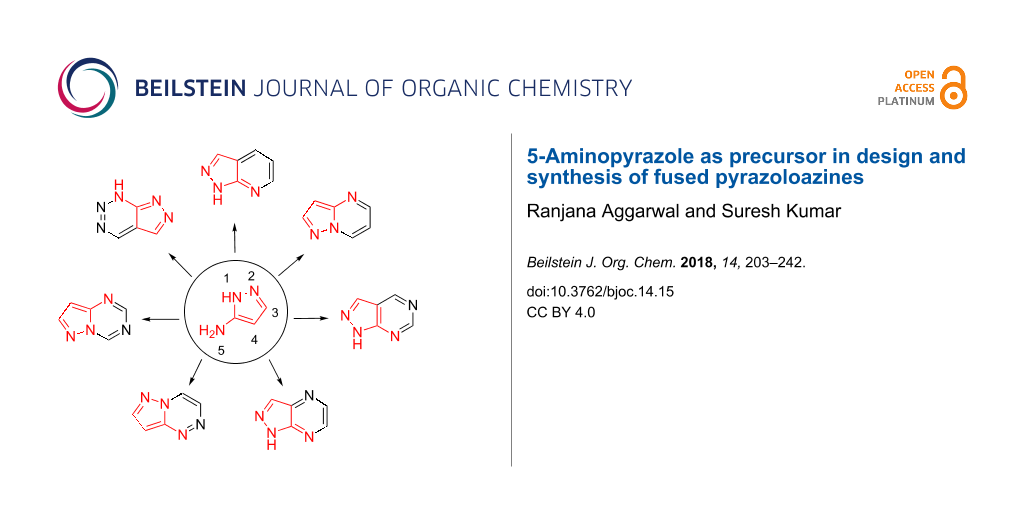
Abstract
The condensation of 5-aminopyrazole with various bielectrophilic moieties results in the formation of pyrazoloazines, an interesting array of fused heterocyclic systems. The development of new synthetic routes towards pyrazoloazines for their biological and medicinal exploration is an attractive area for researchers throughout the world. The present review focuses on various synthetic methods developed in the last decade for the synthesis of differently substituted pyrazoloazines by a broad range of organic reactions by means of 5-aminopyrazole as a precursor.

Graphical Abstract
Review
Pyrazole and its derivatives are known to exhibit significant biological and pharmacological activities such as: anticancer [1,2], anti-inflammatory [3,4], antioxidant [5], antibacterial [6-8], analgesic [9], antiviral [10,11], antimicrobial [12,13], antifungal [6], antiglycemic [14], antiamoebic [15] and antidepressive [16,17]. Considering the immense biological properties pyrazole is one of the most widely studied nitrogen-containing heterocyclic nuclei. Fused pyrazole derivatives are composed of the pyrazole nucleus attached to other heterocyclic moieties which enable them to exhibit improved pharmacological activities compared to the isolated fragments. These compounds are currently used in several marketed drugs like cartazolate (1), zaleplon (2), sildenafil (3), allopurinol (4), indiplon (5), etazolate (6) etc. (Figure 1). Fused pyrazole derivatives, especially pyrazoloazines have been reported to mimic purine bases, present in DNA and RNA, due to close structural resemblance.
![[1860-5397-14-15-1]](/bjoc/content/figures/1860-5397-14-15-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of drugs with fused pyrazole rings.
Figure 1: Selected examples of drugs with fused pyrazole rings.
In addition to the immense biological potential related to fused pyrazoles, their synthetic potential needs to be reviewed for further improvements and extension of interests. Various efforts have been developed for the synthesis of pyrazole-based fused heterocycles. 5-Aminopyrazoles have been extensively employed as useful synthons in designing and constructing a plethora of fused pyrazoloazines of potential synthetic and medicinal interest viz pyrazolo[3,4-b]pyridines 7 [18], pyrazolo[1,5-a]pyrimidines 8 [19], pyrazolo[3,4-d]pyrimidines 9 [20,21], pyrazolo[3,4-b]pyrazines 10 [22], pyrazolo[5,1-c]-1,2,4-triazines 11 [23], pyrazolo[1,5-a]-1,3,5-triazines 12 [24], pyrazolo[3,4-d][1,2,3]triazines 13 [25] (Figure 2).
![[1860-5397-14-15-2]](/bjoc/content/figures/1860-5397-14-15-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Typical structures of some fused pyrazoloazines from 5-aminopyrazoles.
Figure 2: Typical structures of some fused pyrazoloazines from 5-aminopyrazoles.
A number of review articles have been published by us and others highlighting the synthetic and biological aspects of 5-aminopyrazoles [26-28] as well as on the synthesis of fused pyrazole derivatives [25]. However, a perusal of literature reveals that the importance of 5-aminopyrazoles as synthetic precursors for fused heterocycles has not been reported till now to the best of our knowledge. Recent literature shows resurgence of interest in the chemistry and bioactivity of 5-aminopyrazole derivatives leading to improvements in several already known reactions and syntheses of various fused heterocyclic derivatives with various biological activities. Considering the synthetic importance of 5-aminopyrazoles and synthesis of fused pyrazole derivatives with the need for a more general collection, herein we report an exhaustive overview of the main developments in the last decade in the chemistry of 5-aminopyrazoles for the design and synthesis of fused pyrazoloazines.
The typical reactivity of 5-aminopyrazoles
5-Aminopyrazoles are polyfunctional compounds possessing three typical nucleophilic sites: 4-CH, 1-NH and 5-NH2 with the following reactivity order: 5-NH2 > 1-NH > 4-CH. These positions have been used to construct various fused heterocyclic rings where 5-aminopyrazoles undergo cyclization and cycloaddition on reaction with bielectrophiles. Due to the large number of references, reactions of 5-aminopyrazoles with various reagents to construct a six membered ring with pyrazole are discussed. The synthetic methods have been arranged in order of the ascending number of heteroatoms in the azine ring. The systematic arrangement in this review explores the possibility of providing practical guidance to synthetic chemists for further research.
Synthesis of pyrazolo[3,4-b]pyridines
Pyrazolo[3,4-b]pyridines are important fused heterocycles due to their well-known synthetic and medicinal potential as good vasodilators [29], hypotensive [30], HIV reverse transcriptase inhibitors [31], protein kinase inhibitors [32], antiallergic [33], antioxidant [34] and as fungicide [35]. Also, the pyrazolo[3,4-b]pyridine ring system is a key structure in drug discovery and has become the main component in many medicinally important compounds. The large number of synthetic routes to pyrazolo[3,4-b]pyridines and their applications brings great interest in this area. The most commonly applied method for the preparation of pyrazolo[3,4-b]pyridines uses 5-aminopyrazole as synthetic precursor [36-39]. Regardless to substantial studies in this field, researchers are still focused to provide convenient regioselective synthetic methods with mild conditions and good yields of the reactions [40,41].
Aggarwal et al. [42] reported the regiospecific synthesis of 4-trifluoromethyl-1H-pyrazolo[3,4-b]pyridines 18 by the reaction of 5-aminopyrazole 16 with trifluoromethyl-β-diketones 17 in refluxing acetic acid (Scheme 1). In the same report the other regioisomers 6-trifluoromethylpyrazolo[3,4-b]pyridines 20 were obtained under multicomponent solvent-free conditions by the reaction of hydrazine 14, β-ketonitrile 15 and β-diketone 17 as an exclusive product. The structures of both the regioisomers have been confirmed unambiguously by HMBC, HMQC and 19F NMR studies. The authors proposed that trifluoromethyl-β-diketone exists mainly in keto form 17 under solvent-free conditions whereas under solvent-mediated conditions the enolic form 21 towards the carbonyl carbon that carries the CF3 group is predominant. The keto form 17 results in the formation of 6-trifluoromethylpyrazolo[3,4-b]pyridines 20 by attack of the 5-NH2 group (from 5-aminopyrazole 16) on the more electrophilic carbonyl group attached to CF3 (from trifluoromethyl-β-diketones 17) whereas the enolic form 21 reacts with the less nucleophilic C-4 of 5-aminopyrazole and leads to the formation of 4-CF3 product 18. The formation of acetamide 19 as byproduct under solvent-mediated conditions was also observed due to the reaction of NH2 group with acetic acid.
![[1860-5397-14-15-i1]](/bjoc/content/inline/1860-5397-14-15-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Regiospecific synthesis of 4 and 6-trifluoromethyl-1H-pyrazolo[3,4-b]pyridines.
Scheme 1: Regiospecific synthesis of 4 and 6-trifluoromethyl-1H-pyrazolo[3,4-b]pyridines.
Bardajee et al. [43] reported the synthesis of ethyl 1,3,4-triphenyl-1H-pyrazolo[3,4-b]pyridine-6-carboxylate (23) from the reaction of 5-aminopyrazole (R = Ph, 16) and ethyl 2,4-dioxo-4-phenylbutanoate (22, Scheme 2). The presence of an electron-withdrawing group on the aryl ring provided higher yields due to the increased electrophilicity of the carbonyl carbon. Electron-donating groups on the contrary decreased the electrophilicity of the carbonyl carbon and hence resulted in lower yields.
![[1860-5397-14-15-i2]](/bjoc/content/inline/1860-5397-14-15-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of pyrazolo[3,4-b]pyridine-6-carboxylates.
Scheme 2: Synthesis of pyrazolo[3,4-b]pyridine-6-carboxylates.
The synthesis of 3-methyl-1,4,6-triaryl-1H-pyrazolo[3,4-b]pyridines 25 was described by Shi et al. [44] from the reaction of 3-methyl-1-phenyl-1H-pyrazol-5-amine (R = Ph, R1 = Me, 16) and α,β-unsaturated ketones 24 (Scheme 3) in the ionic solvent [bmim]Br at 90 °C with excellent yield. Variation of the aryl substituents on the α,β-unsaturated ketones 24 has no significant effect on the reaction. The reaction was proposed to occur through a sequence of Michael addition, cyclization, dehydration and aromatization reactions. The use of ionic liquids (non-volatile solvents) over toxic organic solvents makes it an environmentally benign process [45,46].
![[1860-5397-14-15-i3]](/bjoc/content/inline/1860-5397-14-15-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1,4,6-triaryl-1H-pyrazolo[3,4-b]pyridines with ionic liquid .
Scheme 3: Synthesis of 1,4,6-triaryl-1H-pyrazolo[3,4-b]pyridines with ionic liquid .
The synthesis of isomeric tetracyclic pyrazolo[3,4-b]pyridine-based coumarin chromophores 27 and 28 was reported by Chen et al. [47] starting from 7-diethylaminocoumarin-3-aldehyde (26) and 5-aminopyrazole derivatives 16 (Scheme 4). The structure of the synthesized compounds was confirmed by X-ray crystallography, 1H and 13C NMR and HRMS studies. The relationships between the structures and chemical properties of these compounds were also investigated by techniques like fluorescence spectroscopy, single photon counting technique, cyclic voltammetry, thermogravimetric analysis, and DFT calculations.
