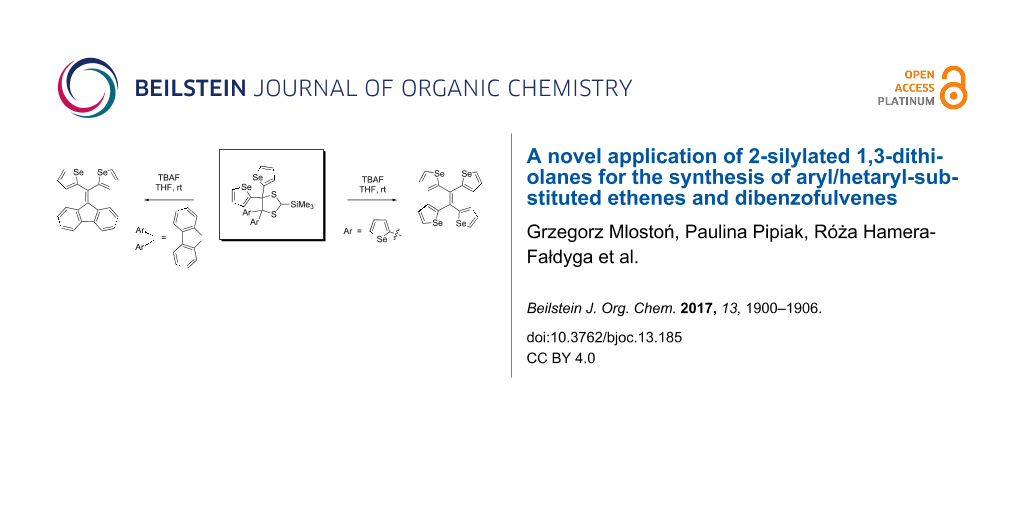
Abstract
Trimethylsilyldiazomethane (TMS-CHN2) reacts readily with hetaryl thioketones to give sterically crowded 2-trimethylsilyl-4,4,5,5-tetrahetaryl-1,3-dithiolanes with complete regioselectivity at −75 °C as well as at rt. Thiofluorenone, a relatively stable and highly reactive aryl thioketone, yields upon treatment with TMS-CHN2 at −60 °C the corresponding 1,3,4-thiadiazoline. This unstable cycloadduct undergoes decomposition at ca. −45 °C and the silylated thiocarbonyl S-methanide generated thereby is trapped with complete regioselectivity by aryl or hetaryl thioketones forming also sterically crowded 2-trimethylsilyl-1,3-dithiolanes. The obtained 1,3-dithiolanes, by treatment with an equimolar amount of TBAF in a one-pot procedure, are converted in high yields into hetaryl/aryl-substituted ethenes or dibenzofulvenes, respectively, via a cycloreversion reaction of the intermediate 1,3-dithiolane carbanion. The presented protocol offers a new, highly efficient approach to tetrasubstituted ethenes and dibenzofulvenes bearing aryl and/or hetaryl substituents.

Graphical Abstract
Introduction
Aryl and hetaryl-substituted ethenes form an important class of organic compounds with a growing number of applications in materials chemistry, crystal engineering, photooptics, etc. Among these, the thiophen-2-yl-substituted ethenes [1-5] as well as dibenzofulvenes-containing compounds [6-9] are of special interest. The development of methods for the synthesis of substituted ethenes is of great importance and the so-called olefination reactions allow for valuable functional-group transformations [10-13]. As a general method for the preparation of tetrasubstituted ethenes, the McMurry reaction is widely applied [14]. Another approach, which opens access to diverse ethenes, is the ‘two-fold extrusion reaction’, which comprises the [3 + 2]-cycloaddition of a diazo compound with a thiocarbonyl dipolarophile and subsequent elimination of N2 followed by sulfur extrusion [15-17].
In our continuing studies on [3 + 2]-cycloadditions with thioketones and diazo compounds, we turned our attention to hetaryl thioketones [18]. It turned out that the presence of the hetaryl groups strongly influences the reactivity of these dipolarophiles in reactions with diazomethane (CH2N2, Schönberg reaction) [19-21]. For example, in contrast to thiobenzophenone (1a), phenyl selenophen-2-yl thioketone (1b) does not form the corresponding 2,5-dihydro-1,3,4-thiadiazole of type 2, and even at −70 °C spontaneous elimination of N2 was observed. As products of this reaction, dimer 4 of the intermediate thiocarbonyl S-methanide 3 and the sterically crowded 4,4,5,5-tetraaryl-1,3-dithiolane 5 were obtained (Scheme 1) [20]. The formation of both products was rationalized by the assumption that the in situ formed 3a reacts as a delocalized diradical species.
![[1860-5397-13-185-i1]](/bjoc/content/inline/1860-5397-13-185-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of diphenyl and phenyl selenophen-2-yl thioketones with diazomethane (CH2N2; Sel = selenophen-2-yl).
Scheme 1: Reactions of diphenyl and phenyl selenophen-2-yl thioketones with diazomethane (CH2N2; Sel = seleno...
