed by intramolecular attack by the amide and release of the iodoarene (Scheme 7).
![[1860-5397-13-177-i7]](/bjoc/content/inline/1860-5397-13-177-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Postulated mechanism for cyclization of β-amidoketones.
Scheme 7: Postulated mechanism for cyclization of β-amidoketones.
These two cyclizations are complementary, however, the reaction with β-amidoketones exhibits a superior substrate scope. In addition, the use of a chiral iodoarene should lead to enantioselective cyclizations of β-amidoketones; this is not possible with the propargylamides.
Conclusion
Two simple and convenient iodoarene-catalyzed methods to prepare substituted 2-oxazolines are reported. One involves the cyclization of propargylamides and the second of β-amidoketones. These two complementary procedures are efficient and showcase the utility of hypervalent iodine in catalytic procedures. Studies concerning the elucidation of reaction mechanisms are ongoing and will be reported in due course.
Experimental
General procedure for 2-iodoanisole-catalyzed cyclization of N-propargylamide 4 or β-amidoketone 5: Propargylamide 4 (1 equiv) or β-amidoketone 5 (1 equiv) was dissolved in acetonitrile (0.14 M) and 2-iodoanisole (0.2 equiv) was added, followed by m-CBPA (3 equiv) and p-TsOH·H2O (3 equiv). The mixture was stirred overnight at room temperature, then saturated aqueous Na2S2O3 solution and saturated aqueous NaHCO3 solution were added and the mixture extracted with CH2Cl2. The organic layers were combined and dried with MgSO4, filtered and concentrated under vacuum. The product was purified by flash chromatography (9:1 petroleum ether/EtOAc) to provide oxazoline 6. See Supporting Information File 1 for full experimental details.
Supporting Information
| Supporting Information File 1: Full experimental details, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 4.7 MB | Download |
References
-
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Zheng, Z.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Sci. China: Chem. 2014, 57, 189. doi:10.1007/s11426-013-5043-1
Return to citation in text: [1] -
Zhdankin, V. V. Hypervalent Iodine Chemistry; Wiley: Chichester, 2014.
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299. doi:10.1021/cr800332c
Return to citation in text: [1] -
Brown, M.; Farid, U.; Wirth, T. Synlett 2013, 24, 424. doi:10.1055/s-0032-1318103
Return to citation in text: [1] -
Yusubov, M. S.; Zhdankin, V. V. Resour.-Effic. Technol. 2015, 1, 49. doi:10.1016/j.reffit.2015.06.001
Return to citation in text: [1] -
Dohi, T.; Kita, Y. Chem. Commun. 2009, 2073. doi:10.1039/b821747e
Return to citation in text: [1] -
Richardson, R. D.; Wirth, T. Angew. Chem., Int. Ed. 2006, 45, 4402. doi:10.1002/anie.200601817
Return to citation in text: [1] -
Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. J. Am. Chem. Soc. 2005, 127, 12244. doi:10.1021/ja0542800
Return to citation in text: [1] -
Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950. doi:10.1002/asia.201301582
Return to citation in text: [1] -
Romero, R. M.; Wöste, T. H.; Muñiz, K. Chem. – Asian J. 2014, 9, 972. doi:10.1002/asia.201301637
Return to citation in text: [1] -
Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453. doi:10.1021/acs.orglett.5b00333
Return to citation in text: [1] [2] -
Rodríguez, A.; Moran, W. J. Org. Lett. 2011, 13, 2220. doi:10.1021/ol200471w
Return to citation in text: [1] -
Rodríguez, A.; Moran, W. J. Synthesis 2012, 44, 1178. doi:10.1055/s-0031-1290590
Return to citation in text: [1] -
Berthiol, F. Synthesis 2015, 47, 587. doi:10.1055/s-0034-1379892
Return to citation in text: [1] -
Parra, A.; Reboredo, S. Chem. – Eur. J. 2013, 19, 17244. doi:10.1002/chem.201302220
Return to citation in text: [1] -
Liang, H.; Ciufolini, M. A. Angew. Chem., Int. Ed. 2011, 50, 11849. doi:10.1002/anie.201106127
Return to citation in text: [1] -
Herkommer, D.; Thiede, S.; Wosniok, P. R.; Dreisigacker, S.; Tian, M.; Debnar, T.; Irschik, H.; Menche, D. J. Am. Chem. Soc. 2015, 137, 4086. doi:10.1021/jacs.5b01894
Return to citation in text: [1] -
Bode, H. B.; Irschik, H.; Wenzel, S. C.; Reichenbach, H.; Müller, R.; Höfle, G. J. Nat. Prod. 2003, 66, 1203. doi:10.1021/np030109v
Return to citation in text: [1] -
Célanire, S.; Wijtmans, M.; Christophe, B.; Collart, P.; de Esch, I.; Dassesse, D.; Delaunoy, C.; Denonne, F.; Durieu, V.; Gelens, E.; Gillard, M.; Lallemand, B.; Lamberty, Y.; Lebon, F.; Nicolas, J.-M.; Quéré, L.; Snip, E.; Vanbellinghen, A.; Van houtvin, N.; Verbois, V.; Timmerman, H.; Talaga, P.; Leurs, R.; Provins, L. ChemMedChem 2009, 4, 1063. doi:10.1002/cmdc.200900055
Return to citation in text: [1] -
Celanire, S.; Talaga, P.; Leurs, R.; Denonne, F.; Timmerman, H.; Lebon, F. Compounds comprising an oxazoline or thiazoline moiety, processes for making them, and their uses. WO Patent WO2006103057 A1, Oct 5, 2006.
