Abstract
Determining the accurate chemical structures of synthesized compounds is essential for biomedical studies and computer-assisted drug design. The unequivocal determination of N-adamantylation or N-arylation site(s) in nitrogen-rich heterocycles, characterized by a low density of hydrogen atoms, using NMR methods at natural isotopic abundance is difficult. In these compounds, the heterocyclic moiety is covalently attached to the carbon atom of the substituent group that has no bound hydrogen atoms, and the connection between the two moieties of the compound cannot always be established via conventional 1H-1H and 1H-13C NMR correlation experiments (COSY and HMBC, respectively) or nuclear Overhauser effect spectroscopy (NOESY or ROESY). The selective incorporation of 15N-labelled atoms in different positions of the heterocyclic core allowed for the use of 1H-15N (JHN) and 13C-15N (JCN) coupling constants for the structure determinations of N-alkylated nitrogen-containing heterocycles in solution. This method was tested on the N-adamantylated products in a series of azolo-1,2,4-triazines and 1,2,4-triazolo[1,5-a]pyrimidine. The syntheses of adamantylated azolo-azines were based on the interactions of azolo-azines and 1-adamatanol in TFA solution. For azolo-1,2,4-triazinones, the formation of mixtures of N-adamantyl derivatives was observed. The JHN and JCN values were measured using amplitude-modulated 1D 1H spin-echo experiments with the selective inversion of the 15N nuclei and line-shape analysis in the 1D 13С spectra acquired with selective 15N decoupling, respectively. Additional spin–spin interactions were detected in the 15N-HMBC spectra. NMR data and DFT (density functional theory) calculations permitted to suggest a possible mechanism of isomerization for the adamantylated products of the azolo-1,2,4-triazines. The combined analysis of the JHN and JCN couplings in 15N-labelled compounds provides an efficient method for the structure determination of N-alkylated azolo-azines even in the case of isomer formation. The isomerization of adamantylated tetrazolo[1,5-b][1,2,4]triazin-7-ones in acidic conditions occurs through the formation of the adamantyl cation.
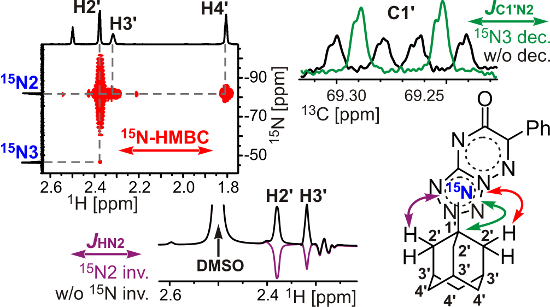
Graphical Abstract
Introduction
The incorporation of an adamantyl moiety in bioactive molecules and analogues of natural compounds is a widely used approach in medicinal chemistry [1]. The increased lipophilicity of adamantane-containing compounds compared with non-adamantylated derivatives [2] leads to considerably higher solubility of these compounds in blood plasma and their easier penetration through cell membranes. The conjugation of adamantane with heterocyclic compounds also provides a method to modify the pharmacological profile and frequently leads to a new type of bioactivity. For example, N-adamantyl tetrazoles 1 and 2 (Figure 1A) demonstrate lower toxicity and, simultaneously, more potent activity against influenza A virus compared with the currently used antiviral drug rimantadine (1-(1-adamantyl)ethanamine) [3]. More recently, Roberge et al. described new inhibitors of the influenza A virus M2 proton channel. Among the studied compounds, adamantyl imidazole 3 showed good activity [4].
![[1860-5397-13-250-1]](/bjoc/content/figures/1860-5397-13-250-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (A) Adamantylated azoles and derivatives of 1,2,4-triazolo[5,1-c][1,2,4]triazine with antiviral activities. (B) Four sites sensitive to N-alkylation in 1,2,4-triazolo[5,1-c][1,2,4]triazin-7-ones are indicated by arrows.
Figure 1: (A) Adamantylated azoles and derivatives of 1,2,4-triazolo[5,1-c][1,2,4]triazine with antiviral act...
An azolo-azine core with a bridgehead nitrogen atom is found in many natural products [5,6] and biologically active synthetic compounds [7,8]. The purine-like scaffold of these nitrogen-containing heterocycles is frequently used in medicinal chemistry and drug design. For example, 6-nitro-1,2,4-triazolo[5,1-c][1,2,4]triazine 4 (Figure 1A, Triazavirin®) was approved in Russia for the treatment of influenza [9]. This drug targets the viral protein haemagglutinin. The incorporation of an adamantyl moiety in azolo-azine structures could lead to the development of new multifunctional antiviral drugs.
Previously, we synthesized N-adamatylated derivatives of 1,2,4-triazolo[5,1-c][1,2,4]triazines 5 and 6 by reaction with the adamantyl cation generated from 1-adamantanol in acidic medium [10]. The azolo-azine scaffold of these compounds has several nitrogen atoms that can react with alkylation reagents [11,12] (Figure 1B). For this reason, the adamantylation of compounds 5 and 6 led to mixtures of N3- and N4-adamantylated isomers, which reisomerized into each other likely via the formation of an adamantyl cation and starting NH-heterocycle. The unambiguous determination of N-adamantylation site(s) in heterocycles 5 and 6 using well-established 1H and 13C NMR methods (such as 1D, 2D COSY, HMQC, HMBC, and INADEQUATE spectra) was difficult because the heterocyclic moiety was covalently attached to the adamantane tertiary carbon that had no bound hydrogen atoms. Nuclear Overhauser effect spectroscopy (NOESY or ROESY) also did not provide unequivocal structures of the N-adamantylated derivatives [13,14]. For example, the attachment of an adamantyl group to the N1 or N3 atom in the azole ring of compounds 5 and 6 could not be distinguished by NOE data. Similar problems with the unambiguous determination of the product structure were also found for N-arylation or N-alkylation with tert-butyl fragments in the series of 1,2,3-triazole [15,16], tetrazole [17-20], and purine [21] derivatives. Meanwhile, knowledge of the accurate chemical structures of N-substituted heterocycles is essential for biomedical studies and computer-assisted drug design, e.g., molecular docking techniques. Thus, the development of effective methods for the unambiguous determination of N-alkylation site(s) in the azolo-azine series is important.
The data that are required to solve this problem could be provided by 15N NMR spectroscopy. For monocyclic derivatives of azoles, the structures of N-alkylated regioisomers can be determined using 2D H-(C)-N multiple bond correlation (HCNMBC) experiments [22,23] using natural isotopic abundance. These experiments rely on the magnetization transfer through 13C-15N J-coupling constants (JCN). However, the fusion of the azine ring to an azolo fragment increases the number of possible alkylation sites and considerably complicates the analysis of the JCN patterns. This issue, together with the inherently low sensitivity of natural abundance 15N NMR spectroscopy, does not always permit the unambiguous positioning of alkyl (N-adamantyl, N-tert-butyl or N-aryl) fragments in azolo-azines.
The incorporation of 15N labels in nitrogen-containing heterocycles greatly facilitates the use of NMR spectroscopy for studies of molecular structures and mechanisms of chemical transformations [10,24-29]. The labelling enhances the sensitivity of detection and permits the quantitative measurements of JCN and 1H-15N J-coupling constants (JHN) even in a mixture of tautomeric forms [24,25]. Additionally, a method based on amplitude-modulated spin-echo experiments was found to be the most efficient way to measure JHN couplings [24]. Previously, the incorporation of a single 15N label in position 1 of the 1,2,4-triazole fragment of compounds 5 and 6 and analysis of the JCN couplings permitted the unambiguous identification of the structures of the N3-adamantylated derivatives (Figure 1B), while the structures of the N4-adamantylated products were determined by 13C NMR spectroscopy via comparison with model compounds, N-methylated azolo-azines [10]. However, this preliminarily study did not evaluate the potential of the incorporation of several 15N-labels and simultaneous analysis of the JCN and JHN coupling constants for the determination of the N-adamantylation site(s) in heterocycles.
Herein, we report the selective incorporation of two 15N-labelled atoms in tetrazolo[1,5-b][1,2,4]triazin-7-one, 1,2,4-triazolo[5,1-c][1,2,4]triazin-7-one, and 1,2,4-triazolo[1,5-a]pyrimidin-7-one and the N-adamantylation of the obtained compounds. The combined analysis of the JCN and JHN couplings permitted the straightforward determination of the adamantylation sites in these azolo-azines, even when a mixture of regioisomers is formed.
Results
Synthesis. Derivatives of 1,2,4-triazolo[1,5-a]pyrimidine [30], 1,2,4-triazolo[5,1-c][1,2,4]triazinone [31] and tetrazolo[1,5-b][1,2,4]triazinone [32] can be obtained by the fusion of an azine ring to an azole fragment. This method can be used for the selective incorporation of 15N atoms in different azolo-azines. Recently, we tested this approach for the syntheses of 15N-labelled tetrazolo[1,5-b][1,2,4]triazines and tetrazolo[1,5-a]pyrimidines [25] starting from 15N-labelled 5-aminotetrazole. However, due to proton tautomerism, the use of single-labelled [2-15N]-5-aminotetrazole led to the formation of isotopomer mixtures, which complicated the subsequent NMR analysis. Meanwhile, the application of [2,3-15N2]-5-aminotetrazole 7-15N2 provided the single double-labelled products in the tetrazolo[1,5-a]pyrimidine series [33]. Thus, in the current work, [2,3-15N2]-5-aminotetrazole 7-15N2 (98% enrichment for each of the labelled 15N atoms) was used to incorporate isotopic labels in the tetrazolo[1,5-b][1,2,4]triazine core (Scheme 1). The interaction of diazonium salt 8-15N2 derived from [2,3-15N2]-5-aminotetrazole 7-15N2 with ethyl α-formylphenylacetate (9) yielded compound 10-15N2. It was expected that the cyclization of 10-15N2 would give [1,2-15N2]-tetrazolo[5,1-c][1,2,4]triazine 11-15N2. Indeed, [2,3-15N2]-tetrazolo[1,5-b][1,2,4]triazin-7-one 13-15N2 was obtained (see below). Most likely, tetrazole 11-15N2 underwent a ring-opening process, yielding azide 12-15N2, and this process was followed by an alternative ring closure. This azido-tetrazole equilibrium has been previously studied in detail [25].