In a recent publication, similar reactions of TMS-CHN2 with 1a and some dihetaryl thioketones, e.g., 1d and 1e, were reported [22]. In contrast to CH2N2, TMS-CHN2 reacted with thiobenzophenone (1a) with evolution of N2 even at −75 °C and led to a mixture of 4,4,5,5-tetraphenyl-1,3-dithiolane 6a and 2,2,3,3-tetraphenyl-1,4-dithiane 7 (Scheme 2). This result demonstrated that TMS-CHN2 does not form a stable [3 + 2]-cycloadduct with 1a, but, in analogy to 1b in the reaction with CH2N2, spontaneous elimination of N2 takes place. In another study, we described a different behaviour of thiofluorenone (1c), which reacted with TMS-CHN2 at −60 °C to yield the expected [3 + 2]-cycloadduct 8, which only at ca. −40 °C extruded N2 [23].
![[1860-5397-13-185-i2]](/bjoc/content/inline/1860-5397-13-185-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of diaryl thioketones with trimethylsilyldiazomethane (TMS-CHN2).
Scheme 2: Reaction of diaryl thioketones with trimethylsilyldiazomethane (TMS-CHN2).
The goal of the present study was the preparation of a series of 2-trimethylsilyl-1,3-dithiolanes of type 6, which, after desilylation, should be applied for nucleophilic additions of the 1,3-dithiolane anion with some electrophilic agents. An analogous sequence of reactions was described earlier for 2-aryl-2-trimethylsilyl-1,3-dithiolanes [24,25].
Results and Discussion
The earlier described protocol for the reaction of thiobenzophenone (1a) with TMS-CHN2 [22] has been slightly modified and the reaction was performed at −70 °C using the reagents in a ratio of 3:1. Under these conditions, 1,3-dithiolane 6a was formed almost exclusively with only traces of 1,4-dithiane 7 as revealed by 1H NMR analysis of the crude reaction mixture. Desilylation of 6a occurred quantitatively and the known tetraphenylethylene (9a) [26] was obtained in 90% yield.
Two symmetric dihetaryl thioketones, 1d and 1e, were reacted with TMS-CHN2 in THF at −75 °C, and after the spontaneous elimination of N2, the sterically crowded 4,4,5,5-tetrahetaryl-1,3-dithiolanes 6b and 6c, respectively, were obtained as exclusive products in high yields [22]. The isolated pure products were treated with equimolar amounts of TBAF in THF at room temperature and the progress of the reaction was monitored by TLC. After ca. 1 h, the crude reaction mixtures obtained after aqueous work-up were analysed by 1H NMR spectroscopy. Unexpectedly, in both cases, only signals of hetaryl rings were observed. The 13C NMR spectra allowed to identify both products as tetrasubstituted ethenes 9b,c (Scheme 3, Table 1). The signals for the ethene C=C atoms appeared at 127.7 and 131.2 ppm, respectively. In addition, the melting point determined for 9b confirmed its identity with the known tetrakis(thiophen-2-yl)ethene [1].
![[1860-5397-13-185-i3]](/bjoc/content/inline/1860-5397-13-185-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of tetraaryl/hetarylethenes 9 from the reaction of TMS-CHN2 with diaryl/hetaryl thioketones 1 (for Ar1, Ar2 see Table 1).
Scheme 3: Formation of tetraaryl/hetarylethenes 9 from the reaction of TMS-CHN2 with diaryl/hetaryl thioketon...
The same products 9b,c were obtained in reactions at 0 °C in comparable yields, when the initially formed 1,3-dithiolanes 6b,c, without isolation, were treated with equimolar amounts of TBAF. Using this protocol, the non-symmetrical thioketones 1b,f,g were smoothly converted into the corresponding tetraarylethenes 9d–f (Table 1). However, in these cases, mixtures of (Z)- and (E)-isomers were formed and isolated in 65–75% yield. In all cases, the ratio of isomers was calculated to ca. 3:2 (1H NMR); however, the attempted chromatographic separation was unsuccessful.
The results obtained with thiofluorenone (1c) prompted us to prepare other 2-trimethylsilylated 1,3-dithiolanes of type 6, available via [3 + 2]-cycloaddition of the in situ generated (at ca. −45 °C) silylated thiocarbonyl S-methanide 10 with thioketones 1a,c,d,h and i. The obtained products of type 6, without isolation, were desilylated at rt to give the expected ethenes 9g–k with a fluorenylidene moiety (Scheme 4, Table 1).
![[1860-5397-13-185-i4]](/bjoc/content/inline/1860-5397-13-185-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of dibenzofulvenes 9g–k.
Scheme 4: Synthesis of dibenzofulvenes 9g–k.