![[1860-5397-14-15-i4]](/bjoc/content/inline/1860-5397-14-15-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of coumarin-based isomeric tetracyclic pyrazolo[3,4-b]pyridines.
Scheme 4: Synthesis of coumarin-based isomeric tetracyclic pyrazolo[3,4-b]pyridines.
Boruah et al. [48,49] developed an efficient method for the construction of regioisomeric 1,3,4-trisubstituted pyrazolo[3,4-b]pyridines 32 and 34 (Scheme 5). In situ cyclocondensation of β-halovinyl aldehydes 29 with 5-aminopyrazoles (R = Ph, 16) under Heck conditions in the presence of Pd(OAc)2 with xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) gave 6-substituted pyrazolo[3,4-b]pyridines 34. On the other hand, isolated imine intermediate 30 under similar conditions provided 4-substituted pyrazolo[3,4-b]pyridine 31 in DMF (Scheme 5). This intramolecular coupling reaction provided highly efficient synthetic procedure for the design and synthesis of pyrazolo[3,4-b]pyridine-nucleus-based pharmacological agents with high regioselectivity.
![[1860-5397-14-15-i5]](/bjoc/content/inline/1860-5397-14-15-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 6-substituted pyrazolo[3,4-b]pyridines under Heck conditions.
Scheme 5: Synthesis of 6-substituted pyrazolo[3,4-b]pyridines under Heck conditions.
Working on similar lines Boruah et al. [49] further explored the reactivity of 5-aminopyrazoles 16 with β-halovinyl/aryl aldehydes 33 under conventional heating and microwave conditions in DMF and DMSO with Pd(OAC)2 (2.5 mol %) catalyst with PPh3 as ligand (Scheme 5). Interestingly, high yields of the corresponding pyrazolo[3,4-b]pyridines 34 were obtained when reactions were carried under solvent-free microwave irradiation. The synthesized pyrazolo[3,4-b]pyridines have shown potential cytotoxic activity against cervical HeLa and prostate DU 205 cancer cell lines.
A similar in situ intramolecular cyclization of 5-aminopyrazole-4-caroxylate 35 with β-haloaldehydes 36 via the corresponding imine derivative was carried out in presence of Pd(PPh3)2Cl2 (1.0 mol %), Cu2O (1.0 mol %), 1,10-phenanthroline (2.0 mol %), TBAI (6 mol %), by Batra et al. [50] to generate the pyrazolo[3,4-b]pyridine nucleus 37 (Scheme 6).
![[1860-5397-14-15-i6]](/bjoc/content/inline/1860-5397-14-15-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Microwave-assisted palladium-catalyzed synthesis of pyrazolo[3,4-b]pyridines.
Scheme 6: Microwave-assisted palladium-catalyzed synthesis of pyrazolo[3,4-b]pyridines.
Aziz et al. [51] developed an acid-catalyzed synthesis of pyrazolo[3,4-b]pyridine derivatives 40 through the reaction of enaminone 38 with 5-aminopyrazole (R = Ph, 16) in acetic acid (Scheme 7). The proposed reaction mechanism involves the generation of new enaminone intermediate 39 which underwent condensation and cyclization within C-4 of 5-aminopyrazole and the carbonyl group of the enaminone to generate pyrazolo[3,4-b]pyridine derivatives 40. However, the formation of pyrazolo[1,5-a]pyrimidine 41, a structural isomer of 40 was obtained when 1-NH-5-aminopyrazole (R = H, 16) was condensed with 38. It was attributed to cyclocondensation between 1-NH (5-aminopyrazole) and the carbonyl carbon of the enaminone. The compounds were found to have cytotoxicity against the normal fibroblast (BHK) cell line and antitumor activity against the colon cancer cell line CaCO-2.
![[1860-5397-14-15-i7]](/bjoc/content/inline/1860-5397-14-15-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Acid-catalyzed synthesis of pyrazolo[3,4-b]pyridines via enaminones.
Scheme 7: Acid-catalyzed synthesis of pyrazolo[3,4-b]pyridines via enaminones.
Lin et al. [52] developed the synthesis of pyrazolo[3,4-b]pyridine derivatives 45 via aza-Diels–Alder reaction of pyrazolylimines 43 with maleimides 44 (Scheme 8). Pyrazolylimines 43 were in turn obtained from the reaction of 5-aminopyrazole 16 with diisopropylformamide dimethyl acetal (R’ = isopropyl, 42). The reactions were carried out with various metal catalysts in acetic acid and acetonitrile solvents but reactions carried in acetic acid in presence of silica gel impregnated with indium trichloride provided the best results. Júnior et al. [53] also used N,N-dimethylpyrazoylimines 42 with N-arylmaleimides 44 in a solvent-free methodology based on microwave-assisted (80 W, 80 °C, 1.5 h) hetero-Diels–Alder reaction for the synthesis of pyrazolo[3,4-b]pyridine derivatives 45 (Scheme 8).
![[1860-5397-14-15-i8]](/bjoc/content/inline/1860-5397-14-15-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of pyrazolo[3,4-b]pyridines via aza-Diels–Alder reaction.
Scheme 8: Synthesis of pyrazolo[3,4-b]pyridines via aza-Diels–Alder reaction.
Jiang et al. [54] described the synthesis of macrocyclane-fused pyrazolo[3,4-b]pyridine derivatives 49 by the reaction of 5-aminopyrazole derivative 46, arylaldehydes 47 and cyclic ketones 48 in various solvents like acetonitrile, ethylene glycol, acetic acid, DMF under MW conditions at 80 °C (Scheme 9). The best results (72–80% yields) were obtained by carrying out the reaction in acetic acid with the addition of TFA as promoter at 80 °C to 140 °C.
![[1860-5397-14-15-i9]](/bjoc/content/inline/1860-5397-14-15-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of macrocyclane fused pyrazolo[3,4-b]pyridine derivatives.
Scheme 9: Synthesis of macrocyclane fused pyrazolo[3,4-b]pyridine derivatives.
A three-component reaction of 5-aminopyrazole 16, 4-hydroxycoumarin (50) and aldehydes 47 was studied by Liu et al. [55] in various solvents like acetonitrile, dichloromethane, toluene and DMSO in the presence of catalysts like ZrCl4, InCl3, FeCl3, L-proline etc. (Scheme 10). Whereas the reaction in acetic acid/acetonitrile (1:5) provided 4,7-dihydro-1H-pyrazolo[3,4-b]pyridine derivatives 51, dimethyl sulfoxide/acetic acid (5:1) yielded the corresponding aromatized pyrazolo[3,4-b]pyridine derivatives 52 exclusively. In acetic acid/ethanol combination an unexpected product 4,5-dihydro-1H-pyrazolo[3,4-b]pyridine-6(7H)-one 53 was formed due to C–O bond cleavage from cyclic ester 51.
![[1860-5397-14-15-i10]](/bjoc/content/inline/1860-5397-14-15-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Three-component synthesis of 4,7-dihydro-1H-pyrazolo[3,4-b]pyridine derivatives.
Scheme 10: Three-component synthesis of 4,7-dihydro-1H-pyrazolo[3,4-b]pyridine derivatives.
Bazgir et al. [56] described the synthesis of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine]-2,6'(1'H)-diones 55 by an efficient three-component procedure from the reaction of 5-aminopyrazole 16 and 4-hydroxycoumarin (50) with isatin 54 under ultrasound irradiation in water (Scheme 11). Solvent and catalytic screening for the reaction have shown that water in presence of p-TSA at 60 °C on heating for 6 hours provide best results with excellent yields.
![[1860-5397-14-15-i11]](/bjoc/content/inline/1860-5397-14-15-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Ultrasonicated synthesis of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine]-2,6'(1'H)-diones.
Scheme 11: Ultrasonicated synthesis of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine]-2,6'(1'H)-diones.
Recently, Wang et al. [57] also described the construction of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine] derivatives 57 from the multicomponent reaction of 5-amino-3-hydroxy-1-phenyl-1H-pyrazole (46), ketones 56 and isatin 54 in water/acetic acid (3:1) at 90 °C (Scheme 12).
![[1860-5397-14-15-i12]](/bjoc/content/inline/1860-5397-14-15-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Synthesis of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine] derivatives under conventional heating conditions.
Scheme 12: Synthesis of spiro[indoline-3,4'-pyrazolo[3,4-b]pyridine] derivatives under conventional heating co...
Quiroga et al. [58] reported the synthesis of the pyrazolo[3,4-b]pyridine-spiroindolinone nucleus 59 with a high degree of regioselectivity without formation of the regioisomeric pyrazolo[1,5-a]pyrimidine 60 involving three-component reaction of 5-aminopyrazole 16, isatin 54 and cyclic β-diketones 58 in aqueous ethanol with p-TSA as catalyst (Scheme 13). Bhaumik et al. [59] carried out a similar reaction of 5-aminopyrazole (R = H, R1 = Me, 16), isatin 54 and cyclic-1,3-diones 58 in aqueous ethanol using aluminosilicate nanoparticles catalyst to yield pyrazolo[3,4-b]pyridines 61 (Scheme 13).
![[1860-5397-14-15-i13]](/bjoc/content/inline/1860-5397-14-15-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Nanoparticle-catalyzed synthesis of pyrazolo[3,4-b]pyridine-spiroindolinones.
Scheme 13: Nanoparticle-catalyzed synthesis of pyrazolo[3,4-b]pyridine-spiroindolinones.
Dandia et al. [60] carried out the multicomponent synthesis of spiropyrazolo[3,4-b]pyridines 63 and 64 starting from 5-aminopyrazole (R = H, R1 = Me, 16), isatin 54 and α-cyanoacetic ester 62 or 15 in aqueous-mediated reaction in presence of NaCl. Regioisomeric pyrazolo[1,5-a]pyrimidines 65 were not formed in any of the tried reaction conditions. An increase in the amount of NaCl from 2.5 to 10 mol % resulted in gradual increase of the yield of the desired product 63 from 85% to 89% and 93%, respectively (Scheme 14). Recently, Jiang et al. [61] have also developed a microwave-assisted synthesis of spiropyrazolo[3,4-b]pyridines 66 via a similar type of three-component reaction of 5-aminopyrazole 16, isatin 54 and 3-oxo-3-phenylpropanenitriles 15 in acetic acid under microwave irradiation at 80 °C in just 20 minutes (Scheme 14).