![[1860-5397-13-250-i1]](/bjoc/content/inline/1860-5397-13-250-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis and adamantylation of 15N-labelled 13-15N2 and JHN and JCN data confirming the structures of adamantylated derivatives 15a,b-15N2. The JHN couplings measured by amplitude-modulated 1D 1H spin-echo experiments and detected in the 2D 15N-HMBC spectra are shown by blue, magenta, and red arrows (see the legend in the figure). The measured JHN values (blue and magenta) are classified into three categories: J ≥ 0.8 Hz, 0.8 > J ≥ 0.1 Hz, and J < 0.1 Hz (bold solid, thin solid, and dashed arrows, respectively). The 1H-15N cross-peaks observed in the 2D HMBC spectrum for the unlabelled and labelled nitrogen atoms are classified into three categories: strong, medium and weak (bold solid, thin solid, and dashed red arrows, respectively). The JCN couplings with adamantane carbons measured in the 1D 13С spectra are classified into three categories: J ≥ 2 Hz, 2 > J ≥ 1 Hz, and J < 1 Hz (bold solid, thin solid, and dashed green arrows, respectively). The 1JNN couplings observed in the 1D 15N NMR spectra of 13-15N2 (16.4 Hz), 15a-15N2 (14.7 Hz) and 15b-15N2 (15.3 Hz) are not shown.
Scheme 1: Synthesis and adamantylation of 15N-labelled 13-15N2 and JHN and JCN data confirming the structures...
The coupling between compound 13-15N2 and 1-adamantanol (14) was conducted in trifluoroacetic acid (TFA) solution under reflux. A general and convenient approach to the N-adamantylation of heterocycles involves a reaction with the adamantyl cation generated from 1-adamantanol in acidic medium [34-37]. The adamantylation of 13-15N2 led to N2- and N1-regioisomers (15a-15N2 and 15b-15N2, respectively, Scheme 1). Interestingly, according to the possible resonance structures, compound 15a-15N2 should represent a mesoionic (betaine-like) structure with positive and negative charges located at the tetrazole and triazine rings, respectively. The relative concentration of regioisomers 15a-15N2 and 15b-15N2 was determined from the integral intensity of the corresponding signals in the 1D 1H and 15N NMR spectra. The regioselectivity of adamantylation depends on the reaction time. Refluxing of the 13-15N2/14 mixture (1:1.5 mol/mol) in TFA over 5 min led to the predominant formation of N2-adamantylated derivative 15a-15N2. The 1:2 15a-15N2/15b-15N2 mixture was obtained after 2 h of refluxing. This phenomenon could be explained by the reisomerization of the initially formed N2-adamantylated product (15a-15N2). Indeed, 2 h of refluxing of isolated 15a-15N2 in TFA with 1.5 molar equivalents of 14 yielded a mixture of compounds 15a-15N2 and 15b-15N2 in the same (1:2) ratio (Scheme 1).
The use of [1-15N]-3-amino-1,2,4-triazole 16-15N (98%, 15N) and labelled sodium nitrite (98%, 15N) in acidic medium allowed for the in situ production of diazonium salt 17-15N2, which reacted with ethyl nitroacetate (18) in a sodium carbonate solution (Scheme 2). This reaction led to the formation of [1,5-15N2]-1,2,4-triazolo[5,1-c][1,2,4]triazinone 19-15N2. Previously, the same approach was described for the incorporation of 15N atoms in azole and azine rings of compound 4 [38]. Heterocycle 20-15N2 was obtained by the treatment of 19-15N2 with hydrobromic acid according to a procedure described for 6-nitro-1,2,4-triazolo[5,1-c][1,2,4]triazin-7-ones [39]. In this case, hydrogen bromide was obtained in situ by the reaction between acetyl bromide and ethanol. The adamantylation of 20-15N2 first occurred on the N3 atom of the azole ring. It was found that 5 min reflux of 20-15N2 in TFA with a 1.5 molar excess of 14 led to the structure 21a-15N2 (Scheme 2). However, prolonged (6 h) refluxing of the N3-regioisomer with 1.5 molar equivalents of 1-adamantanol (14) in a TFA solution led to complete isomerization of the compound and re-attachment of adamantane to the N4-atom of the azine ring (compound 21b-15N2).
![[1860-5397-13-250-i2]](/bjoc/content/inline/1860-5397-13-250-i2.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and adamantylation of 15N-labelled 20-15N2 and JHN and JCN data confirming the structures of adamantylated derivatives 21a,b-15N2.The JHN couplings measured either in the 1D 1H spectra or by amplitude-modulated 1D 1H spin-echo experiments and detected in the 2D 15N-HMBC spectra are shown by grey, magenta, and red arrows (see the legend in the figure). The measured JHN values (gray and magenta) with magnitudes J ≥ 14 Hz and J < 0.1 Hz are indicated by the bold solid and dashed arrows, respectively. The 1H-15N cross-peaks observed in the 2D HMBC spectrum for the unlabelled nitrogen atoms are classified into three categories: strong, medium and weak (bold solid, thin solid, and dashed red arrows, respectively). The JCN couplings with adamantane carbons measured in the 1D 13С spectra are classified into three categories: J ≥ 2 Hz, 2 > J ≥ 1 Hz, and J < 1 Hz (bold solid, thin solid, and dashed green arrows, respectively).
Scheme 2: Synthesis and adamantylation of 15N-labelled 20-15N2 and JHN and JCN data confirming the structures...
Double-labelled [1,2-15N2]-3-amino-1,2,4-triazole 16-15N2 was synthesized by the interaction of 15N2-hydrazine sulphate (98%, 15N) with S-methyl isothiourea sulphate and consecutive cyclization with formic acid (see the Supporting Information File 1). The use of 16-15N2 in a reaction with ethyl 4,4,4-trifluoroacetoacetate (22) yielded azolo-azine 23-15N2 containing two isotopic labels in the 1,2,4-triazole fragment (Scheme 3). The adamantylation of [1,8-15N2]-1,2,4-triazolo[1,5-a]pyrimidine 23-15N2 was regioselective and led to the formation of the N3-isomer 24-15N2 only. This compound did not undergo further isomerization.
![[1860-5397-13-250-i3]](/bjoc/content/inline/1860-5397-13-250-i3.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis and adamantylation of 15N-labelled 23-15N2 and JHN and JCN data confirming the structure of adamantylated derivative 24-15N2. The JHN couplings measured in the 1D 1H spectra and detected in the 2D 15N-HMBC spectra are shown by grey, cyan, and red arrows (see the legend in the figure). The measured JHN values (gray and cyan) with magnitudes J ≥ 3 Hz and J < 1 Hz are indicated by the bold and thin solid arrows, respectively. The correlation between H2' and the unlabelled N3 atom observed in the HMBC spectrum of 24-15N2 is shown by a red arrow. The JCN couplings with the H1' adamantane carbon measured in the 1D 13С spectrum of 24-15N2 have magnitudes < 1 Hz and are shown by dashed green arrows. The 1JNN couplings observed in the 1D 15N NMR spectra of 23-15N2 (13.6 Hz) and 24-15N2 (13.4 Hz) are not shown.
Scheme 3: Synthesis and adamantylation of 15N-labelled 23-15N2 and JHN and JCN data confirming the structure ...
Isomerization of adamantylated derivatives. The adamantylation of 1,2,4-triazolo[5,1-c][1,2,4]triazin-7-one derivatives in acidic medium is a thermodynamically controlled reaction [10], which could explain the rearrangement of N3-isomer 21a into N4-isomer 21b and the formation of the 15a/15b mixture from compound 15a. To evaluate the relative thermodynamic stabilities of isomers 15a,b and 21a,b, we performed DFT calculations with the RB3LYP/6-31-G(d,p) approximation in the gas phase using the Gaussian 09 package [40]. Isomers 15b and 21b are thermodynamically more stable than counterparts 15a and 21a. The calculated relative energy differences were 8.3 kcal/mol and 6.4 kcal/mol for the 15a–15b and 21a–21b pairs, respectively (see the Supporting Information File 1).
To further study the mechanism of isomerization between compounds 15a and 15b, equimolar quantities of unlabelled N2-isomer 15a and its double-labelled non-adamantylated precursor 13-15N2 (isotopic enrichment 98%) were refluxed for 2 h in TFA without the addition of 1-adamantanol (14, Scheme 4). NMR analysis of the resulting mixture revealed the compounds 13*-15N2, 15a*-15N2, and 15b*-15N2 in a 5:2:3 ratio. The observed equal 15N-isotopic enrichment (≈49%) in compounds 13*-15N2, 15a*-15N2, and 15b*-15N2 indicated that the equilibrium 15a![[Graphic 1]](/bjoc/content/inline/1860-5397-13-250-i6.svg?max-width=637&scale=0.472728) 15b was reached in the isomerization process. The obtained ratio between the adamantylated products confirmed the higher thermodynamic stability of compound 15b relative to isomer 15a.
15b was reached in the isomerization process. The obtained ratio between the adamantylated products confirmed the higher thermodynamic stability of compound 15b relative to isomer 15a.
![[1860-5397-13-250-i4]](/bjoc/content/inline/1860-5397-13-250-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Isomerization of 15a in the presence of tetrazolo[1,5-b][1,2,4]triazin-7-one 13-15N2 and isotopic enrichment of the reactants and products. The starting level of 15N-isotopic enrichment (98%) of compound 13-15N2 is shown in blue. The levels of 15N enrichment (≈49%) of the obtained compounds 13*-15N2, 15a*-15N2 and 15b*-15N2 are shown in red. The levels of isotopic enrichment were determined by mass spectrometry. In addition, ≈50% excess of the 15N isotopes in compounds 15a*-15N2 and 15b*-15N2 after reaction was confirmed by 13C NMR spectroscopy. In this case, the C1' signals of the labelled and unlabelled components demonstrated approximately equal integral intensities (Figure S24 in Supporting Information File 1).