Return to citation in text: [1] -
Onishi, H. R.; Pelak, B. A.; Gerckens, L. S.; Silver, L. L.; Kahan, F. M.; Chen, M.-H.; Patchett, A. A.; Galloway, S. M.; Hyland, S. A.; Anderson, M. S.; Raetz, C. R. H. Science 1996, 274, 980. doi:10.1126/science.274.5289.980
Return to citation in text: [1] -
Ilkgul, B.; Gunes, D.; Sirkecioglu, O.; Bicak, N. Tetrahedron Lett. 2010, 51, 5313. doi:10.1016/j.tetlet.2010.07.167
Return to citation in text: [1] -
Brandstätter, M.; Roth, F.; Luedtke, N. W. J. Org. Chem. 2015, 80, 40. doi:10.1021/jo5016695
Return to citation in text: [1] -
Garg, P.; Chaudhary, S.; Milton, M. D. J. Org. Chem. 2014, 79, 8668. doi:10.1021/jo501430p
Return to citation in text: [1] -
Goud, D. R.; Pathak, U. Synthesis 2012, 44, 3678. doi:10.1055/s-0032-1317341
Return to citation in text: [1] -
Takahashi, S.; Togo, H. Synthesis 2009, 2329. doi:10.1055/s-0029-1216843
Return to citation in text: [1] -
Sayama, S. Synlett 2006, 1479. doi:10.1055/s-2006-941597
Return to citation in text: [1] -
Schwekendiek, K.; Glorius, F. Synthesis 2006, 2996. doi:10.1055/s-2006-950198
Return to citation in text: [1] -
Asari, N.; Takemoto, Y.; Shinomoto, Y.; Yagyu, T.; Yoshimura, A.; Zhdankin, V. V.; Saito, A. Asian J. Org. Chem. 2016, 5, 1314. doi:10.1002/ajoc.201600383
Return to citation in text: [1] -
Saito, A.; Matsumoto, A.; Hanzawa, Y. Tetrahedron Lett. 2010, 51, 2247. doi:10.1016/j.tetlet.2010.02.096
Return to citation in text: [1] -
Senadi, G. C.; Guo, B.-C.; Hu, W.-P.; Wang, J.-J. Chem. Commun. 2016, 52, 11410. doi:10.1039/C6CC05138C
Return to citation in text: [1] -
Liu, G.-Q.; Yang, C.-H.; Li, Y.-M. J. Org. Chem. 2015, 80, 11339. doi:10.1021/acs.joc.5b01832
Return to citation in text: [1] -
Wang, N.; Chen, B.; Ma, S. Adv. Synth. Catal. 2014, 356, 485. doi:10.1002/adsc.201300959
Return to citation in text: [1] -
Senadi, G. C.; Hu, W.-P.; Hsiao, J.-S.; Vandavasi, J. K.; Chen, C.-Y.; Wang, J.-J. Org. Lett. 2012, 14, 4478. doi:10.1021/ol301980g
Return to citation in text: [1] -
Hashmi, A. S. K.; Blanco Jaimes, M. C.; Schuster, A. M.; Rominger, F. J. Org. Chem. 2012, 77, 6394. doi:10.1021/jo301288w
Return to citation in text: [1] -
Minakata, S.; Morino, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2006, 8, 3335. doi:10.1021/ol061182q
Return to citation in text: [1] -
Gao, W.-C.; Hu, F.; Huo, Y.-M.; Chang, H.-H.; Li, X.; Wei, W.-L. Org. Lett. 2015, 17, 3914. doi:10.1021/acs.orglett.5b01933
Return to citation in text: [1] -
Bukšnaitienė, R.; Čikotienė, I. Synlett 2015, 26, 479. doi:10.1055/s-0034-1379320
Return to citation in text: [1] -
Chao, M.; Hao, A.; Wang, H. Org. Process Res. Dev. 2009, 13, 645. doi:10.1021/op800313t