![[1860-5397-14-15-i14]](/bjoc/content/inline/1860-5397-14-15-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Microwave-assisted multicomponent synthesis of spiropyrazolo[3,4-b]pyridines.
Scheme 14: Microwave-assisted multicomponent synthesis of spiropyrazolo[3,4-b]pyridines.
Hao et al. [62] described the unexpected synthesis of naphthoic acid substituted pyrazolo[3,4-b]pyridine derivatives 70 via a three-component reaction of 5-aminopyrazole (R = Me, 16) with acenaphthenequinone 67 and β-ketonitrile derivative 68 in glacial acetic acid instead of expected spiropyrazolo[3,4-b]pyridines 69 (Scheme 15). The structures of the products were confirmed by spectral and X-ray crystallographic data. This method provides the first direct conversion of acenaphthenequinone to a naphthoic acid fragment via C–C bond cleavage in a single step.
![[1860-5397-14-15-i15]](/bjoc/content/inline/1860-5397-14-15-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Unexpected synthesis of naphthoic acid-substituted pyrazolo[3,4-b]pyridines.
Scheme 15: Unexpected synthesis of naphthoic acid-substituted pyrazolo[3,4-b]pyridines.
Recently, D. Anand et al. [63] have reported the synthesis of pyrazolo[3,4-b]pyridine derivatives 71 and 72 through the multicomponent reaction of 1-aryl-3-indolyl-5-aminopyrazoles 16, cyclic β-diketones 58 and aryl aldehydes 47 (Scheme 16). The reaction resulted in good yields of pyrazolo[3,4-b]pyridines 72 but in few cases 4,7-dihydropyrazolo[3,4-b]pyridines 71 were formed as major product even after prolonged heating. 4,7-Dihydropyrazolo[3,4-b]pyridines 71 were dehydrogenated to their aromatic counterparts 72 in presence of 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in acetonitrile.
![[1860-5397-14-15-i16]](/bjoc/content/inline/1860-5397-14-15-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Multicomponent synthesis of variously substituted pyrazolo[3,4-b]pyridine derivatives.
Scheme 16: Multicomponent synthesis of variously substituted pyrazolo[3,4-b]pyridine derivatives.
Insuasty et al. [64] adapted a similar synthetic strategy for the construction of 4,7-dihydropyrazolo[3,4-b]pyridines 73 and pyrazolo[3,4-b]pyridines 74 by a three-component reaction of 5-aminopyrazoles 16, cyclic β-diketones 58 and heteroaryl aldehydes 47 (Scheme 17). The reaction under conventional heating in DMF provided best results with high yields of the corresponding pyrazolo[3,4-b]pyridines 74.
![[1860-5397-14-15-i17]](/bjoc/content/inline/1860-5397-14-15-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Three-component synthesis of 4,7-dihydropyrazolo[3,4-b]pyridines and pyrazolo[3,4-b]pyridines.
Scheme 17: Three-component synthesis of 4,7-dihydropyrazolo[3,4-b]pyridines and pyrazolo[3,4-b]pyridines.
The multicomponent reactions of 5-(4-substituted-benzylamino)pyrazoles 75, cyclic-β-diketones 58 and formaldehyde (R’= H, 47) were performed under microwave and conventional heating conditions by Quiroga et al. [65] (Scheme 18). Both the reaction conditions resulted in the formation of pyrazolo[3,4-b]pyridine-5-spirocycloalkanediones 76 but an additional compound 3-tert-butyl-1-phenylindeno[2,3-e]pyrazolo[3,4-b]pyridine 77 was formed in the reaction when indandione 58 was used as β-diketone which was attributed to the loss of the benzyl fragment from 5-aminopyrazole derivative 75. Microwave-assisted reactions went to completion in very short time (5 min) compared to reactions under conventional heating conditions (24 hours).
![[1860-5397-14-15-i18]](/bjoc/content/inline/1860-5397-14-15-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of pyrazolo[3,4-b]pyridine-5-spirocycloalkanediones.
Scheme 18: Synthesis of pyrazolo[3,4-b]pyridine-5-spirocycloalkanediones.
A three-component reaction of 5-aminopyrazole 16, arylaldehydes 47 and indandione 58 under ultrasonic irradiation in ethanol was developed by Nikpassand et al. [66] to synthesize pyrazolo[3,4-b]pyridine derivatives 78 (Scheme 19). Ultrasound-mediated reactions yielded the corresponding pyrazolo[3,4-b]pyridine derivatives 78 in 4–5 minutes with 88–97% yields.
![[1860-5397-14-15-i19]](/bjoc/content/inline/1860-5397-14-15-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Ultrasound-mediated three-component synthesis of pyrazolo[3,4-b]pyridines.
Scheme 19: Ultrasound-mediated three-component synthesis of pyrazolo[3,4-b]pyridines.
Yao et al. [67] demonstrated that 4-aryl-3-methyl-1-phenyl-4,6,8,9-tetrahydropyrazolo[3,4-b]thiopyrano[4,3-e]pyridin-5(1H)-one derivatives 80 could be synthesized from a three-component reaction of 5-aminopyrazole 16, arylaldehyde 47, and 2H-thiopyran-3,5(4H,6H)-dione (79) in glacial acetic acid in presence of ammonium acetate (Scheme 20).
![[1860-5397-14-15-i20]](/bjoc/content/inline/1860-5397-14-15-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Multicomponent synthesis of 4-aryl-3-methyl-1-phenyl-4,6,8,9-tetrahydropyrazolo [3,4-b]thiopyrano[4,3-e]pyridin-5(1H)-ones.
Scheme 20: Multicomponent synthesis of 4-aryl-3-methyl-1-phenyl-4,6,8,9-tetrahydropyrazolo [3,4-b]thiopyrano[4...
The multicomponent reaction of 5-amino-3-hydroxypyrazoles 82, substituted salicylic aldehydes 83 and acetylacetic ester 81 in acetic acid with few drops of piperidine was reported to give 2,3-dihydrochromeno[4,3-d]pyrazolo[3,4-b]pyridine-1,6-diones 84 by Frolova et al. [68] (Scheme 21). The reactions with ethyl benzoylacetate as ketoester component had not provided the corresponding pyrazolo[3,4-b]pyridines which was attributed to the change in the electronic and steric environments. All the synthesized compounds were reported as good antimicrobial agents.
![[1860-5397-14-15-i21]](/bjoc/content/inline/1860-5397-14-15-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Synthesis of 2,3-dihydrochromeno[4,3-d]pyrazolo[3,4-b]pyridine-1,6-diones.
Scheme 21: Synthesis of 2,3-dihydrochromeno[4,3-d]pyrazolo[3,4-b]pyridine-1,6-diones.
Recently the reaction of β-ketoesters 81 as in the three-component reaction with 5-aminopyrazoles 16 and substituted salicylic aldehydes 83 was also studied by Fan et al. [69]. An extensive survey of catalysts and solvents identified 0.2 equivalents of FeCl3 and ethanol as optimal catalyst and solvent, respectively, with which o-hydroxyphenylpyrazolo[3,4-b]pyridine derivatives 85 were obtained in 89% yields with no formation of the cyclized isomer chromenopyrazolo[3,4-b]pyridine 86. The reaction in the presence of other catalysts like L-proline, InCl3 and ZrCl4 also resulted in the formation of o-hydroxyphenylpyrazolo[3,4-b]pyridine derivatives 85 but no product was formed in iodine- and acetic acid-catalyzed reactions (Scheme 22).
![[1860-5397-14-15-i22]](/bjoc/content/inline/1860-5397-14-15-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: FeCl3-catalyzed synthesis of o-hydroxyphenylpyrazolo[3,4-b]pyridine derivatives.
Scheme 22: FeCl3-catalyzed synthesis of o-hydroxyphenylpyrazolo[3,4-b]pyridine derivatives.
Huang et al. [70] investigated a three-component reaction of β-ketonitriles 15, 5-aminopyrazole 16 and aldehydes 47 in various organic solvents and ionic liquids to synthesize pyrazolo[3,4-b]pyridine derivative 87 (Scheme 23). Ionic liquids provided high yields of 87 in very short time with the best results obtained in [bmim]Br whereas organic solvents resulted in low yields and took longer time for the completion of reaction.
![[1860-5397-14-15-i23]](/bjoc/content/inline/1860-5397-14-15-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Ionic liquid-mediated synthesis of pyrazolo[3,4-b]pyridines.
Scheme 23: Ionic liquid-mediated synthesis of pyrazolo[3,4-b]pyridines.
El-borai et al. [71] accomplished the synthesis of pyrazolo[3,4-b]pyridine derivatives 88 in which the multicomponent reactions of β-ketonitriles 15, 5-aminopyrazole 16 and anisaldehyde (47) were carried out in acetic acid under conventional heating and microwave assistance (Scheme 24). The microwave-assisted reaction provided better yields of pyrazolo[3,4-b]pyridine derivatives 88 as compared to reactions under conventional heating conditions in short time.
![[1860-5397-14-15-i24]](/bjoc/content/inline/1860-5397-14-15-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Microwave-assisted synthesis of pyrazolo[3,4-b]pyridines.
Scheme 24: Microwave-assisted synthesis of pyrazolo[3,4-b]pyridines.
Hill et al. [72,73] reported the synthesis of pyrazolo[3,4-b]pyridines 89 from the reaction β-ketonitriles 15 with 5-aminopyrazole 16 and aldehydes 47 (1 equiv each) in presence of triethylamine (2 equiv) by heating the reaction mixture at 90 °C in DMF for 16 hours followed by treatment with sodium nitrite (3 equiv) in acetic acid at ambient temperature. In addition, when the R1 group has significant bulk (R1 = tert-butyl) the reaction results in the formation of pyrazolo[1,5-a]pyrimidine derivative 90 as an additional product. The authors proposed that the bulky group had significantly slowed down the rate of electrophilic aromatic substitution at C-4 on 1H-pyrazol-5-amine due to which the aza-Michael addition becomes competitive at N-1 which ultimately provides pyrazolo[1,5-a]pyrimidine derivative 90 as additional product (Scheme 25). The synthesized pyrazolo[3,4-b]pyridines 89 were found to be good mGluR5 positive allosteric modulators (PAMs) and therefore can be used to develop antipsychotic drugs to treat schizophrenia.
![[1860-5397-14-15-i25]](/bjoc/content/inline/1860-5397-14-15-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Multicomponent synthesis of pyrazolo[3,4-b]pyridine-5-carbonitriles.
Scheme 25: Multicomponent synthesis of pyrazolo[3,4-b]pyridine-5-carbonitriles.