Table 1: Synthesis of tetraaryl/hetarylethenes 9a−f and dibenzofulvenes 9g−k from diaryl thioketones 1 and TMS-CHN2.a
| 1 | Ar1 | Ar2 | 6 | Ar1 | Ar2 | 9 | Ar1 | Ar2 | Yield [%]b |
|---|---|---|---|---|---|---|---|---|---|
| a | Ph | Ph | a | Ph | Ph | 90 | |||
| b | Ph | Sel | – | f | Ph | Sel | 71 | ||
| d | Thi | Thi | b [8] | Thi | Thi | b | Thi | Thi | 89 |
| e | Sel | Sel | c [8] | Sel | Sel | c | Sel | Sel | 87 |
| f | Sel | Fur | – | d | Sel | Fur | 65 | ||
| g | Ph | Thi | – | e | Ph | Thi | 75 | ||
| c | fluorenylidene | – | g | Ph | Ph | 72 | |||
| c | fluorenylidene | – | h | fluorenylidene | 70 | ||||
| c | fluorenylidene | – | i | Thi | Thi | 77 | |||
| c | fluorenylidene | – | j | dibenzosuberenylidene | 66 | ||||
| c | fluorenylidene | – | k | dibenzosuberylidene | 62 | ||||
aPh = Phenyl, Thi = thiophen-2-yl, Sel = selenophen-2-yl, Fur = furan-2-yl. bYield of isolated ethenes 9 from the one-pot reaction.
The unexpected formation of tetrasubstituted ethenes 9 from 1,3-dithiolanes of type 6 requires a mechanistic explanation. The behaviour of these dithiolanes under desilylation conditions differs significantly from that of 4,5-unsubstituted 1,3-dithiolane 13c (Scheme 5). In the latter case, the desilylation leads to the corresponding carbanion, which can be trapped with an aldehyde to give 14 [24,25]. In contrast, the carbanion 11 undergoes a spontaneous cycloreversion to yield tetraaryl/hetarylethene 9 and dithioformic acid anion (12).
![[1860-5397-13-185-i5]](/bjoc/content/inline/1860-5397-13-185-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: a) Mechanistic explanation for formation of ethenes 9 from dithiolanes of type 6 and b) desilylation reactions of 2-trimethylsilyl-1,3-dithiolanes 13.
Scheme 5: a) Mechanistic explanation for formation of ethenes 9 from dithiolanes of type 6 and b) desilylatio...
For comparison reasons, two other 2-trimethylsilyl 1,3-dithiolanes 13a,b with no substituents at C4 and C5 were prepared and tested in desilylation reactions performed with TBAF in THF solutions. In both cases, quenching of the intermediate carbanions with water led to the corresponding 1,3-dithiolanes 15a and 15b, and no [3 + 2]-cycloreversion of the heterocyclic ring was observed (Scheme 5). As mentioned before, 1,3-dithiolane 13c has been reported as a convenient source of a reactive carbanion, which subsequently was trapped with aromatic or aliphatic aldehydes as electrophilic agents yielding alcohols 14, which should be considered as protected forms of corresponding α-hydroxyaldehydes [24,25]. Also in this series, no [3 + 2]-cycloreversion of the intermediate carbanion leading to the destruction of the heterocyclic ring was observed. These experiments demonstrate that the 4,5-unsubstituted 1,3-dithiolane carbanions generated under mild conditions through desilylation of the appropriate precursors 13 do not require an electron-withdrawing substituent for their stabilization [29].
The preparation of 1,3-dithianes and 1,3-dithiolanes, as well as the generation of the corresponding carbanions are widely applied in the umpolung chemistry [27,28] and in the chemistry of protective groups [29,30], but tetrasubstituted ethenes have never been prepared by using this approach.
Conclusion
The presented study showed that 2-trimethylsilyl-4,4,5,5-tetraaryl-1,3-dithiolanes, readily available by treatment of hetaryl thioketones with trimethylsilyl diazomethane (TMS-CHN2), are superior substrates for the preparation of tetraarylethenes. Another group of 2-trimethylsilylated 1,3-dithiolanes, obtained through the [3 + 2]-cycloaddition of trimethylsilylated thiofluorenone S-methanide with aryl and hetaryl thioketones can also be used for this transformation. The described protocol opens a new, straightforward access to a series of new dibenzofulvenes [7] containing hetaryl rings, which can be of great interest in materials chemistry and related sciences. The key intermediate in the reaction is the 1,3-dithiolane carbanion, which, in contrast to the 4,5-unsubstituted analogues, undergoes a spontaneous cycloreversion reaction to give tetraarylethene and dithioformate anion. This method supplements the list of preparatively useful olefination reactions and is another proof for the utility of aryl and hetaryl thioketones in organic synthesis [18]. Another important feature of the presented system with a potential, practical application to materials and coordination chemistry is the release of strictly controlled amounts of the dithioformate anion under mild, neutral conditions [31].
Experimental
General information: Solvents and chemicals were purchased and used as received without further purification. Products were purified by standard column chromatography on silica gel (230–400 mesh, Merck). Unless stated otherwise, yields refer to analytically pure samples. NMR spectra were recorded with a Bruker Avance III 600 MHz (1H NMR [600 MHz]; 13C NMR [151 MHz]) instrument. Chemical shifts are reported relative to solvent residual peaks (1H NMR: δ 7.26 ppm [CHCl3]; 13C NMR: δ 77.0 ppm [CDCl3]). IR spectra were registered with a FTIR NEXUS spectrometer (as film or KBr pellets). High resolution MS measurements were performed with a GCT Premier Waters instrument. Melting points were determined in capillaries with a Stuart SMP30 apparatus with automatic temperature monitoring. Microwave-supported syntheses of thioketones 1 were performed using the CEM-focused Microwave-type Discover SPD reactor.