Return to citation in text: [1] -
Kaku, H.; Imai, T.; Kondo, R.; Mamba, S.; Watanabe, Y.; Inai, M.; Nishii, T.; Horikawa, M.; Tsunoda, T. Eur. J. Org. Chem. 2013, 8208. doi:10.1002/ejoc.201300936
Return to citation in text: [1]
| 1. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328. doi:10.1021/acs.chemrev.5b00547 |
| 2. | Zheng, Z.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Sci. China: Chem. 2014, 57, 189. doi:10.1007/s11426-013-5043-1 |
| 3. | Zhdankin, V. V. Hypervalent Iodine Chemistry; Wiley: Chichester, 2014. |
| 4. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299. doi:10.1021/cr800332c |
| 5. | Brown, M.; Farid, U.; Wirth, T. Synlett 2013, 24, 424. doi:10.1055/s-0032-1318103 |
| 14. | Rodríguez, A.; Moran, W. J. Synthesis 2012, 44, 1178. doi:10.1055/s-0031-1290590 |
| 39. | Bukšnaitienė, R.; Čikotienė, I. Synlett 2015, 26, 479. doi:10.1055/s-0034-1379320 |
| 40. | Chao, M.; Hao, A.; Wang, H. Org. Process Res. Dev. 2009, 13, 645. doi:10.1021/op800313t |
| 41. | Kaku, H.; Imai, T.; Kondo, R.; Mamba, S.; Watanabe, Y.; Inai, M.; Nishii, T.; Horikawa, M.; Tsunoda, T. Eur. J. Org. Chem. 2013, 8208. doi:10.1002/ejoc.201300936 |
| 12. | Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453. doi:10.1021/acs.orglett.5b00333 |
| 37. | Minakata, S.; Morino, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2006, 8, 3335. doi:10.1021/ol061182q |
| 38. | Gao, W.-C.; Hu, F.; Huo, Y.-M.; Chang, H.-H.; Li, X.; Wei, W.-L. Org. Lett. 2015, 17, 3914. doi:10.1021/acs.orglett.5b01933 |
| 6. | Yusubov, M. S.; Zhdankin, V. V. Resour.-Effic. Technol. 2015, 1, 49. doi:10.1016/j.reffit.2015.06.001 |
| 7. | Dohi, T.; Kita, Y. Chem. Commun. 2009, 2073. doi:10.1039/b821747e |
| 8. | Richardson, R. D.; Wirth, T. Angew. Chem., Int. Ed. 2006, 45, 4402. doi:10.1002/anie.200601817 |
| 9. | Ochiai, M.; Takeuchi, Y.; Katayama, T.; Sueda, T.; Miyamoto, K. J. Am. Chem. Soc. 2005, 127, 12244. doi:10.1021/ja0542800 |
| 10. | Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950. doi:10.1002/asia.201301582 |
| 11. | Romero, R. M.; Wöste, T. H.; Muñiz, K. Chem. – Asian J. 2014, 9, 972. doi:10.1002/asia.201301637 |
| 12. | Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453. doi:10.1021/acs.orglett.5b00333 |
| 23. | Ilkgul, B.; Gunes, D.; Sirkecioglu, O.; Bicak, N. Tetrahedron Lett. 2010, 51, 5313. doi:10.1016/j.tetlet.2010.07.167 |
| 30. | Asari, N.; Takemoto, Y.; Shinomoto, Y.; Yagyu, T.; Yoshimura, A.; Zhdankin, V. V.; Saito, A. Asian J. Org. Chem. 2016, 5, 1314. doi:10.1002/ajoc.201600383 |
| 31. | Saito, A.; Matsumoto, A.; Hanzawa, Y. Tetrahedron Lett. 2010, 51, 2247. doi:10.1016/j.tetlet.2010.02.096 |