Scheme 4: Isomerization of 15a in the presence of tetrazolo[1,5-b][1,2,4]triazin-7-one 13-15N2 and isotopic e...
NMR spectroscopy and resonance assignment. The synthesized compounds were studied by NMR spectroscopy in a dimethyl sulfoxide (DMSO-d6) solution using samples with concentrations with range of 30–70 mM. The obtained 1D 15N NMR spectra are shown in Figure 2, and the 1D 1H and 13C spectra are presented as Figures S1–S18 in Supporting Information File 1. Two signals corresponding to the 15N-labelled atoms were observed in the 1D 15N NMR spectra of all the starting azolo-azines and adamantylated products (Figure 2). The 15N spectra of compounds 13-15N2, 15a,b-15N2, 23-15N2 and 24-15N2 containing labelled nitrogens in the neighbouring positions also demonstrated 13.4–16.4 Hz splittings due to the direct 1JNN coupling constants (Figure 2, Table 1).
![[1860-5397-13-250-2]](/bjoc/content/figures/1860-5397-13-250-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1D 15N NMR spectra of 30–70 mM 13-15N2, 15a,b-15N2, 20-15N2, 21a,b-15N2, 23-15N2 and 24-15N2 in DMSO-d6 (45 °C). The signal of the impurity (a base formed from salt 19-15N2 in acidic medium) is marked by an asterisk.
Figure 2: 1D 15N NMR spectra of 30–70 mM 13-15N2, 15a,b-15N2, 20-15N2, 21a,b-15N2, 23-15N2 and 24-15N2 in DMS...
Table 1: 1H and 15N chemical shifts (ppm), 1H-15N and 15N-15N J-coupling constants (Hz), and 1H-15N spin–spin interactions observed in the 2D 15N-HMBC spectra of the synthesized compounds.
| compound | δ(15N)a, JNNb, JHNc and 15N-HMBC peaksd | δ(1H)e | |||
|---|---|---|---|---|---|
|
15N-labelled
N2/N1 |
15N-labelled N3/N5/N8 | 15N at natural abundance | Ad | H2,H6/Ph | |
| 13-15N2 |
5.74 (N2)
1JN2-N3 16.4 |
−29.02 (N3)
1JN3-N2 16.4 |
8.036 (H10)
7.563 (H11) 7.622 (H12) |
||
| 15a-15N2 |
−81.81 (N2)
1JN2-N3 14.7 3JH2'-N2 0.83 (s) 4JH3'-N2 0.60 (m) 5JH4'-N2 0.23 (m) |
−46.42 (N3)
1JN3-N2 14.7 4JH2'-N3 0.06 (w) 5JH3'-N3 0.11 (–) |
2.361 (H2')f
2.306 (H3') 1.792 (H4') |
8.141 (H10)
7.535 (H11) 7.597 (H12) |
|
| 15b-15N2 |
−32.69 (N2)
1JN2-N3 15.3 4JH2'-N2 < 0.04g (–) 5JH3'-N2 0.04 (–) |
−42.14 (N3)
1JN3-N2 15.3 5JH2'-N3 < 0.04g (w) |
−159.34 (N1)
3JH2'-N1 (m) |
2.412 (H2')f
2.258 (H3') 1.776 (H4') |
8.088 (H10)
7.538 (H11) 7.589 (H12) |
| 19-15N2 |
−108.06 (N1)
2JH2-N1 15.9h |
19.27 (N5) | 8.309 (H2) | ||
| 20-15N2 |
−110.79 (N1)
2JH2-N1 16.1h |
−51.36 (N5) | 8.359 (H2) | ||
| 21a-15N2 |
−122.55 (N1)
2JH2-N1 14.0h (s) |
2.79 (N5)
5JH2-N5 0.07 (w) |
−212.12 (N3)
3JH2'-N3 (m) 3JH2-N3 (s) −249.60 (N4) 4JH2-N4 (w) −163.08 (N8) 3JH2-N8 (s) |
2.390 (H2')f
2.231 (H3') 1.752 (H4') |
9.039 (H2) |
| 21b-15N2 |
−110.24 (N1)
2JH2-N1 16.0h (s) |
−49.26 (N5)
4JH2'-N5 0.06 (w) |
159.08 (N3)
2JH2-N3 (m) −192.59 (N4) 3JH2'-N4 (m) −155.08 (N8) 3JH2-N8 (m) |
2.392 (H2')f
2.241 (H3') 1.736 (H4') |
8.409 (H2) |
| 23-15N2 |
−115.62 (N1)
1JN1-N8 13.6 2JH2-N1 14.5h 4JH6-N1 0.8h |
−156.75 (N8)
1JN8-N1 13.6 3JH2-N8 6.4h 3JH6-N8 3.5h |
8.945 (H2)
6.482 (H6) |
||
| 24-15N2 |
−120.03 (N1)
1JN1-N8 13.4 2JH2-N1 13.8h (s) 4JH6-N1 0.9h (m) |
−155.21 (N8)
1JN8-N1 13.4 3JH2-N8 6.6h (s) 3JH6-N8 3.4h (s) |
−206.85 (N3)
3JH2'-N3 (m) |
2.373 (H2')f
2.210 (H3') 1.729 (H4') |
8.996 (H2)
6.521 (H6) |
aThe 15N chemical shifts were referenced indirectly relative to MeNO3. The 15N-signals of the labelled atoms were observed in the 1D 15N NMR spectra, and the 15N-signals at natural isotopic abundance were observed in the 2D 15N-HMBC spectra. bThe JNN coupling constants were measured in the 1D 15N NMR spectra. The estimated error in the JNN values is ≈0.1 Hz. cUnless otherwise stated, the JHN values were measured using amplitude-modulated 1D 1H spin-echo experiments with delays for the evolution of JHN up to 1 s. The estimated error in the JHN values is 0.02 Hz, and the lower limit of reliable JHN measurements is 0.04 Hz. dThe cross-peaks in the 2D 15N-HMBC spectra were classified into three categories (weak – w; medium – m; strong – s). Weak peaks approximately correspond to JHN < 0.5 Hz, strong peaks approximately correspond to JHN > 2 Hz and medium peaks correspond to the other values. The degree of isotopic enrichment was accounted for. It was assumed that the intensity of the HMBC cross-peak is proportional to the sin2(π·JHN·Δ), where Δ is the delay used for the magnetization transfer (62–125 ms). (–) Indicates unobserved HMBC cross-peaks. eThe 1H chemical shifts were referenced relative to the residual signal of DMSO-d6 at 2.50 ppm. fThe signal demonstrated additional splitting, which is likely related to the slow exchange between the rotamers of adamantane substituents (see text for details). gThe measurement of the JHN values was impossible due to the fast transverse relaxation of the corresponding 1H nuclei. hThe JHN coupling constants were measured in the 1D 1H NMR spectra. The estimated error is 0.1 Hz.
The assignments of the 13C and 15N signals in the synthesized compounds were obtained by analysing the 2D 13C-HMQC, 13C-HMBC and 15N-HMBC spectra and observing the 13C-15N and 1H-15N spin–spin interactions (see below). The 13C assignment procedure for 19-15N2, 20-15N2, and 21a,b-15N2 was aided by the data from a previous study of unlabelled derivatives of compound 19 [12]. The 13C-19F J-coupling constants (nJCF, Table 2) observed in the 1D 13C spectra facilitated the assignment of the 13C nuclei for the heterocyclic moieties of compounds 23-15N2 and 24-15N2. The observations of the 3JH2-C3a coupling constants (9.2 Hz) in the 1D 13C spectra of 19-15N2 and 20-15N2 measured without proton decoupling confirmed the assignment of C3a to the signals at 160.23 ppm and 152.32 ppm, respectively. The obtained NMR assignments are collected in Table 1 (1H, 15N) and Table 2 (13C).
Table 2: 13С Chemical shifts (ppm) and 1H-13C, 13C-15N and 13C-19F J-coupling constants (Hz) of the studied compoundsa.