In an interesting report Aggarwal et al. [74] described the synthesis of 4,7-dihydropyrazolo[3,4-b]pyridine-5-nitriles 92 from the reaction of β-ketonitriles 15 with several aryl/heteroaryl hydrazines 14 in ethanol with a catalytic amount of conc. HNO3 (Scheme 26). The authors carried out the reaction under acidic conditions expecting the formation of the regioisomeric 3/5-aminopyrazoles 16/91 but the reaction under the influence of conc. HNO3 resulted in the formation of an unexpected product which was characterized as 4,7-dihydropyrazolo[3,4-b]pyridine 92 through rigorous spectroscopic studies. However, X-ray crystallographic studies indicated that the 4,7-dihydropyrazolo[3,4-b]pyridine-5-nitriles 92 underwent aerial oxidation to its aromatic counterpart pyrazolo[3,4-b]pyridine 93 during crystallization and is propeller in shape. Additionally, non-planar rings due to propeller shape of compound 93 makes it chiral in nature. It was proposed that there is in situ oxidation of ethanol to ethanal by conc. HNO3 which turned the reaction into a multi-component domino assembly of reactants hydrazine 14, β-ketonitriles 15 and acetaldehyde.
![[1860-5397-14-15-i26]](/bjoc/content/inline/1860-5397-14-15-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Unusual domino synthesis of 4,7-dihydropyrazolo[3,4-b]pyridine-5-nitriles.
Scheme 26: Unusual domino synthesis of 4,7-dihydropyrazolo[3,4-b]pyridine-5-nitriles.
Rahmati [75] carried out a reaction of 5-aminopyrazole 16 with aldehydes 47 and ethyl cyanoacetate (94) in ethanol in presence of p-toluenesulfonic acid which resulted in a diastereomeric mixture of cis- and trans-4,5,6,7-tetrahydro-2H-pyrazolo[3,4-b]pyridines 95. Benzaldehydes 47 with electron withdrawing groups provided better yields of the cis-isomer in slightly higher amounts than the trans-isomer. A four-component reaction having ethyl acetoacetate (81) as fourth component resulted in the formation of the same pyrazolo[3,4-b]pyridine derivative 95 showing no involvement of any additional fourth component (Scheme 27).
![[1860-5397-14-15-i27]](/bjoc/content/inline/1860-5397-14-15-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Synthesis of 4,5,6,7-tetrahydro-4H-pyrazolo[3,4-b]pyridines under conventional heating and ultrasound irradiation.
Scheme 27: Synthesis of 4,5,6,7-tetrahydro-4H-pyrazolo[3,4-b]pyridines under conventional heating and ultrasou...
Dandia et al. [76] also reported a similar reaction of 5-aminopyrazole 16, arylaldehyde 47 with ethyl cyanoacetate (94) under ultrasound irradiation in presence of p-TSA in water for the synthesis of 3-methyl-6-oxo-4-aryl-4,5,6,7-tetrahydro-4H-pyrazolo[3,4-b]pyridine-5-carbonitrile derivatives 95 (Scheme 27). All the synthesized compounds were tested for their effect on corrosion of mild steel (MS) in 1.0 M HCl with various experimental techniques like weight loss, electrochemical impedance spectroscopy (EIS), and potentiodynamic polarization techniques.
A three-component reaction of 5-aminopyrazole 16, arylaldehyde 47 and N-methyl-1-(methylthio)-2-nitroethenamine (96) was studied by Gunasekaran et al. [77] (Scheme 28) in ethanol in presence of 30 mol % L-proline as catalyst at 78 °C which resulted in the production of pyrazolo[3,4-b]pyridine derivatives 97 in excellent yields.
![[1860-5397-14-15-i28]](/bjoc/content/inline/1860-5397-14-15-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: L-Proline-catalyzed synthesis of of pyrazolo[3,4-b]pyridine.
Scheme 28: L-Proline-catalyzed synthesis of of pyrazolo[3,4-b]pyridine.
Jiang et al. [78] have investigated a microwave-irradiated reaction of 5-aminopyrazoles 16, 2,2-dihydroxy-1-phenylethanone (98) and p-toluidine (99) under various polar and non-polar solvents with bronsted and lewis acid catalysts (Scheme 29). The reaction in dimethylformamide in presence of p-TSA resulted in the formation of azepino[5,4,3-cd]indole 100 instead of expected pyrazolo[3,4-b]pyridine derivatives 101. However, the reactions of arylglyoxal 98 having an electron-donating group at C-4 position of the phenyl ring resulted in the formation of the desired pyrazolo[3,4-b]pyridines 101.
![[1860-5397-14-15-i29]](/bjoc/content/inline/1860-5397-14-15-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Microwave-assisted synthesis of 5-aminoarylpyrazolo[3,4-b]pyridines.
Scheme 29: Microwave-assisted synthesis of 5-aminoarylpyrazolo[3,4-b]pyridines.
Wang et al. [79] studied the base-catalyzed multicomponent domino reaction of 5-aminopyrazoles 16, cyclic 1,3-diones 58 and arylglyoxals 98 under microwave irradiation. Triethylamine (20 mol %) as base and DMSO as solvent at 120 °C provided best results with high yields of pyrazolo[3,4-e]indolizines (a derivative of pyrazolo[3,4-b]pyridines) 102 (Scheme 30).
![[1860-5397-14-15-i30]](/bjoc/content/inline/1860-5397-14-15-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Microwave-assisted multi-component synthesis of pyrazolo[3,4-e]indolizines.
Scheme 30: Microwave-assisted multi-component synthesis of pyrazolo[3,4-e]indolizines.
Synthesis of pyrazolo[1,5-a]pyrimidines
Pyrazolo[1,5-a]pyrimidines, structural isomers of pyrazolo[3,4-b]pyridines, are of interest because they constitute an important class of heterocycles which display biological and pharmacological activities and are useful precursors in the synthesis of many biologically active compounds [80-83]. Consequently, there has been an ongoing interest in the synthesis of pyrazolo[1,5-a]pyrimidines [84-86].
Navarrete et al. [87] reported the reaction of acetylacetone (104) with 5-amino-3-(4-iodophenyl)pyrazole 103 in ethanol that gives pyrazolo[1,5-a]pyrimidine derivatives 105 which were subsequently used to prepare alkynyl alcohol 111 derivatives of pyrazolo[1,5-a]pyrimidines 106 by a Sonogashira coupling in 69–94% yields. Fluorodeoxygention of 106 using deoxofluor afforded fluoropropynyl-substituted pyrazolo[1,5-a]pyrimidine 107 with variable efficiency in terms of yields. Alternatively, by shuffling the steps of acetylacetone condensation and fluoroalkynylation, via 5-amino-3-(4-(fluoroalkynyl)phenyl)pyrazole 108 intermediate, a better and efficient route to synthesize 107 was developed (Scheme 31). Recently, the same research group [19] also reported the synthesis of fluoroalkyl-substituted pyrazolo[1,5-a]pyrimidine derivatives 112 and 114 using similar synthetic strategies (Scheme 31). Alkynyl alcohol derivatives of pyrazolo[1,5-a]pyrimidines 106 were hydrogenated to give alkyl alcohol-substituted pyrazolo[1,5-a]pyrimidines 113 which on treatment with deoxofluor resulted in the formation of fluoroalkyl-pyrazolo[1,5-a]pyrimidines 114. Additionally, reaction of 104 with 103 in refluxing ethanol resulted in the formation of pyrazolo[1,5-a]pyrimidines 109 which on treatment with TBAF provided alkyl alcohol derivatives of pyrazolo[1,5-a]pyrimidines 110 which were later on converted to fluoroalkyl pyrazolo[1,5-a]pyrimidines 112 by treatment with deoxofluor.
![[1860-5397-14-15-i31]](/bjoc/content/inline/1860-5397-14-15-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Synthesis of fluoropropynyl and fluoroalkyl substituted pyrazolo[1,5-a]pyrimidine.
Scheme 31: Synthesis of fluoropropynyl and fluoroalkyl substituted pyrazolo[1,5-a]pyrimidine.
Marjani et al. [88] have described the synthesis of pyrazolo[1,5-a]pyrimidine 116 and 4,7-dihydropyrazolo[1,5-a]pyrimidinone derivatives 117 by condensing 4-cyano/carboxylate-5-aminopyrazole derivatives 115 with acetylacetone (104) and various β-ketoesters 81, respectively, in refluxing acetic acid with catalytic amount of suphuric acid (Scheme 32).
![[1860-5397-14-15-i32]](/bjoc/content/inline/1860-5397-14-15-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Acid-catalyzed synthesis of pyrazolo[1,5-a]pyrimidine derivatives.
Scheme 32: Acid-catalyzed synthesis of pyrazolo[1,5-a]pyrimidine derivatives.
The reaction of 3(5)-amino-5(3)-hydrazinopyrazole dihydrochloride (118) with symmetrical and unsymmetrical diketones 119 was studied by Aggarwal et al. [80,89] under aqueous conditions. The reaction exhibited a high level of chemoselectivity and regiospecificity yielding 2-(3-methylpyrazol-1-yl)-5-methylpyrazolo[1,5-a]pyrimidines 120 out of the four possible isomers (Scheme 33). In the case of arylbutadiones, formation of two more products in small amounts namely 3(5)-methyl-5(3)-phenyl-1H-pyrazole (121) obtained by CN bond cleavage and benzoic acid (122) was also observed (Scheme 33). The structure of the regioisomer was established unequivocally by performing 1H,13C-HMQC, 1H,13C- and 1H,15N-HMBC experiments. Aqueous mediated conditions makes it a sought after the procedure for the synthesis of pyrazolo[1,5-a]pyrimidines.
![[1860-5397-14-15-i33]](/bjoc/content/inline/1860-5397-14-15-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Chemoselective and regiospecific synthesis of 2-(3-methylpyrazol-1’-yl)-5-methylpyrazolo[1,5-a]pyrimidines.
Scheme 33: Chemoselective and regiospecific synthesis of 2-(3-methylpyrazol-1’-yl)-5-methylpyrazolo[1,5-a]pyri...
In another report, Aggarwal et al. [90] have described a regioselective synthesis of 2-H/methyl-3-phenyl-7-trifluoromethylpyrazolo[1,5-a]pyrimidines 124 by condensing 4-aryl-5-aminopyrazoles 123 with an equimolar amount of trifluoromethyl-β-diketones 17. To gain an insight of the reaction mechanism, the intermediate, 5-methyl-3-phenyl-7-trifluoromethyl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-5,7-diol 125 was isolated by performing the reaction in DCM at −15 °C for 6 h which was later converted to 7-trifluoromethylpyrazolo[1,5-a]pyrimidine derivative 124 by dehydration on refluxing with acetic anhydride (Scheme 34). All the synthesized compounds were screened for their anti-inflammatory activity.
![[1860-5397-14-15-i34]](/bjoc/content/inline/1860-5397-14-15-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Regioselective synthesis of 7-trifluoromethylpyrazolo[1,5-a]pyrimidines.