Starting materials: Trimethylsilyldiazomethane (TMS-CHN2) was a commercial reagent which was used in all reactions as 1 M THF solution without further purification. Thiobenzophenone (1a), thiodibenzosuberenone (1h), and thiodibenzosuberone (1i) were prepared from the corresponding ketones by treatment with Lawesson’s reagent (L.R.) in toluene upon irradiation with microwaves (150 W) over 2 min [32]. The most efficient method for the preparation of thiofluorenone (1c) was the thionation of fluorenone by simultaneous passing of dry hydrogen chloride and hydrogen sulfide streams through the ethanolic solution at 0–5 °C (ice bath cooling) [33]. In analogy to 1a, hetaryl thioketones 1b,d–g were prepared from the corresponding ketones [34] by treatment with L.R. in toluene solution upon irradiation with microwaves over 2 min [32].
General Procedure for the one-pot synthesis of ethenes 9a–f: A hetaryl thioketone 1 (1 mmol) was dissolved in THF (2–3 mL) and the solution was cooled to 0 °C (ice bath). Then, the mixture was treated with small portions of an ethereal solution of TMS-CHN2 (2 M, 0.5 mL, 1 mmol). The reaction was complete after ca. 15 min (TLC, petroleum ether/CH2Cl2 8:2). Then, a solution of TBAF (1 M in THF, 1 mL, 1 mmol) was added. After complete reaction (TLC, petroleum ether/CH2Cl2 8:2), the solvent was removed under vacuum, and the residue was subjected to 1H NMR analysis in CDCl3 solution. The crude products 9a–f were purified by column chromatography (Et2O/CH2Cl2 8:2).
1,1,2,2-Tetrakis(selenophen-2-yl)ethene (9c): Yield: 236 mg (87%); chromatographic purification (petroleum ether/CHCl3 8:2). Orange crystals; mp 199–202 °C; IR (KBr) ν: 3085 (w), 1445 (m), 1429 (m), 1236 (s), 1182 (m), 1116 (m), 1024 (m), 849 (m), 840 (m), 748 (s), 687 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.14–7.15 (m, 4 Harom), 7.20–7.21 (m, 4 Harom), 8.09 (dd, J = 6.0 Hz, 1.2 Hz, 4 Harom) ppm; 13C NMR (150 MHz, CDCl3) δ 129.1, 133.0, 134.2 (12 CHarom), 131.2 (2 C=), 149.6 (4 Carom) ppm; anal. calcd for C18H12Se4 (544.13): C, 39.73; H, 2.22; found: C, 39.68; H, 2.25.
1,2-Diphenyl-1,2-bis(selenophen-2-yl)ethene (9f, mixture of E/Z isomers, ratio 2:1.2). Yield: 156 mg (71%); chromatographic purification (petroleum ether/CHCl3 8:2). Yellow crystals; mp 221–223 °C; IR (KBr) ν: 3044 (w), 1483 (w), 1439 (m), 1233 (m), 1201 (w), 1071 (w), 1021 (w), 770 (m), 706 (s), 685 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 6.53 (d, J = 3.6 Hz, 2 Harom), 6.96 (dd, J = 6.0 Hz, 2.4 Hz, 2 Harom), 7.06 (d, J = 3.6 Hz, 2 Harom), 7.11–7.15 (m, 12 Harom), 7.45–7.50 (m, 10 Harom), 7.80 (d, J = 6.0 Hz, 2 Harom), 8.03 (d, J = 5.4 Hz, 2 Harom) ppm; 13C NMR (150 MHz, CDCl3) δ 126.8, 127.6, 128.3, 128.4, 129.1, 129.4, 130.8, 131.4, 132.2, 132.6, 133.0, 133.1 (32 CHarom), 135.0, 136.5, 141.5, 142.9, 151.0, 151.7 (8 Carom, 4 C=) ppm; anal. calcd for C22H16Se2 (438.28): C, 60.29; H, 3.68; found: C, 60.09; H, 3.67.
Synthesis of dibenzofulvenes 9g–k: Thiofluorenone (1c, 98 mg, 0.5 mmol) was dissolved in THF (1.5 mL) and the solution was cooled to −78 °C (dry ice/acetone). Then, the mixture was treated with small portions of an ethereal solution of TMS-CHN2 (2 M, 0.25 mL, 0.5 mmol) until the intense colour of 1c vanished. Next, the mixture was allowed to warm slowly to −45 °C, whereby elimination of N2 was observed. Then, a solution of thiofluorenone (1c) or di(thiophen-2-yl) thioketone (1d, 0.5 mmol) in 1.5 mL of THF was added. After ca. 10 min, the cooling bath was replaced by an ice bath (0 °C) and a solution of TBAF (1 M in THF, 1 mL, 1 mmol) was added to the mixture. After removal of the solvent under vacuum, the residue was subjected to 1H NMR analysis in CDCl3 solution. The crude products were purified by column chromatography.