| compound | C2/Ph | C3a/C8a | C6 | C7 | Ad, C5, CF3 | |
|---|---|---|---|---|---|---|
| 13-15N2 |
131.35 (C9)
129.99 (C10) 128.76(C11) 132.01 (C12) |
145.99 (C8a)
2JC-N2 2.0 2JC-N3 3.3 |
152.15
4JC-N2 0.8 3JC-N3 1.5 |
154.31
4JC-N2 0.2 |
||
| 15a-15N2 |
132.70 (C9)
130.12 (C10) 128.58 (C11) 131.85 (C12) |
154.61 (C8a)
2JC-N2 0.9 2JC-N3 2.4 |
154.47
4JC-N2 0.6 3JC-N3 1.1 |
161.03
4JC-N3 0.3 |
69.26 (C1')b
1JC-N2 6.5 2JC-N3 3.8 29.44 (C3') 3JC-N2 1.6 4JC-N3 0.2 |
41.30 (C2')b
2JC-N2 0.4 3JC-N3 1.2 35.39 (C4') 4JC-N2 0.3 |
| 15b-15N2 |
132.47 (C9)
129.86 (C10) 128.65 (C11) 131.62 (C12) |
144.56 (C8a)
2JC-N2 0.7 |
151.04
4JC-N2 0.8 3JC-N3 1.8 |
160.72
4JC-N2 0.2 |
63.52 (C1')b
2JC-N2 2.7 3JC-N3 0.3 29.30 (C3') 4JC-N2 0.3 |
39.93 (C2')b,c
3JC-N2 1.1 35.67 (C4') |
| 19-15N2 |
154.98 (C2)
1JC-N1 3.7 4JC-N5 0.2 1JH2-C 206.9d |
160.23 (C3a)
2JC-N1 0.3 2JC-N5 2.0 3JH2-C 9.2d |
144.84e,f |
144.41
2JC-N1 3.6 2JC-N5 1.3 |
||
| 20-15N2 |
154.23 (C2)
1JC-N1 3.3 1JH2-C 211.0d |
152.32 (C3a)
2JC-N5 2.3 3JH2-C 9.2d |
126.82
3JC-N1 1.3 1JC-N5 1.9 |
147.62
2JC-N1 3.4 2JC-N5 1.3 |
||
| 21a-15N2 |
142.93 (C2)
1JC-N1 1.4 |
149.56 (C3a)
2JC-N1 1.8 2JC-N5 2.8 |
133.40
3JC-N1 1.3 1JC-N5 7.3 |
145.65
2JC-N1 3.1 2JC-N5 1.4 |
60.58 (C1')b
3JC-N1 0.4 29.40 (C3')b |
40.11 (C2')b,c
35.70 (C4') |
| 21b-15N2 |
153.34 (C2)
1JC-N1 3.2 |
151.10 (C3a)
2JC-N1 ≤ 0.2f 2JC-N5 2.3 |
123.80
3JC-N1 1.3 1JC-N5 2.7 |
147.04
2JC-N1 3.4 2JC-N5 1.1 |
68.15 (C1')b
4JC-N1 ≤ 0.2f 2JC-N5 5.0 29.89 (C3')b 4JC-N5 0.4 |
39.82 (C2')b,c
3JC-N5 1.7 35.92 (C4')b |
| 23-15N2 |
143.27 (C2)
1JC-N1 1.4 2JC-N8 1.0 |
151.09 (C3a)g
2JC-N1 1.8 1JC-N8 11.4 4JC-F 0.5 |
100.46
3JC-N1 1.1 2JC-N8 8.3 3JC-F 3.0 |
155.84
2JC-N1 3.0 1JC-N8 10.7 4JC-F 0.6 |
151.08 (C5)g
3JC-N8 1.1 2JC-F 34.0 |
121.63 (CF3)
4JC-N8 0.3 1JC-F 275.0 |
| 24-15N2 |
142.18 (C2)
1JC-N1 1.2 2JC-N8 1.2 |
149.36 (C3a)
2JC-N1 1.9 1JC-N8 12.0 4JC-F 0.7 |
100.88
3JC-N1 1.1 2JC-N8 8.1 3JC-F 2.8 |
156.04
2JC-N1 3.2 1JC-N8 10.4 4JC-F 0.4 |
60.58 (C1')
3JC-N1 0.4 3JC-N8 0.6 29.51 (C3') 150.48 (C5) 3JC-N8 1.2 2JC-F 34.1 |
40.14 (C2')c
35.86 (C4') 121.64 (CF3) 4JC-N8 ≤ 0.3f 1JC-F 275.0 |
aThe 13C chemical shifts were referenced indirectly relative to tetramethylsilane (TMS). Using this indirect scale, the 13C signal of DMSO-d6 was observed at 40.155 ppm. The 13C-15N and 13C-19F J-coupling constants (JCN and JCF, respectively) were measured by line-shape analysis in the 1D 13С spectra acquired with selective 15N decoupling and broadband 1H decoupling. The estimated error in the JCN values is 0.1 Hz, and the lower limit of reliable JCN measurements is 0.2 Hz. bThe signal demonstrated additional splitting, which is likely related to slow exchange between the rotamers of the adamantane substituents (see text for details). The fitted intensity ratio for the two components was 10:7. сThe signal is overlapped with the 13C signal of DMSO-d6. dThe 1H-13C J-coupling constants (JHC) were measured by line-shape analysis in the 1D 13С spectra acquired without 1H decoupling. eThe signal demonstrated additional broadening, which was not related to the JCN couplings. fPrecise measurements of JCN couplings were impossible due to low intensity of the corresponding 13С resonance. gThe C5 and C3a signals overlap.
13C-15N couplings for the structure determination of N-adamantylated azoloazines. The incorporation of 15N labels into the synthesized compounds led to the appearance of 1H-15N and 13C-15N J-coupling constants (JCN and JHN couplings, respectively). The JCN couplings became evident from the additional splitting of the corresponding signals in the 1D 13C NMR spectra and were measured by nonlinear fits of the 13С line shapes in the 1D spectra acquired with band-selective decoupling from 15N nuclei [25] (Figure 3). This method allowed for the measurement of the 13C-15N spin-spin interactions of different magnitudes and ranges starting from the direct 1JCN couplings (magnitudes of 1.2–12.0 Hz) to long-range 4JCN couplings (magnitudes of 0.2–0.8 Hz). The full list of measured JCN couplings is collected in Table 2. The couplings between adamantane carbons and the nitrogens of the heterocycles are shown in Schemes 1–3.
![[1860-5397-13-250-3]](/bjoc/content/figures/1860-5397-13-250-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Signals of the C1' and C6 atoms in the proton-decoupled 1D 13C NMR spectra of 30–42 mM 15a,b-15N2, 21a,b-15N2 and 24-15N2 in DMSO-d6 (45 ºC). The spectra were measured without (black traces) and with band-selective decoupling from the 15N1/15N2 or 15N3/15N5/15N8 nuclei (blue or red traces, respectively). The values of JCN obtained by line-shape analysis are listed. The additional splittings of the C1' signals are due to the presence of two structural forms of adamantane substituents (see text for details).
Figure 3: Signals of the C1' and C6 atoms in the proton-decoupled 1D 13C NMR spectra of 30–42 mM 15a,b-15N2, ...
The JCN couplings observed for the C6, C7 and C8a atoms in the heterocyclic moieties of compounds 13-15N2 and 15a,b-15N2 confirmed the [1,5-b]-type fusion between the azole and azine rings in these structures (Table 2, Figure 3). The observation of the direct 1JC1'-N2 (6.5 Hz) and other 13C-15N interactions for the C1' (2JC-N3 3.8 Hz), C2' (2JC-N2 0.4 Hz and 3JC-N3 1.2 Hz), C3' (3JC-N2 1.6 Hz and 4JC-N3 0.2 Hz) and C4' (4JC-N2 0.3 Hz) atoms of the adamantane group in 15a-15N2 indicated that the initial adamantylation of 13-15N2 underwent a reaction with the N2 atom of the tetrazole ring (Scheme 1). However, the detection of geminal (2JC1'-N2 2.7 Hz), vicinal (3JC2'-N2 1.1 Hz and 3JC1'-N3 0.3 Hz) and long-range (4JC3'-N2 0.3 Hz) couplings in the 1D 13C NMR spectra of 15b-15N2 revealed the attachment of the adamantane fragment to the N1 atom of the tetrazole ring.
The structures of compounds 19-15N2, 20-15N2, 21a,b-15N2, 23-15N2 and 24-15N2 were also confirmed by the measured nJCN patterns (Table 2). The presence of characteristic vicinal 3JC6-N1 coupling (magnitudes of 1.1-1.3 Hz) and other coupling constants revealed that the fusions of the triazole rings with the triazine (compounds 19, 20 and 21) or pyrimidine rings (compounds 23 and 24) have [5,1-c] or [5,1-a] configurations, respectively.
The detection of a single 3JC1'-N1 coupling (0.4 Hz) with the adamantane carbons in compound 21a-15N2 indicated that the substituent group is attached to the N3 atom of the 1,2,4-triazole ring (Figure 2, Scheme 2). Similarly, the N3-adamantylation in compound 24-15N2 was characterized by two weak 3JC1'-N1/N8 couplings (0.4/0.6 Hz) detected for the C1' atom (Figure 2, Scheme 3). In contrast, the attachment of the adamantane fragment to the N4 atom of the triazine ring in compound 21b-15N2 led to a large set of observable JCN couplings, including geminal (2JC1'-N5 5.0 Hz), vicinal (3JC2'-N5 1.7 Hz) and long-range (4JC1'-N1 ≤ 0.2 Hz and 4JC3'-N5 0.4 Hz) couplings (Figure 2, Scheme 2).
1H-15N couplings for the characterization of N-adamantylation sites in fused azolo-azines. The signal splittings due to the JHN couplings were observed in the 1D 1H spectra only in a limited number of cases (compounds 20-15N2, 21a,b-15N2, 23-15N2 and 24-15N2, see Scheme 2 and Scheme 3). In the other cases, the JHN couplings were measured by amplitude-modulated 1D 1H spin-echo experiments with selective inversion of the 15N nuclei [24] (Figure 4A, Table 1) or detected using the conventional 2D 15N-HMBC spectra (Figure 4B and Figures S19–S23 in Supporting Information File 1). These methods allowed for the straightforward detection and measurements of the geminal 2JHN (values of 13.8–16.1 Hz), vicinal 3JHN (values of 0.83–6.6 Hz) and long-range 4/5JHN (values of 0.04–0.9 Hz) couplings for the isotopically enriched nitrogen atoms. The 1H-15N spin–spin interactions with the unlabelled 15N nuclei (at natural abundance) were also detected in the 15N-HMBC experiments (Table 1). The intensities of the HMBC cross-peaks for the 15N-labelled nuclei demonstrated an approximate correlation with the measured JHN values (see Table 1). This provides a way to qualitatively estimate the JHN magnitudes for unlabelled and 15N-labelled nuclei using the relative intensities of the HMBC cross-peaks, corrected for the degree of the isotope enrichment. The measured JHN couplings and HMBC 1H-15N spin–spin interactions are shown in Schemes 1–3.