Scheme 34: Regioselective synthesis of 7-trifluoromethylpyrazolo[1,5-a]pyrimidines.
Mulakayala and co-workers [91] synthesized 7-trifluoromethylpyrazolo[1,5-a]pyrimidine carboxylates 127 by the reaction of 5-aminopyrazole-4/3-ethylcarboxylates 126 with trifluoromethyl-β-diketones 17 in acetic acid under microwave heating, which were subsequently hydrolyzed to the corresponding pyrazolo[1,5-a]pyrimidine carboxylic acids 128 by treating with sodium hydroxide in ethanol at 65 °C. The compounds were screened for their cytotoxic activity against human colon carcinoma (Colo-205) cell lines (Scheme 35).
![[1860-5397-14-15-i35]](/bjoc/content/inline/1860-5397-14-15-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Microwave-assisted synthesis of 7-trifluoromethylpyrazolo[1,5-a]pyrimidine carboxylates.
Scheme 35: Microwave-assisted synthesis of 7-trifluoromethylpyrazolo[1,5-a]pyrimidine carboxylates.
Buriol et al. [92] described the reaction of 5-aminopyrazole 16 with 4-alkoxy-1,1,1-trifluoro-3-alken-2-ones 129 to yield pyrazolo[1,5-a]pyrimidine derivatives 130 in acetic acid and ethanol using conventional heating, ultrasound and microwave conditions (Scheme 36). The reaction in ethanol provided best results with high yields of pyrazolo[1,5-a]pyrimidines 130. The effect of microwave irradiation was found to be as efficient as of ultrasound radiations with better yields and shorter reaction times than conventional heating methods.
![[1860-5397-14-15-i36]](/bjoc/content/inline/1860-5397-14-15-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Microwave and ultrasound-assisted synthesis of 7-trifluoromethylpyrazolo[1,5-a]pyrimidines.
Scheme 36: Microwave and ultrasound-assisted synthesis of 7-trifluoromethylpyrazolo[1,5-a]pyrimidines.
Recently, Boruah et al. [93] reported an unprecedented synthesis of pyrazolo[1,5-a]pyrimidines 132 involving a C–C bond cleavage through KOt-Bu-catalyzed condensation of 1,3,5-trisubstituted pentane-1,5-diones 131 with substituted 5-aminopyrazoles 16 in ethanol. Symmetrical 1,5-dicarbonyls reacted efficiently with 5-aminopyrazoles 16 to give the corresponding substituted pyrazolo[1,5-a]pyrimidines 132 (Scheme 37). Moreover, the reaction of 1,5-dicarbonyls 131 with 5-amino-3-methylpyrazole 16 provided a mixture of two regioisomeric pyrazolo[1,5-a]pyrimidines.
![[1860-5397-14-15-i37]](/bjoc/content/inline/1860-5397-14-15-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Base-catalyzed unprecedented synthesis of pyrazolo[1,5-a]pyrimidines via C–C bond cleavage.
Scheme 37: Base-catalyzed unprecedented synthesis of pyrazolo[1,5-a]pyrimidines via C–C bond cleavage.
Kamal et al. [94] reported the synthesis of aminobenzothiazole linked pyrazolo[1,5-a]pyrimidine conjugates (benzothiazolyl derivatives, 136). Methyl-2,7-diphenylpyrazolo[1,5-a]pyrimidine-5-carboxylates, obtained by the reactions of 5-aminopyrazole 16 with aryl-β-diketoesters 133 in ethanol, were hydrolyzed in presence of methanolic sodium hydroxide to give corresponding carboxylic acids 134. Aminobenzothiazoles 135 were linked with carboxylic acids 134 to provide the amide derivatives (benzothiazolyl derivatives, 136) using amide coupling reagent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide–hydroxybenzotriazole (Scheme 38). The compounds were found to possess good anticancer activity against human colon, leukemia, melanoma cancer cell lines. In another report, Kamal et al. [95] synthesized similar pyrazolo[1,5-a]pyrimidinyl amide derivatives (piperazinyl derivatives, 136) by condensing piperazin-1-yl(pyridin-3-yl)methanone (piperazinyl derivatives, 135) with 134 which were evaluated for their cytotoxic potential against MCF-7, HeLa, IMR 32 and SiHa cancer cell lines. Pyrazolo[1,5-a]pyrimidinyl amide derivatives 136 having piperazinyl derivatives, R1 = H, R2 = F and OCH3, R3 = H and OCH3, respectively, were found to be the most active compounds showing a minimum survival of the cancer cells.
![[1860-5397-14-15-i38]](/bjoc/content/inline/1860-5397-14-15-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Synthesis of aminobenzothiazole/piperazine linked pyrazolo[1,5-a]pyrimidines.
Scheme 38: Synthesis of aminobenzothiazole/piperazine linked pyrazolo[1,5-a]pyrimidines.
Griffith et al. [96] described the synthesis of 7-hydroxypyrazolo[1,5-a]pyrimidine derivatives 137 by cyclocondensation of 5-aminopyrazole 126 (X = Me, Y = Ar) with ethyl acetoacetate 81. Pyrazolo[1,5-a]pyrimidine derivatives 137 thus obtained were treated with POCl3 to give the 7-chloropyrazolo[1,5-a]pyrimidine derivative which on coupling with the appropriately substituted diamine derivatives provided aminoalkylpyrazolo[1,5-a]pyrimidine-7-amines 139. Substituted ethylenediamines resulted in the product formed by addition from the less sterically hindered amino group. The free amino group was alkylated by reductive amination on reaction with substituted aldehyde or ketones to provide the corresponding pyrazolo[1,5-a]pyrimidine derivatives 139 (Scheme 39). The pyrazolo[1,5-a]pyrimidine derivatives 139 were evaluated as neuropeptide NPY Y1R antagonists with high binding affinity and selectivity.
![[1860-5397-14-15-i39]](/bjoc/content/inline/1860-5397-14-15-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Synthesis of aminoalkylpyrazolo[1,5-a]pyrimidine-7-amines.
Scheme 39: Synthesis of aminoalkylpyrazolo[1,5-a]pyrimidine-7-amines.
Using a similar approach Dwyer et al. [97] reported the synthesis of various pyrazolo[1,5-a]pyrimidinyl derivatives 142, 143, 145 and 146 following a sequence of reactions as depicted in Scheme 40 and Scheme 41. 7-Chloropyrazolo[1,5-a]pyrimidines 140 obtained by 4-H/cyano/carboxylate-5-aminopyrazoles 126 (X = H, Y = R) and β-ketoesters 81 followed by chlorination with POCl3, were converted to 7-aminopyrazolo[1,5-a]pyrimidines 142 and 7-methoxy/thiomethoxypyrazolo[1,5-a]pyrimidines 143 on treatment of NIS, NH3 141 in propanol and NaOMe/NaSMe in THF, respectively. Further, compound 143 was coupled with 3-pyrazolylboronate to give 3-pyrazolylpyrazolo[1,5-a]pyrimidines 145 and subsequently converted to 7-amino-3-pyrazolylpyrazolo[1,5-a]pyrimidines 146 (Scheme 41). The synthesized pyrazolo[1,5-a]pyrimidine derivatives were evaluated for their CHK1 kinase inhibitory activity. Pyrazolo[1,5-a]pyrimidine derivative 142 with R1 = 3-(1-methylpyrazolyl), R2 = H, R = 3-pyridyl and R´ = 5-(3-methylthiazolyl) was found to be the most potent, selective CHK1 inhibitor.
![[1860-5397-14-15-i40]](/bjoc/content/inline/1860-5397-14-15-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Synthesis of pyrazolo[1,5-a]pyrimidines from condensation of 5-aminopyrazole 126 and ethyl acetoacetate.
Scheme 40: Synthesis of pyrazolo[1,5-a]pyrimidines from condensation of 5-aminopyrazole 126 and ethyl acetoace...
![[1860-5397-14-15-i41]](/bjoc/content/inline/1860-5397-14-15-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Synthesis of 7-aminopyrazolo[1,5-a]pyrimidines.
Scheme 41: Synthesis of 7-aminopyrazolo[1,5-a]pyrimidines.
Azeredo et al. [98] reported a similar synthesis of 7-arylaminopyrazolo[1,5-a]pyrimidines 143 by the coupling reaction of 7-chloropyrazolo[1,5-a]pyrimidines 140 with various aryl amines 141 in ethanol, which were evaluated for their anti-Plasmodium falciparum, antimalarial, and Pf-dihydroorotate dehydrogenase inhibitory activity (Scheme 40). 7-Arylaminopyrazolo[1,5-a]pyrimidine derivative 142 with R = CF3, R1 = H, R2 = CH3 and R´ = 7-β-naphthyl was found to be having highest selectivity and inhibition with IC50 = 0.16 ± 0.01 mM.
Synthesis of 7-aminopyrazolo[1,5-a]pyrimidines 146 was also reported by Hylsov et al. [99] using an almost similar synthetic procedure by coupling 7-chloropyrazolo[1,5-a]pyrimidines 140 with 3-picolylamine in acetonitrile at reflux temperature (Scheme 40).
Recently, Aggarwal et al. [100] reported an unexpected synthesis of 7-aminopyrazolo[1,5-a]pyrimidine (R’ = R, 148) from the reaction of 3(5)-amino-5(3)-hydrazinopyrazole dihydrochloride (147) with 3-oxo-3-arylpropanenitrile 15 under solvent-free grinding conditions. The reaction was proposed to proceed via formation of hydrazine by C–N bond cleavage which under reaction conditions provided 7-aminopyrazolo[1,5-a]pyrimidines 148 on coupling with 3-oxo-3-arylpropanenitrile 15 (Scheme 42). The structure of compounds 148 was established by the combined use of NMR and DFT calculations.
![[1860-5397-14-15-i42]](/bjoc/content/inline/1860-5397-14-15-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: Unexpected synthesis of 7-aminopyrazolo[1,5-a]pyrimidines under solvent free and solvent-mediated conditions.
Scheme 42: Unexpected synthesis of 7-aminopyrazolo[1,5-a]pyrimidines under solvent free and solvent-mediated c...
In another report Aggarwal et al. [101] synthesized similar 7-aminopyrazolo[1,5-a]pyrimidine 148 from the reaction of hydrazine hydrate with two different 3-oxo-3-arylpropanenitriles 15 which are successively added one after the other in toluene/ethanol (9:1) at reflux temperature in presence of p-TSA. The reaction carried out in pure ethanol provided a mixture of 5-aminopyrazoles (Scheme 42). The synthesized 7-aminopyrazolo[1,5-a]pyrimidines 148 were found to be good anti-inflammatory agents.