2,2’-[(9H-Fluoren-9-ylidene)methylene]dithiophene (9i): Yield: 132 mg (77%); chromatographic purification (petroleum ether/CHCl3 7:3). Orange crystals; mp 175–177 °C; IR (KBr) ν: 3094 (w), 3066(w), 1556 (w), 1445 (m), 1416 (m), 1252 (w), 1220 (w), 837 (w), 777 (m), 713 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 6.68–6.94 (m, 4 Harom), 7.02–7.04 (m, 2 Harom), 7.14–7.18 (m, 4 Harom), 7.42–7.43 (m, 2 Harom), 7.59–7.61 (m, 2 Harom) ppm; 13C NMR (150 MHz, CDCl3) δ 119.3, 124.8, 126.6, 127.4, 128.1, 128.5, 129.2 (14 CHarom), 137.7, 138.4, 140.6, 144.6 (6 Carom, 2 C=) ppm; HRMS–ESI+ (TOF): [M]+ calcd for C22H14S2, 342.0537; found, 342.0527.
Preparation of 1,3-dithiolanes 13a,b: Both compounds were prepared from commercial methyl (trimethylsilyl) ketone or phenyl (trimethylsilyl) ketone [35], respectively, by treatment with 1,2-ethanedithiol in the presence of BF3·Et2O following a literature protocol [36].
2-Phenyl-2-(trimethyl)silyl-1,3-dithiolane (13b): Yield: 191 mg (75%); colourless solid; mp 52–53 °C; IR (KBr) ν: 2952 (m), 2920 (m), 1591 (m), 1496 (m), 1480 (m), 1435 (m), 1245 (vs), 1078 (m), 1036 (m), 922 (m), 840 (vs), 736 (s), 698 (vs), 609 (m), 504 (s) cm–1; 1H NMR (600 MHz, CDCl3) δ 0.10 (s, 9H), 2.99–3.05 (m, 2H), 3.20–3.25 (m, 2H), 7.12–7.15 (m, 1Harom), 7.23–7.26 (m, 2Harom), 7.65–7.68 (m, 2Harom) ppm; 13C NMR (150 MHz, CDCl3) δ 2.1 (3 CH3), 38.9 (2 CH2), 58.7 (C(2)), 125.7, 127.4, 127.8 (5 CHarom), 144.6 (1 Carom) ppm; anal. calcd for C12H18S2Si (254.49): C, 56.63; H, 7.13; S, 25.20; found: C, 56.64; H, 7.37; S, 25.31.
Desilylation of 1,3-dithiolanes 13a,b. General procedure: The corresponding 1,3-dithiolane (0.5 mmol) was dissolved in THF (2 mL) and the solution was cooled to 0 °C (ice bath). Then, a solution of TBAF (0.5 mL, 1 M in THF, 0.5 mmol) was added portion-wise. The progress of the desilylation was controlled by TLC (petroleum ether/CH2Cl2 7:3). After removal of the solvent under vacuum, the residue was subjected to 1H NMR analysis in CDCl3 solution. The crude products were purified by column chromatography (petroleum ether/CH2Cl2, increasing polarity to CH2Cl2).
Supporting Information
| Supporting Information File 1: Experimental data for compounds 9, 13, 15 and copies of the original 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.6 MB | Download |
References
-
Suzuki, T.; Shiohara, H.; Monobe, M.; Sakimura, T.; Tanaka, S.; Yamashita, Y.; Miyashi, T. Angew. Chem., Int. Ed. Engl. 1992, 31, 455–458. doi:10.1002/anie.199204551
Return to citation in text: [1] [2] -
Fischer, E.; Larsen, J.; Christensen, J. B.; Fourmigué, M.; Madsen, H. G.; Harrit, N. J. Org. Chem. 1996, 61, 6997–7005. doi:10.1021/jo960022x
Return to citation in text: [1] -
Bolzoni, A.; Viglianti, L.; Bossi, A.; Mussini, P. R.; Canteruccio, S.; Baldoli, C.; Licandro, E. Eur. J. Org. Chem. 2013, 7489–7499. doi:10.1002/ejoc.201300745
Return to citation in text: [1] -
Zhang, G.-F.; Wang, H.; Aldred, M. P.; Chen, T.; Chen, Z.-Q.; Meng, X.; Zhu, M.-Q. Chem. Mater. 2014, 26, 4433–4446. doi:10.1021/cm501414b
Return to citation in text: [1] -
Yamamoto, A.; Matsui, Y.; Asada, T.; Kumeda, M.; Takagi, K.; Suenaga, Y.; Nagae, K.; Ohta, E.; Sato, H.; Koseki, S.; Naito, H.; Ikeda, H. J. Org. Chem. 2016, 81, 3168–3176. doi:10.1021/acs.joc.6b00117
Return to citation in text: [1] -
Hopf, H.; Sherburn, M. S., Eds. Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry; Wiley VCH Verlag GmbH: Weinheim, 2016.