![[1860-5397-13-250-4]](/bjoc/content/figures/1860-5397-13-250-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Detection and quantification of the 1H-15N spin–spin interactions in compound 15a-15N2 (DMSO-d6, 45 °C). (A) Fragment of the 1D 1H amplitude-modulated spin-echo spectrum measured without (black trace) or with selective inversion of the 15N2 (blue trace) or 15N3 (red trace) nuclei. The spectrum was measured using a spin-echo delay (delay for the evolution of JHN) of 1 s. The measured JHN values are listed. The signals of 13C-DMSO at natural isotope abundance and the signals of impurities are marked by # and *, respectively. The concentration of these impurities relative to the concentration of 15a-15N2 does not exceed 2%. The up-field region of the spectrum is drawn with increased scaling. (B) Fragment of the 2D 15N-HMBC spectrum of 15a-15N2. The 1H-15N cross-peaks between the adamantane protons and 15N-labelled atoms are shown.
Figure 4: Detection and quantification of the 1H-15N spin–spin interactions in compound 15a-15N2 (DMSO-d6, 45...
Compound 15a-15N2 was characterized by a set of JHN couplings detected for the H2' (3/4JH2'-N2/N3 0.83/0.06 Hz), H3' (4/5JH3'-N2/N3 0.60/0.11 Hz) and H4' (5JH4'-N2 0.23 Hz) atoms of the adamantane group (Figure 4A, Table 1, Scheme 1). These spin–spin interactions, with the exception of 5JH3'-N3, were also observed in the 2D 15N-HMBC spectrum (Figure 4B). The observed JHN pattern indicated that the adamantane substituent is attached to the 15N-labelled atom (N2) of the tetrazole ring. Similarly, the observation of the 5JH3'-N2 coupling constant (≈0.04 Hz) and the medium intensity H2'-N1 HMBC cross-peak at natural 15N abundance revealed that compound 15b-15N2 contains an N-adamantane moiety attached to the unlabelled N1 atom (Scheme 1). Note that the weak cross-peak corresponding to the long-range 5JH2'-N3 coupling was observed in the HMBC spectrum of 15b-15N2 (Figure S20 in Supporting Information File 1), but the magnitude of this J coupling was under the limit of reliable JHN measurements (0.04 Hz). Thus, if the JHN couplings are too small to be measured quantitatively, the 15N-HMBC experiment could provide useful information about the position of the adamantane substituent. However, the assignment of the 15N-labelled atoms in compounds 15a,b-15N2 (differentiation between N2 and N3 resonances) could not be achieved using the JHN and 15N-HMBC data alone. The absence of protons in the tetrazolo[1,5-b][1,2,4]triazine core of these compounds dictates the necessity of JCN analysis for the unambiguous assignment of 15N-labelled nuclei.
In contrast to the situation observed for compounds 15a,b-15N2, the JHN interactions with the H2 proton in the 1,2,4-triazolo[5,1-c][1,2,4]triazines 19-15N2, 20-15N2, and 21a,b-15N2 and the H2 and H6 protons in the 1,2,4-triazolo[1,5-a]pyrimidines 23-15N2 and 24-15N2 permitted the straightforward assignments of the labelled 15N atoms (see Scheme 2 and Scheme 3). The attachment of an adamantyl substituent to the N4 atom in compound 21b-15N2 was confirmed by the measured long-range 4JH2'-N5 coupling constant (0.06 Hz) and the medium intensity H2'-N4 HMBC cross-peak observed at natural 15N abundance (Table 1, Scheme 2). Notably, the weak cross-peak corresponding to the 4JH2'-N5 coupling was also detected in the 15N-HMBC spectrum (Figure S22D in Supporting Information File 1).
For the adamantylated heterocycles 21a-15N2 and 24-15N2, the JHN interactions between the adamantane protons and the labelled N1, N5 or N8 atoms were not detected by amplitude-modulated 1H spin-echo or 15N-HMBC experiments. Meanwhile, the interactions between the H2' proton of the adamantane and the unlabelled N3 atom of the heterocyclic moieties of the compounds were observed in the 15N-HMBC spectra (Scheme 2 and Scheme 3). These results confirmed the coupling of the adamantane bridgehead C1' carbon with the N3 nitrogen of the azole ring in 21a-15N2 and 24-15N2.
The identification of adamantylation sites based on 15N-HMBC data requires the preliminary assignment of the nitrogen atoms at natural isotopic abundance. For compounds 21a,b-15N2 and 24-15N2, the required 15N assignment could be obtained by observing the 15N-HMBC correlations from the H2 and H6 protons. However, the detection of the corresponding cross-peaks was hindered by the presence of large (>3 Hz) JHN couplings with the isotopically enriched 15N-nuclei. The suppression of the magnetization transfer through the geminal 2JH2-N1 couplings by setting a delay in the 15N-HMBC experiment to 1/JHN (62.5–71.4 ms) permitted the observation of the correlations between H2 and the unlabelled N3 and N8 atoms in compounds 21a,b-15N2 (Figures S21 and S22 in Supporting Information File 1). Meanwhile, the presence of additional large vicinal couplings (3JH2-N8 and 3JH6-N8) made this strategy not applicable for compound 24-15N2. In this case, the supposed assignment of the N3 resonance was indirectly confirmed by the similarity of its chemical shifts in compounds 21a-15N2 and 24-15N2.
NMR and Х-ray diffraction data revealing several rotameric configurations of adamantane substituents. The 13C signals of the N-adamantyl substituents in compounds 15a,b-15N2 and 21a,b-15N2 measured at 45 °C demonstrated additional splitting, which was not connected to the 1H-13C and 13C-15N J-couplings. The C1' and C2' signals of adamantane (and C3' for 21a-15N2) were split into two components with a relative intensity ratio of ≈10:7 and a frequency difference of 0.5–1.2 Hz (Figure 3, Table 2). This revealed the presence of the two structural forms of the N-adamantylated heterocycles in solution with a slow (characteristic time ≥ 1 s) exchange between them. The rotation of the N-adamantyl substituents around the N–C1' bond in the bulky bicyclic heterocycles is likely hindered, and the observed conformational heterogeneity corresponds to the different rotameric configurations of the substituent. To test this hypothesis, additional NMR measurements at elevated temperature were carried out for compound 21a-15N2. The 13C 1D NMR spectrum measured at 70 °C with 1H and 15N decoupling did not demonstrate additional splitting (Figure S25 in Supporting Information File 1). This confirmed that the studied NMR samples contained unique and chemically pure compounds, while the heterogeneity observed at 45 °C was connected to the presence of different rotameric states.
To confirm the determined positions of the N-adamantane substitutions, compounds 15a and 15b were studied by X-ray crystallography. Suitable crystals of 15a and 15b were obtained by slow evaporation from ethyl acetate solutions. The solved X-ray structures were in a full agreement with the results of the JCN and JHN analysis and confirmed the N2-substituted mesoionic form for compound 15a as well as the attachment of adamantane to the N1 atom in compound 15b. In accordance with expectations, the adamantane groups in the crystals of 15a and 15b were found disordered between two conformations with different rotameric configurations around the N–C1' bond (Figure 5 and Supporting Information Files 2 and 3). These forms differ by the rotation around the N–C1' bond by 40–60°; thus, in each of them, one of the C2' atoms of the adamantane substituent is located approximately in plane with the heterocyclic moiety of the compound. The populations of the two conformational forms in the single crystals of 15a and 15b (4:1 and 17:3, respectively) differ from the populations of the conformers observed by NMR spectroscopy in DMSO solution (≈10:7). Interestingly, for 15a, the major conformer corresponds to a rotameric state with a screened N1 atom, but in the major conformer of 15b, the N2 atom of the heterocycle is screened. Notably, similar structural disorder was previously observed in the crystals of adamantylated tetrazolylpyrazole derivatives [37,41].
![[1860-5397-13-250-5]](/bjoc/content/figures/1860-5397-13-250-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP diagrams of the X-ray structures of compounds 15a-15N2 (a) and 15b-15N2 (b). For clarity, the H atoms are omitted. The observed disorder of the adamantane fragment is shown by black ellipsoids and dashed bonds (the carbon atoms are unlabelled).
Figure 5: ORTEP diagrams of the X-ray structures of compounds 15a-15N2 (a) and 15b-15N2 (b). For clarity, the...
Discussion
Comparison of different NMR approaches for the determination of N-alkylation sites in fused heterocycles. The obtained data permit a comparison of the abilities of different NMR parameters (13C and 15N chemical shifts, JHN and JCN) to provide structural information about the N-adamantylation sites in bicyclic heterocycles. The previous studies of azolo[5,1-c][1,2,4]triazin-7-ones, 1,2,4-triazolo[1,5-a]pyrimidin-7-ones and tetrazolo-azines revealed that the 13C chemical shifts of the nearest carbon atoms to N-alkyl fragments could be used as indicators for the formation of N-alkylated azolo-azines [12,42]. For the presently studied compounds, we can expect considerable changes in the chemical shifts of the bridgehead C3a and C8a atoms. The shifts of the other carbon atoms from the heterocyclic parts moieties of the compounds (C2, C5, C6, and C7) may also provide useful structural information.
In the studied tetrazolo-triazines and tetrazolo-pyrimidines (compounds 13-15N2 and 15a,b-15N2 and compounds from work [25]), the resonances of the bridgehead C8a atom were observed over a relatively narrow spectral range (144–155 ppm). In the triazolo-triazines and triazolo-pyrimidines 19-15N2, 20-15N2, 21a,b-15N2, 23-15N2, and 24-15N2, the similar bridgehead C3a atoms are shifted slightly downfield (149–160 ppm). Here, we observed that N-adamantylation of the nitrogen atom directly attached to the C3a, C8a or C2 atoms induced up-field shifts of the corresponding 13C signal. The majority of these shifts had relatively small magnitudes (Δδ −0.9 to −2.8 ppm), and a large shift was only observed for the C2 resonance of 21a-15N2 (Δδ −11.3 ppm (Table 2)). However, these up-field shifts could not be used to determine the N-adamantylation site. The attachment of the adamantane moiety to other nitrogen atoms could lead to similar 13C shifts. For example, similar Δδ values (−0.9 and −1.1 ppm) were observed for the C2 resonances in compounds 21b-15N2 and 24-15N2, where the adamantane fragments are attached to N3 and N4, respectively. Note that the C2 and N4 atoms are separated by three covalent bonds.