Tian et al. [102] reported a protocol for the efficient synthesis of pyrazolo[1,5-a]pyrimidine-5,7-dione (150) by the reaction of 5-aminopyrazole (126) with diethyl malonate (149). Pyrazolo[1,5-a]pyrimidine-5,7-dione (150) was chlorinated to give 5,7-dichloropyrazolo[1,5-a]pyrimidine (151) which subsequently coupled with various phenols 152 at the more reactive 7-position under mild reaction conditions in presence of K2CO3 in acetic acid/DMF to give pyrazolo[1,5-a]pyrimidine derivative 153. Various aromatic amines 154 were then coupled at 5-postion under Buchwald–Hartwig conditions to get the desired 5-aminopyrazolo[1,5-a]pyrimidine derivatives 155 (Scheme 43). All the synthesized compounds were screened for their anti-HIV activities in MT4 cell cultures and compound 155 (R1 = 2,4,6-trimethyl and R2 = 4-cyano) was found as most inhibiting for HIV-1 replication having an EC50 = 0.070 μM and the SI (selectivity index) = 3999, which were better than the drugs NVP (nevirapine) (EC50 = 0.17 μM) and DLV (delavirdine) (EC50 = 0.16 μM).
![[1860-5397-14-15-i43]](/bjoc/content/inline/1860-5397-14-15-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 43: Synthesis of N-(4-aminophenyl)-7-aryloxypyrazolo[1,5-a]pyrimidin-5-amines.
Scheme 43: Synthesis of N-(4-aminophenyl)-7-aryloxypyrazolo[1,5-a]pyrimidin-5-amines.
Kaswan et al. [103] reported the reaction of 5-aminopyrazoles 16/126 with chalcones 156 in DMF in the presence of inorganic base KOH for the synthesis of 5,7-diarylpyrazolo[1,5-a]pyrimidines 157. Chalcones 156 with electron-withdrawing group like nitro, cyano on para-position of aryl or heteroaryl ring and 2-hydroxyphenyl group resulted in lower yields as compared to chalcones with electron-donating groups (Scheme 44).
![[1860-5397-14-15-i44]](/bjoc/content/inline/1860-5397-14-15-i44.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 44: Base-catalyzed synthesis of 5,7-diarylpyrazolo[1,5-a]pyrimidines.
Scheme 44: Base-catalyzed synthesis of 5,7-diarylpyrazolo[1,5-a]pyrimidines.
Chobe et al. [104] reported the reaction of 3,5-diamino-4-diazopyrazole derivative 158 with 4-substituted benzylidene-3-methyl-1H-pyrazol-5(4H)-one 159 in PEG-400. The reaction resulted in the synthesis of 6,7-dihydropyrazolo[1,5-a]pyrimidine derivatives 160 (Scheme 45). Selected compounds were studied for their interaction with calf thymus DNA using various techniques like electronic spectra, viscosity measurement and thermal denaturation.
![[1860-5397-14-15-i45]](/bjoc/content/inline/1860-5397-14-15-i45.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 45: Synthesis of 6,7-dihydropyrazolo[1,5-a]pyrimidines in PEG-400.
Scheme 45: Synthesis of 6,7-dihydropyrazolo[1,5-a]pyrimidines in PEG-400.
Ahmetaj et al. [105] described a simple and efficient protocol for the synthesis of 7-heteroarylpyrazolo[1,5-a]pyrimidine-3-carboxamides 166 from the reaction of 5-aminopyrazole 161 with (E)-3-(dimethylamino)-1-(heteroaryl)prop-2-en-1-one 162 in aqueous ethanol at ambient temperature through the intermediacy of methyl 7-heteroarylpyrazolo[1,5-a]pyrimidine-3-carboxylates 163 which was subsequently hydrolyzed to give the corresponding carboxylic acids 164 followed by coupling with various primary and secondary amines 165 in presence of bis(pentafluorophenyl) carbonate (BPC) as activating agent (Scheme 46).
![[1860-5397-14-15-i46]](/bjoc/content/inline/1860-5397-14-15-i46.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 46: Synthesis of 7-heteroarylpyrazolo[1,5-a]pyrimidine-3-carboxamides.
Scheme 46: Synthesis of 7-heteroarylpyrazolo[1,5-a]pyrimidine-3-carboxamides.
Abdelhamid et al. [106] reported the synthesis of pyrazolo[1,5-a]pyrimidine derivatives 168 from the reaction of 5-aminopyrazoles 16/126 with sodium 3-(5-methyl-1-phenyl-1H-pyrazol-4-yl)-3-oxoprop-1-en-1-olate (167) with high regioselectivity without any traces of other possible regioisomeric pyrazolo[1,5-a]pyrimidines 169. The regioselectivity of the reaction was attributed to the higher nucleophilicity of the exocyclic primary amino group over the endocyclic amino group. Synthesis of pyrazolo[1,5-a]pyrimidine derivatives 168 was also achieved by an alternate route with equal ease by the reaction of 5-aminopyrazoles 16/126 with 3-(dimethylamino)-1-(5-methyl-1-phenyl-1H-pyrazol-4-yl)prop-2-en-1-ones (170) for structural confirmations (Scheme 47).
![[1860-5397-14-15-i47]](/bjoc/content/inline/1860-5397-14-15-i47.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 47: Synthesis of 7-heteroarylpyrazolo[1,5-a]pyrimidine derivatives under conventional heating and microwave assistance.
Scheme 47: Synthesis of 7-heteroarylpyrazolo[1,5-a]pyrimidine derivatives under conventional heating and micro...
Recently, Abdelhamid et al. [107] also reported the synthesis of 7-(furan-2-yl)-2-phenylpyrazolo[1,5-a]pyrimidine (168) using a similar synthetic strategy from the reaction of 5-aminopyrazole 16/126 with sodium 3-(furan-2-yl)-3-oxoprop-1-en-1-olate (167, Scheme 47).
Ren et al. [108] described the synthesis of 6-aminopyrazolo[1,5-a]pyrimidine derivatives 172 involving the condensation of 5-aminopyrazole derivative 16 and sodium nitromalonaldehyde 171 followed by reduction of the nitro group by hydrogenation to give 6-aminopyrazolo[1,5-a]pyrimidines 172. 6-Aminopyrazolo[1,5-a]pyrimidines 172 thus obtained were coupled with variously substituted benzoic acids 173 to give corresponding amide derivatives of pyrazolo[1,5-a]pyrimidines 174 (Scheme 48). Some of the compounds were found to be potent, selective and orally available B-Raf inhibitors with favorable physicochemical and pharmacokinetic properties.
![[1860-5397-14-15-i48]](/bjoc/content/inline/1860-5397-14-15-i48.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 48: Synthesis of N-aroylpyrazolo[1,5-a]pyrimidine-5-amines.
Scheme 48: Synthesis of N-aroylpyrazolo[1,5-a]pyrimidine-5-amines.
Stepaniuk et al. [109] reported the reaction of 5-aminopyrazole derivatives 16/126 with β,γ-unsaturated-γ-alkoxy-α-ketoesters 175 for the regioselective synthesis of pyrazolo[1,5-a]pyrimidines 177 in refluxing ethanol. The reaction provided high regioselectivity compared to other 1,3-dielectrophiles like 1,3-dicarbonyl compounds. The reaction was proposed to proceed through intermediate 176 which was isolated at −10 °C to 0 °C but was found to be unstable even at room temperature (Scheme 49).
![[1860-5397-14-15-i49]](/bjoc/content/inline/1860-5397-14-15-i49.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 49: Regioselective synthesis of ethyl pyrazolo[1,5-a]pyrimidine-7-carboxylate.
Scheme 49: Regioselective synthesis of ethyl pyrazolo[1,5-a]pyrimidine-7-carboxylate.
Ma et al. [110] reported the synthesis of 3-cyano-6,7-diarylpyrazolo[1,5-a]pyrimidines 179 comprising the reaction of 1.5 equivalents of 5-aminopyrazole 126 with 1 equivalent of isoflavones 178 in the presence of 3 equivalents of sodium methoxide in methanol (Scheme 50). The method has the merits of being simple in operation with mild reaction conditions and good yields of fused pyrazole derivatives.
![[1860-5397-14-15-i50]](/bjoc/content/inline/1860-5397-14-15-i50.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 50: Sodium methoxide-catalyzed synthesis of 3-cyano-6,7-diarylpyrazolo[1,5-a]pyrimidines.
Scheme 50: Sodium methoxide-catalyzed synthesis of 3-cyano-6,7-diarylpyrazolo[1,5-a]pyrimidines.
Synthesis of pyrazolo[3,4-d]pyrimidines
A diversity of biological effects is associated with pyrazolo[3,4-d]pyrimidines. They are known to exhibit antiviral [111,112], pesticidal [113], anti-inflammatory [114], antimicrobial [115-117], antileukemic [118], antitumor [114,119,120], pan-RAF inhibiting [121], tyrosine kinase RET inhibiting [122], CNS [123], cardiovascular [124,125] and tuberculostatic [126,127] activities. The promising therapeutic potential of pyrazolo[3,4-d]pyrimidines prompted researchers to develop novel synthetic strategies to provide this class of compounds.
Ghozlan et al. [128] reported the reactions of ethyl 5-amino-3-phenylpyrazole-4-carboxylate (126) with benzoylisothiocyanate or phenylisothiocyantes for the synthesis of N-thiocarbamoyl pyrazole derivatives 180 and 183 which gave pyrazolo[3,4-d]pyrimidine derivatives 181 and 184 on treatment with ethanolic sodium ethoxide. Pyrazolo[3,4-d]pyrimidine derivatives 181 were also obtained directly by fusion of thiourea/urea with 5-aminopyrazole 126 in an oil bath at 120 °C. N-Thiocarbamoyl pyrazole derivatives 180 and 183 underwent cyclization with hydrazine hydrate to give 5-(N-triazolyl)aminopyrazole derivative 182 and hydrazinopyrazolo[3,4-d]pyrimidines 185, respectively (Scheme 51).
![[1860-5397-14-15-i51]](/bjoc/content/inline/1860-5397-14-15-i51.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 51: Synthesis of various pyrazolo[3,4-d]pyrimidine derivatives.
Scheme 51: Synthesis of various pyrazolo[3,4-d]pyrimidine derivatives.