Return to citation in text: [1] -
Shimizu, M.; Nagao, I.; Kiyomoto, S.-i.; Hiyama, T. Aust. J. Chem. 2012, 65, 1277–1284. doi:10.1071/CH12060
Return to citation in text: [1] [2] -
Gopikrishna, P.; Iyer, P. K. J. Phys. Chem. C 2016, 120, 26556–26568. doi:10.1021/acs.jpcc.6b09689
Return to citation in text: [1] [2] [3] -
Krygowski, T. M.; Szatylowicz, H.; Stasyuk, O. A.; Dominikowska, J.; Palusiak, M. Chem. Rev. 2014, 114, 6383–6422. doi:10.1021/cr400252h
Return to citation in text: [1] -
Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863–927. doi:10.1021/cr00094a007
Return to citation in text: [1] -
Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H
Return to citation in text: [1] -
van Staden, L. F.; Gravestock, D.; Ager, D. J. Chem. Soc. Rev. 2002, 31, 195–200. doi:10.1039/A908402I
Return to citation in text: [1] -
Fustero, S.; Simón-Fuentes, A.; Barrio, P.; Haufe, G. Chem. Rev. 2015, 115, 871–930. doi:10.1021/cr500182a
Return to citation in text: [1] -
Fürstner, A.; Bogdanović, B. Angew. Chem., Int. Ed. Engl. 1996, 35, 2442–2469. doi:10.1002/anie.199624421
Return to citation in text: [1] -
Barton, D. H. R.; Guziec, F. S., Jr.; Shahak, I. J. Chem. Soc., Perkin Trans. 1 1974, 1794–1799. doi:10.1039/P19740001794
Return to citation in text: [1] -
Guziec, F. S., Jr.; Sanfilippo, L. J. Tetrahedron 1988, 44, 6241–6285. doi:10.1016/S0040-4020(01)89815-X
Return to citation in text: [1] -
Mlostoń, G.; Heimgartner, H. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Italian Chemical Society: Rome, 2005; Vol. 9, pp 141–157.
Return to citation in text: [1] -
Mlostoń, G.; Grzelak, P.; Hamera-Fałdyga, R.; Jasiński, M.; Pipiak, P.; Urbaniak, K.; Albrecht, Ł.; Hejmanowska, J.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 204–211. doi:10.1080/10426507.2016.1252368
Return to citation in text: [1] [2] -
Mlostoń, G.; Huisgen, R. Heterocycles 1985, 23, 2201–2203. doi:10.3987/R-1985-09-2201
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050
Return to citation in text: [1] [2] -
McKee, M. L.; Mlostoń, G.; Urbaniak, K.; Heimgartner, H. Beilstein J. Org. Chem. 2017, 13, 410–416. doi:10.3762/bjoc.13.44
Return to citation in text: [1] -
Mlostoń, G.; Pipiak, P.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 716–724. doi:10.3762/bjoc.12.71
Return to citation in text: [1] [2] [3] -
Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Pol. J. Chem. 2007, 81, 1849–1860.
Return to citation in text: [1] -
Degl’Innocenti, A.; Capperucci, A.; Nocentini, T. Tetrahedron Lett. 2001, 42, 4557–4559. doi:10.1016/S0040-4039(01)00771-7
Return to citation in text: [1] [2] [3] -
Capperucci, A.; Cerè, V.; Degl’Innocenti, A.; Nocentini, T.; Pollicino, S. Synlett 2002, 1447–1450. doi:10.1055/s-2002-33521
Return to citation in text: [1] [2] [3] -
Rele, S.; Talukdar, S.; Banerji, A.; Chattopadhyay, S. J. Org. Chem. 2001, 66, 2990–2994. doi:10.1021/jo001586a
Return to citation in text: [1] -
Gröbel, B.-T.; Seebach, D. Synthesis 1977, 357–402. doi:10.1055/s-1977-24412
Return to citation in text: [1] -
Eymur, S.; Göllü, M.; Tanyeli, C. Turk. J. Chem. 2013, 37, 586–609. doi:10.3906/kim-1303-85
Return to citation in text: [1] -
Leung, M.-K.; Luh, T.-Y. Product Subclass 3: 1,3-Dithiolanes. In Science of Synthesis; Otera, J., Ed.; G. Thieme Verlag AG: Stuttgart, New York, 2007; pp 325 ff.
Return to citation in text: [1] [2] -
Green, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 3rd ed.; Wiley-Interscience: New York, 1999.
pp 329–344 and pp 724–727.
Return to citation in text: [1] -
Pipiak, P.; Mlostoń, G.; Glatthor, J.; Mitzel, N. W. Advances in the Chemistry of Heteroorganic Compounds. In Book of abstracts of XIX International Symposium, Łódź, Nov 25, 2016; p 136.