A similar situation was observed for the carbon atoms that are separated from the N-adamantylation site by two covalent bonds (Table 2). The attachment of adamantane to the N2 atom in compound 15a-15N2 induced a large down-field shift (Δδ +8.6 ppm) of the C8a resonance, while modification of the N4 atom in compound 21b-15N2 induced an up-field shift (Δδ −3.0 ppm) of the C6 resonance. Thus, the obtained data did not reveal an easily interpreted correlation between the 13C chemical shifts and the position of N-adamantane substituents. The same issue was previously noted in the study of N-alkylated tetrazolo[1,5-a]pyridine derivatives [43].
Similar to the situation observed for the 13C nuclei, a comparison of the 15N chemical shifts in the starting heterocycles 13-15N2, 20-15N2, and 23-15N2 and their N-adamantylated derivatives 15a,b-15N2, 21a,b-15N2, and 24-15N2 did not reveal a simple correlation with the position of the substituent group (Figure 2, Table 1). Large changes in the 15N resonance position were observed for the N2 atom in compounds 15a-15N2 (Δδ −87.6 ppm) and 15b-15N2 (Δδ −38.4 ppm) and for the N5 atom in compound 21a-15N2 (Δδ +54.2 ppm). According to the data reported for tetrazolo[1,5-a]pyridines [43], the shielding of the N2 nucleus in compound 15b-15N2 can be explained by the adamantylation of the neighbouring N1 atom in the tetrazole fragment. In contrast, the coupling of the adamantyl fragment to the N4 atom in compound 21b-15N2 did not considerable change the chemical shift of the neighbouring 15N5 nucleus (Δδ ≈ +2.1 ppm). For clarity, we should mention that the information that could be obtained from the 15N chemical shifts is restricted by the pattern of the 15N-label incorporation. In some cases, the isotopic labels were located far from the position of the attached adamantane group. This fact could partially explain the lack of correlation between chemical shifts and structure.
The obtained data indicated that changes in the 13C and 15N chemical shifts could not reliably determine the adamantylation sites in azolo-azines. Therefore, we focused our study on the analysis of 1H-15N and 13C-15N spin–spin interactions. Despite the relatively ‘sparse’ placement of 15N labels, in all the synthesized compounds, the bridgehead C1' atom of the adamantyl fragment demonstrated detectable JCN couplings (Schemes 1–3, Figure 2). The observed JCN values greatly varied in magnitude. The direct and vicinal couplings (1,2JCN) were relatively large (6.5–2.7 Hz), while the geminal and long-range interactions (3,4JCN) were small (0.6–0.2 Hz). The fact that the 1JCN and 2JCN as well as the 3JCN and 4JCN couplings for the C1' atom had similar magnitudes indicated that additional data are required for the unambiguous determination of the adamantylation sites. For this purpose, we measured and analysed the 13C-15N and 1H-15N spin–spin interactions for the other atoms of the adamantane groups (Schemes 1–3). These additional sets of 2-4JCN and 2-5JHN data reliably identified the N-adamantylation sites in the all studied compounds. The proposed structures of 15a,b-15N2 were independently confirmed by Х-ray diffraction data.
One of the advantages of JCN and JHN data compared with chemical shift data is the usefulness of ‘negative’ information. In the majority of the cases, the absence of a detectable 13C-15N or 1H-15N spin–spin interaction indicates the remote localization of the adamantane substituent and labelled nitrogen of the heterocycle. The obtained results showed that the structural information provided by the 1H-15N spin–spin interactions (measured by 1D 1H spin-echo experiments or detected in 2D 15N-HMBC experiments) is similar to the information obtained from the JCN couplings. However, these approaches are not equivalent. On one hand, the acquisition of JHN data requires less measurement time and less sophisticated equipment compared with that of JCN data (conventional broadband probe and two-channel NMR spectrometer versus triple-resonance probe and three-channel spectrometer, respectively). On the other hand, the structural characterization of the N-adamantylation site(s) in heterocycles based on the JHN data requires the preliminary assignment of the 15N resonances. Therefore, the combination of these approaches based on the analysis of JCN and JHN couplings represents the most effective NMR tool for the determination of adamantylation sites in azolo-azines.
Possible mechanisms of the isomerization of N-adamantylated derivatives 15a and 15b. The isomerization of unlabelled 15a in the presence of 13a-15N2 (Scheme 4) elucidated the possible mechanism of the isomerization of 15a-15N2 into 15b-15N2. This experiment confirmed that this rearrangement occurs via the formation of adamantyl cation 25 and heterocyclic base 13-15N2 (Scheme 5). Moreover, the equilibration of the isotope composition over the reaction products (15a*-15N2, 15b*-15N2 and 13*-15N2) indicated that the transformation of 15a into 15b is reversible. Note that the protonation of compound 13 and its adamantylated derivatives probably plays an important role in the 15a15b conversion in TFA solution. The precise positions of the attached protons are unknown, and this determination requires additional investigation, but the analysis of calculated Mulliken charges in compounds 15a and 15b (see Supporting Information File 1, Scheme S2) suggests that the most negatively charged atom N8 undergoes the initial protonation. Similar mechanisms can be proposed to describe the isomerization of compounds 21a and 21b.
![[1860-5397-13-250-i5]](/bjoc/content/inline/1860-5397-13-250-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanism of the isomerization of compounds 15a and 15b.
Scheme 5: Mechanism of the isomerization of compounds 15a and 15b.
Conclusion
We reported the selective incorporation of two 15N atoms at different positions of 1,2,4-triazolo[1,5-a]pyrimidine, azolo-1,2,4-triazines and their N-adamantylated derivatives. The selective incorporation of the 15N-labels into the azolo and azine rings of the heterocyclic structures led to the appearance of 1H-15N and 13C-15N J-coupling constants. The combined analysis of the JHN and JCN couplings allowed for the effective determination of the adamantylation sites in the azolo-azine series. To the best of our knowledge, the applicability of this approach for the structural determination of N-substituted heterocycles has not been previously considered. We suggest that the proposed method is generally applicable for the studies of N-alkylated heterocyclic compounds with a high abundance of nitrogen nuclei, where 13C chemical shifts and 1H-1H NOE data cannot provide reliable structural information. The incorporation of the 15N-labels also permitted the study of the mechanism of isomerization of N-adamantylated tetrazolo[1,5-b][1,2,4]triazin-7-one in TFA solution. The formation of an adamantyl cation and NH-tetrazolo-triazine during the isomerization reaction was confirmed.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, the synthesis of labelled compounds, crystallographic information for 15a-15N2 and 15b-15N2, computational data, and 1D 1H, 13C and 2D 15N-HMBC spectra of the synthesized compounds. | ||
| Format: PDF | Size: 3.6 MB | Download |
| Supporting Information File 2: Crystallographic data for 15a-15N2. | ||
| Format: CIF | Size: 20.1 KB | Download |
| Supporting Information File 3: Crystallographic data for 15b-15N2. | ||
| Format: CIF | Size: 20.2 KB | Download |
Acknowledgements
This work was supported by the Russian Ministry of Education and Science (State contract 4.6351.2017/8.9), the Russian Foundation for Basic Research (grant 17-03-01029) and the DAAD (scholarship 4.9988.2017/DAAD). The 1H-15N and 13C-15N J-coupling measurements were carried out using NMR equipment provided by the IBCH core facility (CKP IBCH, supported by the Russian Ministry of Education and Science, grant RFMEFI62117X0018). J. O. Subbotina thanks Compute Canada–Calcul Canada and West Grid for computing resources and Prof. Arvi Rauk (University of Calgary, Canada) for his personal assistance.