El-Moghazy et al. [129] described the synthesis of pyrazolo[3,4-d]pyrimidine derivatives 187 using a similar approach by the reaction of ethyl 5-amino-1-phenyl-1H-pyrazolo-4-carboxylate (186) and phenyl isothiocyanate in pyridine. The pyrazolo[3,4-d]pyrimidine derivative 187 thus obtained was methylated with iodomethane in acetone in the presence of potassium carbonate to give methyl thioether 188 which provided hydrazinopyrazolo[3,4-d]pyrimidines 189 on treatment with an excess of hydrazine hydrate in ethanol. The hydrazine derivative 189 thus obtained was made to react with several carbonyl compounds like aldehydes, benzoyl chloride and ethyl acetoacetate to append hydrazone, carbohydrazide and pyrazolone type moieties on pyrazolo[3,4-d]pyrimidine. Further, hydrazinyl derivative 189 provided various fused triazolylpyrazolo[3,4-d]pyrimidines on treatment with various reagents like aliphatic acids, benzoyl chlorides, chloroacetyl chloride, isothiocyanate and carbon disulfide under appropriate reaction conditions (Scheme 52).
![[1860-5397-14-15-i52]](/bjoc/content/inline/1860-5397-14-15-i52.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 52: Synthesis of hydrazinopyrazolo[3,4-d]pyrimidine derivatives.
Scheme 52: Synthesis of hydrazinopyrazolo[3,4-d]pyrimidine derivatives.
Hassan et al. [130] reported the synthesis of various pyrazolo[3,4-d]pyrimidine derivatives 193 (Scheme 53). 5-Aminopyrazole-4-carboxylic acid 190 on refluxing in acetic anhydride provided pyrazolooxazinones 191 which converted to 5-aminopyrazolo[3,4-d]pyrimidine 192 by reaction with hydrazine hydrate in n-butanol. Further treatment of 192 with aromatic aldehydes provided the corresponding Schiff base 193.
![[1860-5397-14-15-i53]](/bjoc/content/inline/1860-5397-14-15-i53.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 53: Synthesis of N-arylidinepyrazolo[3,4-d]pyrimidin-5-amines.
Scheme 53: Synthesis of N-arylidinepyrazolo[3,4-d]pyrimidin-5-amines.
Singla et al. [131] reported the synthesis of pyrazolo[3,4-d]pyrimidinyl-4-amines 198 possessing 4-(1H-benzimidazol-2-yl)phenylamine moiety at C4 position and primary as well as secondary amines at C6 position starting from 5-aminopyrazole-4-carboxamide (194). Compound 194 was treated with urea to give 1H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione (195) followed by chlorination with POCl3 to furnish 4,6-dichloropyrazolo[3,4-d]pyrimidine (196) which on coupling with the corresponding amines provided [4-(1H-benzimidazol-2-yl)-phenyl]-(6-substituted-1H-pyrazolo[3,4-d]pyrimidin-4-yl)-amines 198 (Scheme 54). Pyrazolo[3,4-d]pyrimidine derivatives 198 were evaluated for their antitumor activities against various human cancer cell lines. The compound 198 with a pyrrolidine moiety was identified as most potent and promising member as it showed superior antitumor activities over other derivatives.
![[1860-5397-14-15-i54]](/bjoc/content/inline/1860-5397-14-15-i54.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 54: Synthesis of pyrazolo[3,4-d]pyrimidinyl-4-amines.
Scheme 54: Synthesis of pyrazolo[3,4-d]pyrimidinyl-4-amines.
Bakavoli et al. [115] reported the synthesis of pyrazolo[3,4-d]pyrimidine derivatives 200 from the cyclocondensation of 5-amino-1-(2,4-dinitrophenyl)-1H-pyrazole-4-carboxamide (199) with aromatic aldehydes in the presence of iodine in acetonitrile (Scheme 55). The synthesized pyrazolo[3,4-d]pyrimidines were evaluated for antibacterial activities.
![[1860-5397-14-15-i55]](/bjoc/content/inline/1860-5397-14-15-i55.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 55: Iodine-catalyzed synthesis of pyrazolo[3,4-d]pyrimidinones.
Scheme 55: Iodine-catalyzed synthesis of pyrazolo[3,4-d]pyrimidinones.
Venkatesan et al. [132] also used 4-carboxamide-5-aminopyrazole 199 for the synthesis of pyrazolo[3,4-d]pyrimidines 207 (Scheme 56). The condensation of 199 with benzoyl isothiocyanate under reflux conditions in dry acetone provided benzoylthioureido derivatives 201 which were converted to methylthio derivative 202 with iodomethane in aqueous sodium hydroxide solution at ambient temperature. The methylthio group was converted to benzoylguanidino derivative 203 by nucleophilic displacement with ammonia in DMF on vigorous heating in a sealed tube. Subsequently, compounds 203 were converted to 6-amino-2-substituted-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one derivatives 204 by refluxing in 1 N sodium hydroxide. Pyrazolo[3,4-d]pyrimidinone 204 were further chlorinated by phosphorus oxychloride and subsequently converted to carboxylic esters 207 via cyanation followed by hydrolysis and esterification.
![[1860-5397-14-15-i56]](/bjoc/content/inline/1860-5397-14-15-i56.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 56: Synthesis of ethyl 6-amino-2H-pyrazolo[3,4-d]pyrimidine-4-carboxylate.
Scheme 56: Synthesis of ethyl 6-amino-2H-pyrazolo[3,4-d]pyrimidine-4-carboxylate.
Kaplan et al. [20] explored the synthesis of pyrazolo[3,4-d]pyrimidine derivatives starting from 5-amino-4-cyanopyrazoles 208 (Scheme 57). 5-Amino-4-cyanopyrazole 208 was benzoylated with p-nitrobenzoyl chloride (209) and subsequently cyclized to pyrazolo[3,4-d]pyrimidine derivative 211 by refluxing in sodium hydroxide and hydrogen peroxide. Chlorination of pyrazolo[3,4-d]pyrimidine derivative 211 with phosphorus oxychloride afforded 4-chloro-1H-pyrazolo[3,4-d]pyrimidine derivative 212. The chloro and nitro groups were manipulated to introduce a 3,6-dihydropyran group at position-4 by Stille reaction which provided 4-(4-(3,6-dihydropyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidin-6-yl)aniline 214 by NO2 group reduction with H2, Pd/C followed by Boc protection, coupling with tributyl(3,6-dihydro-2H-pyran-4-yl)stannane (213) and subsequent Boc deprotection with TFA in DCM. Pyrazolo[3,4-d]pyrimidinylaniline 214 was used to synthesize pyrazolo[3,4-d]pyrimidinylureas 215 on treatment with triphosgene and corresponding amines.
![[1860-5397-14-15-i57]](/bjoc/content/inline/1860-5397-14-15-i57.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 57: Synthesis of 4-substituted-(3,6-dihydropyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidines.
Scheme 57: Synthesis of 4-substituted-(3,6-dihydropyran-4-yl)-1H-pyrazolo[3,4-d]pyrimidines.
Liu et al. [133] reported the synthesis of 4-amino-1-(2,4-dichlorophenyl)pyrazolo[3,4-d]pyrimidine derivatives 217 by the reaction of ethyl N-(4-cyano-1-(2,4-dichlorophenyl)-1H-pyrazol-5-yl)formimidate (216) with ammonia. N-(4-Cyano-1-(2,4-dichlorophenyl)-1H-pyrazol-5-yl)formimidate (216), in turn was obtained by reaction of 5-amino-1-(2,4-dichlorophenyl)-1H-pyrazole-4-carbonitrile (208) with trimethylorthoformate. 4-Amino-1-(2,4-dichlorophenyl)pyrazolo[3,4-d]pyrimidine derivatives 217 were coupled with various carboxylic acids in the presence of EDCl, DMAP and HOBt in N,N-dimethylformamide at room temperature for the synthesis of the corresponding amide derivatives 218 (Scheme 58).
![[1860-5397-14-15-i58]](/bjoc/content/inline/1860-5397-14-15-i58.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 58: Synthesis of 1-(2,4-dichlorophenyl)pyrazolo[3,4-d]pyrimidin-4-yl carboxamides.
Scheme 58: Synthesis of 1-(2,4-dichlorophenyl)pyrazolo[3,4-d]pyrimidin-4-yl carboxamides.
Song et al. [134] explored the synthesis of pyrazolo[3,4-d]pyrimidine derivatives 221 through the intermediacy of amidines 219 obtained by reaction of 5-amino-4-cyanopyrazole 208 with N,N-dimethylformamide dimethyl acetal (DMFDMA) in acetonitrile at reflux temperature. Amidines 219 were condensed with appropriate 2-amino-5-subsitituted-1,3,4-thiadiazoles 220 under microwave irradiation in acetic acid for the generation of the desired pyrazolo[3,4-d]pyrimidine derivatives 221 (Scheme 59). The synthesized pyrazolo[3,4-d]pyrimidines 221 were proved to be good anticancer agents by MTT assay against HL-60 cancer cell lines.
![[1860-5397-14-15-i59]](/bjoc/content/inline/1860-5397-14-15-i59.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 59: Synthesis of 5-(1,3,4-thidiazol-2-yl)pyrazolo[3,4-d]pyrimidine.
Scheme 59: Synthesis of 5-(1,3,4-thidiazol-2-yl)pyrazolo[3,4-d]pyrimidine.
Zhang et al. [135] reported the reaction of 5-amino-4-cyanopyrazole 208 and aliphatic acids (R2COOH) in the presence of POCl3 to afford the respective 1-arylpyrazolo[3,4-d]pyrimidin-4-ones 222 in a one pot single step procedure (Scheme 60). POCl3 acted as chlorinating agent as well as an oxidant in the reaction which in situ generated acyl chlorides from acids making the condensation and cyclization easier and faster.
![[1860-5397-14-15-i60]](/bjoc/content/inline/1860-5397-14-15-i60.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 60: One pot POCl3-catalyzed synthesis of 1-arylpyrazolo[3,4-d]pyrimidin-4-ones.
Scheme 60: One pot POCl3-catalyzed synthesis of 1-arylpyrazolo[3,4-d]pyrimidin-4-ones.
The reaction of 5-amino-4-cyanopyrazole (208) and formamide was carried out by Todorovic et al. [136] under microwave irradiation at 200 °C to give 4-aminopyrazolo[3,4-d]pyrimidine (223) which on iodination with N-iodosuccinimide followed by N1-alkylation (Mitsunobu or substitution) provided corresponding N1-alkyl-3-iodopyrazolo[3,4-d]pyrimidine derivatives 225. The iodinated pyrazolo[3,4-d]pyrimidines were alkylated at C3 with boronic acids (R2-B(OH)2) using Suzuki coupling conditions to give 4-amino-N1,C3-dialkylpyrazolo[3,4-d]pyrimidines 226 (Scheme 61).
![[1860-5397-14-15-i61]](/bjoc/content/inline/1860-5397-14-15-i61.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 61: Synthesis of 4-amino-N1,C3-dialkylpyrazolo[3,4-d]pyrimidines under Suzuki conditions.
Scheme 61: Synthesis of 4-amino-N1,C3-dialkylpyrazolo[3,4-d]pyrimidines under Suzuki conditions.