Return to citation in text: [1] -
Hamera-Fałdyga, R.; Grzelak, P.; Pipiak, P.; Mlostoń, G. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 197–198. doi:10.1080/10426507.2016.1252367
Return to citation in text: [1] [2] -
Campaigne, E.; Reid, W. B., Jr. J. Am. Chem. Soc. 1946, 68, 769–770. doi:10.1021/ja01209a020
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Gębicki, K.; Grzelak, P.; Heimgartner, H. Heteroat. Chem. 2014, 25, 548–555. doi:10.1002/hc.21191
Return to citation in text: [1] -
Tongco, E. C.; Wang, Q.; Prakash, G. K. S. Synth. Commun. 1997, 27, 2117–2123. doi:10.1080/00397919708006819
Return to citation in text: [1] -
Vicha, R.; Rouchal, M.; Kozubková, Z.; Kuřitka, I.; Marek, R.; Branná, P.; Čmelik, R. Supramol. Chem. 2011, 23, 663–677. doi:10.1080/10610278.2011.593628
Return to citation in text: [1]
| 34. | Mlostoń, G.; Urbaniak, K.; Gębicki, K.; Grzelak, P.; Heimgartner, H. Heteroat. Chem. 2014, 25, 548–555. doi:10.1002/hc.21191 |
| 32. | Hamera-Fałdyga, R.; Grzelak, P.; Pipiak, P.; Mlostoń, G. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 197–198. doi:10.1080/10426507.2016.1252367 |
| 35. | Tongco, E. C.; Wang, Q.; Prakash, G. K. S. Synth. Commun. 1997, 27, 2117–2123. doi:10.1080/00397919708006819 |
| 1. | Suzuki, T.; Shiohara, H.; Monobe, M.; Sakimura, T.; Tanaka, S.; Yamashita, Y.; Miyashi, T. Angew. Chem., Int. Ed. Engl. 1992, 31, 455–458. doi:10.1002/anie.199204551 |
| 2. | Fischer, E.; Larsen, J.; Christensen, J. B.; Fourmigué, M.; Madsen, H. G.; Harrit, N. J. Org. Chem. 1996, 61, 6997–7005. doi:10.1021/jo960022x |
| 3. | Bolzoni, A.; Viglianti, L.; Bossi, A.; Mussini, P. R.; Canteruccio, S.; Baldoli, C.; Licandro, E. Eur. J. Org. Chem. 2013, 7489–7499. doi:10.1002/ejoc.201300745 |
| 4. | Zhang, G.-F.; Wang, H.; Aldred, M. P.; Chen, T.; Chen, Z.-Q.; Meng, X.; Zhu, M.-Q. Chem. Mater. 2014, 26, 4433–4446. doi:10.1021/cm501414b |
| 5. | Yamamoto, A.; Matsui, Y.; Asada, T.; Kumeda, M.; Takagi, K.; Suenaga, Y.; Nagae, K.; Ohta, E.; Sato, H.; Koseki, S.; Naito, H.; Ikeda, H. J. Org. Chem. 2016, 81, 3168–3176. doi:10.1021/acs.joc.6b00117 |
| 15. | Barton, D. H. R.; Guziec, F. S., Jr.; Shahak, I. J. Chem. Soc., Perkin Trans. 1 1974, 1794–1799. doi:10.1039/P19740001794 |
| 16. | Guziec, F. S., Jr.; Sanfilippo, L. J. Tetrahedron 1988, 44, 6241–6285. doi:10.1016/S0040-4020(01)89815-X |
| 17. | Mlostoń, G.; Heimgartner, H. In Targets in Heterocyclic Systems; Attanasi, O. A.; Spinelli, D., Eds.; Italian Chemical Society: Rome, 2005; Vol. 9, pp 141–157. |
| 1. | Suzuki, T.; Shiohara, H.; Monobe, M.; Sakimura, T.; Tanaka, S.; Yamashita, Y.; Miyashi, T. Angew. Chem., Int. Ed. Engl. 1992, 31, 455–458. doi:10.1002/anie.199204551 |
| 14. | Fürstner, A.; Bogdanović, B. Angew. Chem., Int. Ed. Engl. 1996, 35, 2442–2469. doi:10.1002/anie.199624421 |
| 8. | Gopikrishna, P.; Iyer, P. K. J. Phys. Chem. C 2016, 120, 26556–26568. doi:10.1021/acs.jpcc.6b09689 |
| 10. | Maryanoff, B. E.; Reitz, A. B. Chem. Rev. 1989, 89, 863–927. doi:10.1021/cr00094a007 |
| 11. | Blakemore, P. R. J. Chem. Soc., Perkin Trans. 1 2002, 2563–2585. doi:10.1039/B208078H |
| 12. | van Staden, L. F.; Gravestock, D.; Ager, D. J. Chem. Soc. Rev. 2002, 31, 195–200. doi:10.1039/A908402I |
| 13. | Fustero, S.; Simón-Fuentes, A.; Barrio, P.; Haufe, G. Chem. Rev. 2015, 115, 871–930. doi:10.1021/cr500182a |
| 26. | Rele, S.; Talukdar, S.; Banerji, A.; Chattopadhyay, S. J. Org. Chem. 2001, 66, 2990–2994. doi:10.1021/jo001586a |
| 6. | Hopf, H.; Sherburn, M. S., Eds. Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry; Wiley VCH Verlag GmbH: Weinheim, 2016. |
| 7. | Shimizu, M.; Nagao, I.; Kiyomoto, S.-i.; Hiyama, T. Aust. J. Chem. 2012, 65, 1277–1284. doi:10.1071/CH12060 |
| 8. | Gopikrishna, P.; Iyer, P. K. J. Phys. Chem. C 2016, 120, 26556–26568. doi:10.1021/acs.jpcc.6b09689 |