References
-
Wanka, L.; Iqbal, K.; Schreiner, P. R. Chem. Rev. 2013, 113, 3516–3604. doi:10.1021/cr100264t
Return to citation in text: [1] -
Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. Eur. J. Med. Chem. 2011, 46, 1949–1963. doi:10.1016/j.ejmech.2011.01.047
Return to citation in text: [1] -
Zarubaev, V. V.; Golod, E. L.; Anfimov, P. M.; Shtro, A. A.; Saraev, V. V.; Gavrilov, A. S.; Logvinov, A. V.; Kiselev, O. I. Bioorg. Med. Chem. 2010, 18, 839–848. doi:10.1016/j.bmc.2009.11.047
Return to citation in text: [1] -
Balgi, A. D.; Wang, J.; Cheng, D. Y. H.; Ma, C.; Pfeifer, T. A.; Shimizu, Y.; Anderson, H. J.; Pinto, L. H.; Lamb, R. A.; DeGrado, W. F.; Roberge, M. PLoS One 2013, 8, e55271. doi:10.1371/journal.pone.0055271
Return to citation in text: [1] -
Blair, L. M.; Sperry, J. J. Nat. Prod. 2013, 76, 794–812. doi:10.1021/np400124n
Return to citation in text: [1] -
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2004, 67, 1216–1238. doi:10.1021/np040031y
Return to citation in text: [1] -
Hajós, G.; Riedl, Z. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Chapter 11.16, Vol. 11; Pergamon Press: Oxford, 2008; pp 671–763. doi:10.1016/B978-008044992-0.01016-6
Return to citation in text: [1] -
Regan, A. C. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Chapter 11.12, Vol. 11; Pergamon Press: Oxford, 2008; pp 551–587. doi:10.1016/B978-008044992-0.01012-9
Return to citation in text: [1] -
Chupakhin, O. N.; Charushin, V. N.; Rusinov, V. L. Herald Russ. Acad. Sci. 2016, 86, 206–212. doi:10.1134/S1019331616030163
Return to citation in text: [1] -
Ulomskii, E. N.; Deev, S. L.; Tkachev, A. V.; Moiseev, I. K.; Rusinov, V. L. Russ. J. Org. Chem. 2002, 38, 272–280. doi:10.1023/A:1015538322029
Return to citation in text: [1] [2] [3] [4] -
Farras, J.; Fos, E.; Ramos, R.; Vilarrasa, J. J. Org. Chem. 1988, 53, 887–891. doi:10.1021/jo00239a042
Return to citation in text: [1] -
Ulomskii, E. N.; Rusinov, V. L.; Chupakhin, O. N.; Rusinov, G. L.; Chernyshev, A. I.; Aleksandrov, G. G. Chem. Heterocycl. Compd. 1987, 23, 1236–1243. doi:10.1007/BF00479378
Return to citation in text: [1] [2] [3] -
Tsypin, V. G.; Kachala, V. V.; Ugrak, B. I.; Golod, E. L. Russ. J. Org. Chem. 2002, 38, 90–94. doi:10.1023/A:1015310926725
Return to citation in text: [1] -
Gavrilov, A. S.; Golod, E. L.; Kachala, V. V.; Ugrak, B. I. Russ. J. Org. Chem. 2001, 37, 1741–1746. doi:10.1023/A:1013930219704
Return to citation in text: [1] -
Le, Z.-G.; Chen, Z.-C.; Hu, Y.; Zheng, Q.-G. Heterocycles 2004, 63, 1077–1081. doi:10.3987/COM-04-10010
Return to citation in text: [1] -
Liu, Y.; Yan, W.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 5389–5392. doi:10.1021/ol802246q
Return to citation in text: [1] -
Onaka, T.; Umemoto, H.; Miki, Y.; Nakamura, A.; Maegawa, T. J. Org. Chem. 2014, 79, 6703–6707. doi:10.1021/jo500862t
Return to citation in text: [1] -
Messmer, A.; Hajós, G.; Fleischer, J.; Czugler, M. Monatsh. Chem. 1985, 116, 1227–1231. doi:10.1007/BF00811256
Return to citation in text: [1] -
Lisakova, A. D.; Ryabukhin, D. S.; Trifonov, R. E.; Ostrovskii, V. A.; Vasilyev, A. V. Tetrahedron Lett. 2015, 56, 7020–7023. doi:10.1016/j.tetlet.2015.11.005
Return to citation in text: [1] -
Sveshnikov, N. N.; Nelson, J. H. Magn. Reson. Chem. 1997, 35, 209–212. doi:10.1002/(SICI)1097-458X(199703)35:3<209::AID-OMR40>3.0.CO;2-6
Return to citation in text: [1] -
Keder, R.; Dvořáková, H.; Dvořák, D. Eur. J. Org. Chem. 2009, 1522–1531. doi:10.1002/ejoc.200801002
Return to citation in text: [1] -
Cheatham, S.; Kline, M.; Kupče, E. Magn. Reson. Chem. 2015, 53, 363–368. doi:10.1002/mrc.4205
Return to citation in text: [1] -
Cheatham, S.; Gierth, P.; Bermel, W.; Kupče, Ē. J. Magn. Reson. 2014, 247, 38–41. doi:10.1016/j.jmr.2014.07.011
Return to citation in text: [1] -
Shestakova, T. S.; Shenkarev, Z. O.; Deev, S. L.; Chupakhin, O. N.; Khalymbadzha, I. A.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2013, 78, 6975–6982. doi:10.1021/jo4008207
Return to citation in text: [1] [2] [3] [4] -
Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lyčka, A.; Frebort, Š.; Almonasy, N. Tetrahedron Lett. 2008, 49, 4213–4215. doi:10.1016/j.tetlet.2008.04.161
Return to citation in text: [1] -
Atzrodt, J.; Beckert, R.; Günther, W.; Görls, H. Eur. J. Org. Chem. 2000, 1661–1668. doi:10.1002/(SICI)1099-0690(200004)2000:8<1661::AID-EJOC1661>3.0.CO;2-1
Return to citation in text: [1] -
Filák, L.; Riedl, Z.; Egyed, O.; Czugler, M.; Hoang, C. N.; Schantl, J. G.; Hajós, G. Tetrahedron 2008, 64, 1101–1113. doi:10.1016/j.tet.2007.10.103
Return to citation in text: [1] -
Holm, А.; Schaumburg, K.; Dahlberg, N.; Christophersen, C.; Snyder, J. P. J. Org. Chem. 1975, 40, 431–436. doi:10.1021/jo00892a010
Return to citation in text: [1] -
Elashry, E. S. H.; Rashed, N. Adv. Heterocycl. Chem. 1998, 72, 127–224. doi:10.1016/S0065-2725(08)60316-5
Return to citation in text: [1] -
El Ashry, E. S. H.; Rashed, N.; Taha, M.; Ramadan, E. Adv. Heterocycl. Chem. 1994, 59, 39–177. doi:10.1016/S0065-2725(08)60007-0
Return to citation in text: [1] -
Shchegol'kov, E. V.; Ivanova, A. E.; Burgart, Y. V.; Saloutin, V. I. J. Heterocycl. Chem. 2013, 50, E80–E86. doi:10.1002/jhet.1068
Return to citation in text: [1] -
Khalymbadzha, I. A.; Shestakova, T. S.; Deev, S. L.; Rusinov, V. L.; Chupakhin, O. N.; Shenkarev, Z. O.; Arseniev, A. S. Russ. Chem. Bull. 2013, 62, 521–528. doi:10.1007/s11172-013-0072-7
Return to citation in text: [1] -
Saraev, V. V.; Kanakina, T. P.; Pevzner, M. S.; Golod, E. L.; Ugrak, B. I.; Kachala, V. V. Chem. Heterocycl. Compd. 1996, 32, 928–936. doi:10.1007/BF01176969
Return to citation in text: [1] -
Saraev, V. V.; Golod, E. L. Russ. J. Org. Chem. 1997, 33, 571–574.
Return to citation in text: [1] -
Amandurdyeva, A. D.; Saraev, V. V.; Kuz’mina, N. E.; Golod, E. L. Russ. J. Gen. Chem. 2004, 74, 1277–1281. doi:10.1007/s11176-005-0151-z
Return to citation in text: [1] -
Gavrilov, A. S.; Kachala, V. V.; Kuz’mina, N. E.; Golod, E. L. Russ. J. Gen. Chem. 2004, 74, 752–762. doi:10.1023/B:RUGC.0000039090.05255.64
Return to citation in text: [1] [2] -
Shestakova, T. S.; Khalymbadzha, I. A.; Deev, S. L.; Eltsov, O. S.; Rusinov, V. L.; Shenkarev, Z. O.; Arseniev, A. S.; Chupakhin, O. N. Russ. Chem. Bull. 2011, 60, 729–732. doi:10.1007/s11172-011-0113-z
Return to citation in text: [1] -
Rusinov, V. L.; Ulomskii, E. N.; Chupakhin, O. N.; Petrov, A. Y.; Sharonov, E. A. Chem. Heterocycl. Compd. 1989, 25, 209–213. doi:10.1007/BF00479921
Return to citation in text: [1] -
Gaussian 09; Gaussian, Inc.: Wallingford CT, 2009.
Return to citation in text: [1] -
Cabildo, P.; Claramunt, R. M.; Sanz, D. Tetrahedron 1985, 41, 473–478. doi:10.1016/S0040-4020(01)96441-5
Return to citation in text: [1] -
Kushnir, M. N.; Rusinov, V. L.; Ulomskii, E. N.; Klyuev, N. A.; Shorshnev, S. V.; Aleksandrov, G. G.; Chupakhin, O. N. Russ. J. Org. Chem. 1993, 29, 525–533.