| 20. | Célanire, S.; Wijtmans, M.; Christophe, B.; Collart, P.; de Esch, I.; Dassesse, D.; Delaunoy, C.; Denonne, F.; Durieu, V.; Gelens, E.; Gillard, M.; Lallemand, B.; Lamberty, Y.; Lebon, F.; Nicolas, J.-M.; Quéré, L.; Snip, E.; Vanbellinghen, A.; Van houtvin, N.; Verbois, V.; Timmerman, H.; Talaga, P.; Leurs, R.; Provins, L. ChemMedChem 2009, 4, 1063. doi:10.1002/cmdc.200900055 |
| 21. | Celanire, S.; Talaga, P.; Leurs, R.; Denonne, F.; Timmerman, H.; Lebon, F. Compounds comprising an oxazoline or thiazoline moiety, processes for making them, and their uses. WO Patent WO2006103057 A1, Oct 5, 2006. |
| 22. | Onishi, H. R.; Pelak, B. A.; Gerckens, L. S.; Silver, L. L.; Kahan, F. M.; Chen, M.-H.; Patchett, A. A.; Galloway, S. M.; Hyland, S. A.; Anderson, M. S.; Raetz, C. R. H. Science 1996, 274, 980. doi:10.1126/science.274.5289.980 |
| 32. | Senadi, G. C.; Guo, B.-C.; Hu, W.-P.; Wang, J.-J. Chem. Commun. 2016, 52, 11410. doi:10.1039/C6CC05138C |
| 33. | Liu, G.-Q.; Yang, C.-H.; Li, Y.-M. J. Org. Chem. 2015, 80, 11339. doi:10.1021/acs.joc.5b01832 |
| 34. | Wang, N.; Chen, B.; Ma, S. Adv. Synth. Catal. 2014, 356, 485. doi:10.1002/adsc.201300959 |
| 35. | Senadi, G. C.; Hu, W.-P.; Hsiao, J.-S.; Vandavasi, J. K.; Chen, C.-Y.; Wang, J.-J. Org. Lett. 2012, 14, 4478. doi:10.1021/ol301980g |
| 36. | Hashmi, A. S. K.; Blanco Jaimes, M. C.; Schuster, A. M.; Rominger, F. J. Org. Chem. 2012, 77, 6394. doi:10.1021/jo301288w |
| 18. | Herkommer, D.; Thiede, S.; Wosniok, P. R.; Dreisigacker, S.; Tian, M.; Debnar, T.; Irschik, H.; Menche, D. J. Am. Chem. Soc. 2015, 137, 4086. doi:10.1021/jacs.5b01894 |
| 19. | Bode, H. B.; Irschik, H.; Wenzel, S. C.; Reichenbach, H.; Müller, R.; Höfle, G. J. Nat. Prod. 2003, 66, 1203. doi:10.1021/np030109v |
| 15. | Berthiol, F. Synthesis 2015, 47, 587. doi:10.1055/s-0034-1379892 |
| 16. | Parra, A.; Reboredo, S. Chem. – Eur. J. 2013, 19, 17244. doi:10.1002/chem.201302220 |
| 17. | Liang, H.; Ciufolini, M. A. Angew. Chem., Int. Ed. 2011, 50, 11849. doi:10.1002/anie.201106127 |
| 24. | Brandstätter, M.; Roth, F.; Luedtke, N. W. J. Org. Chem. 2015, 80, 40. doi:10.1021/jo5016695 |
| 25. | Garg, P.; Chaudhary, S.; Milton, M. D. J. Org. Chem. 2014, 79, 8668. doi:10.1021/jo501430p |
| 26. | Goud, D. R.; Pathak, U. Synthesis 2012, 44, 3678. doi:10.1055/s-0032-1317341 |
| 27. | Takahashi, S.; Togo, H. Synthesis 2009, 2329. doi:10.1055/s-0029-1216843 |
| 28. | Sayama, S. Synlett 2006, 1479. doi:10.1055/s-2006-941597 |
| 29. | Schwekendiek, K.; Glorius, F. Synthesis 2006, 2996. doi:10.1055/s-2006-950198 |
© 2017 Kamouka and Moran; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)