| 9. | Krygowski, T. M.; Szatylowicz, H.; Stasyuk, O. A.; Dominikowska, J.; Palusiak, M. Chem. Rev. 2014, 114, 6383–6422. doi:10.1021/cr400252h |
| 22. | Mlostoń, G.; Pipiak, P.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 716–724. doi:10.3762/bjoc.12.71 |
| 22. | Mlostoń, G.; Pipiak, P.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 716–724. doi:10.3762/bjoc.12.71 |
| 24. | Degl’Innocenti, A.; Capperucci, A.; Nocentini, T. Tetrahedron Lett. 2001, 42, 4557–4559. doi:10.1016/S0040-4039(01)00771-7 |
| 25. | Capperucci, A.; Cerè, V.; Degl’Innocenti, A.; Nocentini, T.; Pollicino, S. Synlett 2002, 1447–1450. doi:10.1055/s-2002-33521 |
| 20. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050 |
| 22. | Mlostoń, G.; Pipiak, P.; Heimgartner, H. Beilstein J. Org. Chem. 2016, 12, 716–724. doi:10.3762/bjoc.12.71 |
| 19. | Mlostoń, G.; Huisgen, R. Heterocycles 1985, 23, 2201–2203. doi:10.3987/R-1985-09-2201 |
| 20. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050 |
| 21. | McKee, M. L.; Mlostoń, G.; Urbaniak, K.; Heimgartner, H. Beilstein J. Org. Chem. 2017, 13, 410–416. doi:10.3762/bjoc.13.44 |
| 36. | Vicha, R.; Rouchal, M.; Kozubková, Z.; Kuřitka, I.; Marek, R.; Branná, P.; Čmelik, R. Supramol. Chem. 2011, 23, 663–677. doi:10.1080/10610278.2011.593628 |
| 18. | Mlostoń, G.; Grzelak, P.; Hamera-Fałdyga, R.; Jasiński, M.; Pipiak, P.; Urbaniak, K.; Albrecht, Ł.; Hejmanowska, J.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 204–211. doi:10.1080/10426507.2016.1252368 |
| 23. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Pol. J. Chem. 2007, 81, 1849–1860. |
| 24. | Degl’Innocenti, A.; Capperucci, A.; Nocentini, T. Tetrahedron Lett. 2001, 42, 4557–4559. doi:10.1016/S0040-4039(01)00771-7 |
| 25. | Capperucci, A.; Cerè, V.; Degl’Innocenti, A.; Nocentini, T.; Pollicino, S. Synlett 2002, 1447–1450. doi:10.1055/s-2002-33521 |
| 8. | Gopikrishna, P.; Iyer, P. K. J. Phys. Chem. C 2016, 120, 26556–26568. doi:10.1021/acs.jpcc.6b09689 |
| 24. | Degl’Innocenti, A.; Capperucci, A.; Nocentini, T. Tetrahedron Lett. 2001, 42, 4557–4559. doi:10.1016/S0040-4039(01)00771-7 |
| 25. | Capperucci, A.; Cerè, V.; Degl’Innocenti, A.; Nocentini, T.; Pollicino, S. Synlett 2002, 1447–1450. doi:10.1055/s-2002-33521 |
| 32. | Hamera-Fałdyga, R.; Grzelak, P.; Pipiak, P.; Mlostoń, G. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 197–198. doi:10.1080/10426507.2016.1252367 |
| 33. | Campaigne, E.; Reid, W. B., Jr. J. Am. Chem. Soc. 1946, 68, 769–770. doi:10.1021/ja01209a020 |
| 18. | Mlostoń, G.; Grzelak, P.; Hamera-Fałdyga, R.; Jasiński, M.; Pipiak, P.; Urbaniak, K.; Albrecht, Ł.; Hejmanowska, J.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 204–211. doi:10.1080/10426507.2016.1252368 |
| 31. | Pipiak, P.; Mlostoń, G.; Glatthor, J.; Mitzel, N. W. Advances in the Chemistry of Heteroorganic Compounds. In Book of abstracts of XIX International Symposium, Łódź, Nov 25, 2016; p 136. |
| 29. | Leung, M.-K.; Luh, T.-Y. Product Subclass 3: 1,3-Dithiolanes. In Science of Synthesis; Otera, J., Ed.; G. Thieme Verlag AG: Stuttgart, New York, 2007; pp 325 ff. |
| 30. |
Green, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, 3rd ed.; Wiley-Interscience: New York, 1999.
pp 329–344 and pp 724–727. |
| 7. | Shimizu, M.; Nagao, I.; Kiyomoto, S.-i.; Hiyama, T. Aust. J. Chem. 2012, 65, 1277–1284. doi:10.1071/CH12060 |
| 29. | Leung, M.-K.; Luh, T.-Y. Product Subclass 3: 1,3-Dithiolanes. In Science of Synthesis; Otera, J., Ed.; G. Thieme Verlag AG: Stuttgart, New York, 2007; pp 325 ff. |
| 27. | Gröbel, B.-T.; Seebach, D. Synthesis 1977, 357–402. doi:10.1055/s-1977-24412 |
| 28. | Eymur, S.; Göllü, M.; Tanyeli, C. Turk. J. Chem. 2013, 37, 586–609. doi:10.3906/kim-1303-85 |
© 2017 Mlostoń et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)