Return to citation in text: [1] -
Cmoch, P.; Wiench, J. W.; Stefaniak, L.; Sitkowski, J. J. Mol. Struct. 1999, 477, 119–125. doi:10.1016/S0022-2860(98)00589-4
Return to citation in text: [1] [2]
| 10. | Ulomskii, E. N.; Deev, S. L.; Tkachev, A. V.; Moiseev, I. K.; Rusinov, V. L. Russ. J. Org. Chem. 2002, 38, 272–280. doi:10.1023/A:1015538322029 |
| 12. | Ulomskii, E. N.; Rusinov, V. L.; Chupakhin, O. N.; Rusinov, G. L.; Chernyshev, A. I.; Aleksandrov, G. G. Chem. Heterocycl. Compd. 1987, 23, 1236–1243. doi:10.1007/BF00479378 |
| 1. | Wanka, L.; Iqbal, K.; Schreiner, P. R. Chem. Rev. 2013, 113, 3516–3604. doi:10.1021/cr100264t |
| 5. | Blair, L. M.; Sperry, J. J. Nat. Prod. 2013, 76, 794–812. doi:10.1021/np400124n |
| 6. | Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2004, 67, 1216–1238. doi:10.1021/np040031y |
| 10. | Ulomskii, E. N.; Deev, S. L.; Tkachev, A. V.; Moiseev, I. K.; Rusinov, V. L. Russ. J. Org. Chem. 2002, 38, 272–280. doi:10.1023/A:1015538322029 |
| 24. | Shestakova, T. S.; Shenkarev, Z. O.; Deev, S. L.; Chupakhin, O. N.; Khalymbadzha, I. A.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2013, 78, 6975–6982. doi:10.1021/jo4008207 |
| 25. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 26. | Lyčka, A.; Frebort, Š.; Almonasy, N. Tetrahedron Lett. 2008, 49, 4213–4215. doi:10.1016/j.tetlet.2008.04.161 |
| 27. | Atzrodt, J.; Beckert, R.; Günther, W.; Görls, H. Eur. J. Org. Chem. 2000, 1661–1668. doi:10.1002/(SICI)1099-0690(200004)2000:8<1661::AID-EJOC1661>3.0.CO;2-1 |
| 28. | Filák, L.; Riedl, Z.; Egyed, O.; Czugler, M.; Hoang, C. N.; Schantl, J. G.; Hajós, G. Tetrahedron 2008, 64, 1101–1113. doi:10.1016/j.tet.2007.10.103 |
| 29. | Holm, А.; Schaumburg, K.; Dahlberg, N.; Christophersen, C.; Snyder, J. P. J. Org. Chem. 1975, 40, 431–436. doi:10.1021/jo00892a010 |
| 43. | Cmoch, P.; Wiench, J. W.; Stefaniak, L.; Sitkowski, J. J. Mol. Struct. 1999, 477, 119–125. doi:10.1016/S0022-2860(98)00589-4 |
| 4. | Balgi, A. D.; Wang, J.; Cheng, D. Y. H.; Ma, C.; Pfeifer, T. A.; Shimizu, Y.; Anderson, H. J.; Pinto, L. H.; Lamb, R. A.; DeGrado, W. F.; Roberge, M. PLoS One 2013, 8, e55271. doi:10.1371/journal.pone.0055271 |
| 24. | Shestakova, T. S.; Shenkarev, Z. O.; Deev, S. L.; Chupakhin, O. N.; Khalymbadzha, I. A.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2013, 78, 6975–6982. doi:10.1021/jo4008207 |
| 25. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 3. | Zarubaev, V. V.; Golod, E. L.; Anfimov, P. M.; Shtro, A. A.; Saraev, V. V.; Gavrilov, A. S.; Logvinov, A. V.; Kiselev, O. I. Bioorg. Med. Chem. 2010, 18, 839–848. doi:10.1016/j.bmc.2009.11.047 |
| 21. | Keder, R.; Dvořáková, H.; Dvořák, D. Eur. J. Org. Chem. 2009, 1522–1531. doi:10.1002/ejoc.200801002 |
| 25. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 2. | Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. Eur. J. Med. Chem. 2011, 46, 1949–1963. doi:10.1016/j.ejmech.2011.01.047 |
| 22. | Cheatham, S.; Kline, M.; Kupče, E. Magn. Reson. Chem. 2015, 53, 363–368. doi:10.1002/mrc.4205 |
| 23. | Cheatham, S.; Gierth, P.; Bermel, W.; Kupče, Ē. J. Magn. Reson. 2014, 247, 38–41. doi:10.1016/j.jmr.2014.07.011 |
| 43. | Cmoch, P.; Wiench, J. W.; Stefaniak, L.; Sitkowski, J. J. Mol. Struct. 1999, 477, 119–125. doi:10.1016/S0022-2860(98)00589-4 |
| 11. | Farras, J.; Fos, E.; Ramos, R.; Vilarrasa, J. J. Org. Chem. 1988, 53, 887–891. doi:10.1021/jo00239a042 |
| 12. | Ulomskii, E. N.; Rusinov, V. L.; Chupakhin, O. N.; Rusinov, G. L.; Chernyshev, A. I.; Aleksandrov, G. G. Chem. Heterocycl. Compd. 1987, 23, 1236–1243. doi:10.1007/BF00479378 |
| 15. | Le, Z.-G.; Chen, Z.-C.; Hu, Y.; Zheng, Q.-G. Heterocycles 2004, 63, 1077–1081. doi:10.3987/COM-04-10010 |
| 16. | Liu, Y.; Yan, W.; Chen, Y.; Petersen, J. L.; Shi, X. Org. Lett. 2008, 10, 5389–5392. doi:10.1021/ol802246q |
| 37. | Gavrilov, A. S.; Kachala, V. V.; Kuz’mina, N. E.; Golod, E. L. Russ. J. Gen. Chem. 2004, 74, 752–762. doi:10.1023/B:RUGC.0000039090.05255.64 |
| 41. | Cabildo, P.; Claramunt, R. M.; Sanz, D. Tetrahedron 1985, 41, 473–478. doi:10.1016/S0040-4020(01)96441-5 |
| 10. | Ulomskii, E. N.; Deev, S. L.; Tkachev, A. V.; Moiseev, I. K.; Rusinov, V. L. Russ. J. Org. Chem. 2002, 38, 272–280. doi:10.1023/A:1015538322029 |
| 17. | Onaka, T.; Umemoto, H.; Miki, Y.; Nakamura, A.; Maegawa, T. J. Org. Chem. 2014, 79, 6703–6707. doi:10.1021/jo500862t |
| 18. | Messmer, A.; Hajós, G.; Fleischer, J.; Czugler, M. Monatsh. Chem. 1985, 116, 1227–1231. doi:10.1007/BF00811256 |
| 19. | Lisakova, A. D.; Ryabukhin, D. S.; Trifonov, R. E.; Ostrovskii, V. A.; Vasilyev, A. V. Tetrahedron Lett. 2015, 56, 7020–7023. doi:10.1016/j.tetlet.2015.11.005 |
| 20. | Sveshnikov, N. N.; Nelson, J. H. Magn. Reson. Chem. 1997, 35, 209–212. doi:10.1002/(SICI)1097-458X(199703)35:3<209::AID-OMR40>3.0.CO;2-6 |
| 12. | Ulomskii, E. N.; Rusinov, V. L.; Chupakhin, O. N.; Rusinov, G. L.; Chernyshev, A. I.; Aleksandrov, G. G. Chem. Heterocycl. Compd. 1987, 23, 1236–1243. doi:10.1007/BF00479378 |
| 42. | Kushnir, M. N.; Rusinov, V. L.; Ulomskii, E. N.; Klyuev, N. A.; Shorshnev, S. V.; Aleksandrov, G. G.; Chupakhin, O. N. Russ. J. Org. Chem. 1993, 29, 525–533. |
| 9. | Chupakhin, O. N.; Charushin, V. N.; Rusinov, V. L. Herald Russ. Acad. Sci. 2016, 86, 206–212. doi:10.1134/S1019331616030163 |
| 25. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 7. | Hajós, G.; Riedl, Z. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Chapter 11.16, Vol. 11; Pergamon Press: Oxford, 2008; pp 671–763. doi:10.1016/B978-008044992-0.01016-6 |
| 8. | Regan, A. C. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Chapter 11.12, Vol. 11; Pergamon Press: Oxford, 2008; pp 551–587. doi:10.1016/B978-008044992-0.01012-9 |
| 13. | Tsypin, V. G.; Kachala, V. V.; Ugrak, B. I.; Golod, E. L. Russ. J. Org. Chem. 2002, 38, 90–94. doi:10.1023/A:1015310926725 |
| 14. | Gavrilov, A. S.; Golod, E. L.; Kachala, V. V.; Ugrak, B. I. Russ. J. Org. Chem. 2001, 37, 1741–1746. doi:10.1023/A:1013930219704 |
| 24. | Shestakova, T. S.; Shenkarev, Z. O.; Deev, S. L.; Chupakhin, O. N.; Khalymbadzha, I. A.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2013, 78, 6975–6982. doi:10.1021/jo4008207 |
| 30. | Elashry, E. S. H.; Rashed, N. Adv. Heterocycl. Chem. 1998, 72, 127–224. doi:10.1016/S0065-2725(08)60316-5 |
| 24. | Shestakova, T. S.; Shenkarev, Z. O.; Deev, S. L.; Chupakhin, O. N.; Khalymbadzha, I. A.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2013, 78, 6975–6982. doi:10.1021/jo4008207 |
| 10. | Ulomskii, E. N.; Deev, S. L.; Tkachev, A. V.; Moiseev, I. K.; Rusinov, V. L. Russ. J. Org. Chem. 2002, 38, 272–280. doi:10.1023/A:1015538322029 |
| 38. | Shestakova, T. S.; Khalymbadzha, I. A.; Deev, S. L.; Eltsov, O. S.; Rusinov, V. L.; Shenkarev, Z. O.; Arseniev, A. S.; Chupakhin, O. N. Russ. Chem. Bull. 2011, 60, 729–732. doi:10.1007/s11172-011-0113-z |
| 39. | Rusinov, V. L.; Ulomskii, E. N.; Chupakhin, O. N.; Petrov, A. Y.; Sharonov, E. A. Chem. Heterocycl. Compd. 1989, 25, 209–213. doi:10.1007/BF00479921 |
| 25. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 34. | Saraev, V. V.; Kanakina, T. P.; Pevzner, M. S.; Golod, E. L.; Ugrak, B. I.; Kachala, V. V. Chem. Heterocycl. Compd. 1996, 32, 928–936. doi:10.1007/BF01176969 |
| 35. | Saraev, V. V.; Golod, E. L. Russ. J. Org. Chem. 1997, 33, 571–574. |
| 36. | Amandurdyeva, A. D.; Saraev, V. V.; Kuz’mina, N. E.; Golod, E. L. Russ. J. Gen. Chem. 2004, 74, 1277–1281. doi:10.1007/s11176-005-0151-z |
| 37. | Gavrilov, A. S.; Kachala, V. V.; Kuz’mina, N. E.; Golod, E. L. Russ. J. Gen. Chem. 2004, 74, 752–762. doi:10.1023/B:RUGC.0000039090.05255.64 |
| 25. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 33. | Khalymbadzha, I. A.; Shestakova, T. S.; Deev, S. L.; Rusinov, V. L.; Chupakhin, O. N.; Shenkarev, Z. O.; Arseniev, A. S. Russ. Chem. Bull. 2013, 62, 521–528. doi:10.1007/s11172-013-0072-7 |
| 31. | El Ashry, E. S. H.; Rashed, N.; Taha, M.; Ramadan, E. Adv. Heterocycl. Chem. 1994, 59, 39–177. doi:10.1016/S0065-2725(08)60007-0 |
| 32. | Shchegol'kov, E. V.; Ivanova, A. E.; Burgart, Y. V.; Saloutin, V. I. J. Heterocycl. Chem. 2013, 50, E80–E86. doi:10.1002/jhet.1068 |
© 2017 Deev et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)