Synthesis of pyrazolo[3,4-b]pyrazines
Pyrazolo[3,4-b]pyrazines have received great attention because of their interesting biological activities such as inhibition of protein kinases [137], blood platelet aggregation [138], bone metabolism improvers [139] as well as antifungal [140], antibacterial [141], antiparasitic [142] and antiviral [143] activity. There are several methods reported in literature for the construction of pyrazolo[3,4-b]pyrazine nucleus.
Quiroga et al. [144] studied the reaction of o-aminonitrosopyrazoles 227 and cyclic β-diketones 58 in various solvents like pyridine, acetic acid and N,N-dimethylformamide for the synthesis of pyrazolo[3,4-b]pyrazines 228 (Scheme 62). No measurable product was observed in acetic acid and pyridine but reaction in DMF provided promising results with good yields of the pyrazolo[3,4-b]pyrazines 228 in short reaction time. The reaction under microwave irradiation (100 W at 80 °C) in DMF provided the desired product in 85% yield in just 9 min. Easy work-up, mild reaction conditions and good yields makes this protocol a simple procedure for the synthesis of pyrazolo[3,4-b]pyrazines.
![[1860-5397-14-15-i62]](/bjoc/content/inline/1860-5397-14-15-i62.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 62: Microwave-assisted synthesis of pyrazolo[3,4-b]pyrazines.
Scheme 62: Microwave-assisted synthesis of pyrazolo[3,4-b]pyrazines.
Emary et al. [145] described the cyclocondensation of 5-amino-4-nitrosopyrazoles 229 and β-ketonitriles 15 in pyridine to give 1,3,6-trisubstitutedpyrazolo[3,4-b]pyrazine-5-carbonitriles 230 in good yields. 5-Carbonitrilepyrazolo[3,4-b]pyrazines 230 were hydrolyzed to corresponding pyrazolo[3,4-b]pyrazine-5-carboxylic acids 231 and subsequently converted to acid chloride 232 at reflux temperature with thionyl chloride (SOCl2) which underwent intramolecular Friedel–Crafts reaction in presence of Lewis acid to give 3-methyl-1-phenyl-1H-indeno[2,1-e]pyrazolo[3,4-b]pyrazin-5-one (233). Compound 233 was used to synthesize several other indenopyrazolopyrazinone derivatives by reaction with active methylene compounds, aromatic amines, hydroxylamine hydrochloride, semicarbazide hydrochloride, thiosemicarbazide, hydrazine hydrate and phenyl hydrazine (Scheme 63).
![[1860-5397-14-15-i63]](/bjoc/content/inline/1860-5397-14-15-i63.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 63: Synthesis and derivatization of pyrazolo[3,4-b]pyrazine-5-carbonitriles.
Scheme 63: Synthesis and derivatization of pyrazolo[3,4-b]pyrazine-5-carbonitriles.
Similarly, Farghley et al. [146] reported the reaction of 5-amino-4-nitrosopyrazoles 229 and β-ketonitriles 15 in pyridine for the synthesis of pyrazolo[3,4-b]pyrazines 230 which was used as synthetic precursor to generate several other substituted pyrazolo[3,4-b]pyrazine derivatives via amidoxime and carbohydrazide intermediates obtained from the reaction of appropriate substrates with nitrile group (Scheme 63).
Synthesis of pyrazolo[1,5-a][1,3,5]triazine
Pyrazolo[1,5-a][1,3,5]triazine is a well-known class of fused pyrazole derivatives with a broad spectrum of biological activities such as anticancer [147], anti-inflammatory [148], anxiolytic [149], anticonvulsant [150] and antidepressant [151]. Accordingly, a large number of synthetic methods have been reported for the construction of pyrazolo[1,5-a][1,3,5]triazine derivatives, out of which condensation of the 5-aminopyrazoles with ethoxycarbonyl isothiocyanate/ethoxycarbonyl isocyanates is the most common method for their synthesis.
Bera et al. [152] reported the synthesis of 2-thioxo-pyrazolo[1,5-a][1,3,5]triazin-4-ones 236 and 238 via annulation of 1,3,5-triazine ring onto 5-aminopyrazoles. The reactions of 5-aminopyrazoles 16 with ethoxycarbonyl isothiocyanate 234 was carried out in DMF to give thiourea derivatives 235 which on treatment with NaOH in ethanol underwent cyclization to give 2-thioxo-pyrazolo[1,5-a][1,3,5]triazin-4-ones 236 (Scheme 64). 3,5-Diaminopyrazoles (R = NH2, 15) following the same reaction sequence led to the formation of 2-thioxopyrazolo[1,5-a][1,3,5]triazin-4-one-6-thiourea derivative 238 through the intermediacy of bithiourea 237 (Scheme 64).
![[1860-5397-14-15-i64]](/bjoc/content/inline/1860-5397-14-15-i64.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 64: Synthesis of 2-thioxo-pyrazolo[1,5-a][1,3,5]triazin-4-ones.
Scheme 64: Synthesis of 2-thioxo-pyrazolo[1,5-a][1,3,5]triazin-4-ones.
Sun et al. [24] reported the synthesis of 7/8-substituted-2-oxo/thioxo-2,3-dihydropyrazolo[1,5-a][1,3,5]triazin-4(1H)-one 241 from the reaction of 3/4-substituted-5-aminopyrazoles 16/126 with ethoxycarbonyl isothiocyanate/ethoxycarbonyl isocyanate 234/239, respectively (Scheme 65). This two-step procedure involves the intermediacy of ethoxycarbonyl isocyanate to give N-ethoxycarbonyl-N’-(pyrazol-3-yl)ureas/thioureas 240 followed by their intramolecular cyclization with sodium ethoxide. The reaction of ethoxycarbonyl isothiocyanate provided higher yields in short time as compared to the reaction of ethoxycarbonyl isocyanate.
![[1860-5397-14-15-i65]](/bjoc/content/inline/1860-5397-14-15-i65.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 65: Synthesis of 2,3-dihydropyrazolo[1,5-a][1,3,5]triazin-4(1H)-one.
Scheme 65: Synthesis of 2,3-dihydropyrazolo[1,5-a][1,3,5]triazin-4(1H)-one.
Bakr et al. [153] reported the synthesis of pyrazolo[1,5-a][1,3,5]triazine-8-carboxylic acid ethyl ester 244 from the reaction of aminopyrazolylurea derivative 242 and N-bis(methylthio)methylenecyanamide (243) out in DMF in presence of potassium carbonate (Scheme 66).
![[1860-5397-14-15-i66]](/bjoc/content/inline/1860-5397-14-15-i66.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 66: Synthesis of pyrazolo[1,5-a][1,3,5]triazine-8-carboxylic acid ethyl ester.
Scheme 66: Synthesis of pyrazolo[1,5-a][1,3,5]triazine-8-carboxylic acid ethyl ester.
Insuasty et al. [154] reported the reaction of 5-aminopyrazoles 246 with thiocarbonates (245, X = O) and dithiocarbonates (245, X = S) under solvent-free conditions using microwave irradiation (300 W, 160–180 °C, 10–20 min) to yield a mixture of two products which were characterized as 2-ethylthio/ethoxy-4,7-dihetarylpyrazolo[1,5-a][1,3,5]triazines 247 and 248. When the reaction was carried out only for 3 min under similar reaction conditions two isomeric intermediates namely ethyl N’-(heteroaryl-1-carbonyl)-N-(3-heteroaryl-1H-pyrazol-5-yl)carbamimidothioate/carbamimidate 249 were isolated successfully in 10% and 24% yields, respectively (Scheme 67). The reactions were also carried under solvent mediated conventional heating conditions which required longer time for completion and provided lower yields of the products.
![[1860-5397-14-15-i67]](/bjoc/content/inline/1860-5397-14-15-i67.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 67: Microwave-assisted synthesis of 4,7-dihetarylpyrazolo[1,5-a][1,3,5]triazines.
Scheme 67: Microwave-assisted synthesis of 4,7-dihetarylpyrazolo[1,5-a][1,3,5]triazines.
In an alternative two step path, thiourea derivatives 251 obtained by reaction of 5-aminopyrazoles 246 with heteroarylisothiocyanates 250 were treated with ethyl bromide in presence of sodium hydride in DMF to generate the intermediate isothioureas 252 which on in situ heating provided target diheteroarylpyrazolo[1,5-a][1,3,5]triazines 247 (Scheme 68).
![[1860-5397-14-15-i68]](/bjoc/content/inline/1860-5397-14-15-i68.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 68: Alternative synthetic route to 4,7-diheteroarylpyrazolo[1,5-a][1,3,5]triazines.
Scheme 68: Alternative synthetic route to 4,7-diheteroarylpyrazolo[1,5-a][1,3,5]triazines.
In another report, Insuasty et al. [155] utilized S,S-diethyl aroyliminodithiocarbonates 245 for condensation with 5-amino-3-methylpyrazole (16) to afford 4-aryl-2-ethylthio-7-methylpyrazolo[1,5-a][1,3,5]triazines 247/253 (Scheme 69). Synthesized pyrazolo[1,5-a][1,3,5]triazines 247/253 were evaluated for their anticonvulsant profile by exposing on to electrical and chemical experimental seizures induced in ICR albino mice. Pyrazolo[1,5-a][1,3,5]triazines 247 having R1,R2 = 2-thienyl, showed a good dose-dependent response in the 50, 150 and 300 mg/kg, p.o., range (3/7, 4/7, 5/7; p < 0.05).
![[1860-5397-14-15-i69]](/bjoc/content/inline/1860-5397-14-15-i69.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 69: Synthesis of 4-aryl-2-ethylthio-7-methylpyrazolo[1,5-a][1,3,5]triazines.
Scheme 69: Synthesis of 4-aryl-2-ethylthio-7-methylpyrazolo[1,5-a][1,3,5]triazines.
Lim et al. [156] studied the reaction of 3,5-diaminopyrazole derivative 254, cyanamide and triethyl orthoformate in methanol under microwave irradiation (Scheme 70). The reaction resulted in exclusive formation of 4-aminopyrazolo[1,5-a][1,3,5]triazine-8-carbonitriles 255 barring the possibilities of isomeric 2-aminopyrazolo[1,5-a][1,3,5]triazine 257 and pyrazolo[3,4-d]pyrimidine derivatives 258 or 259. The formation of 4-aminopyrazolo[1,5-a][1,3,5]triazine derivative 255 was also confirmed by step-wise annulation of a triazine ring with a predisposed position of the amino group by converting 5-aminopyrazole 254 into the corresponding formamidine 256 on treatment with N,N-dimethylformamide dimethyl acetal (DMFDMA) and their subsequent condensation with cyanamide in the presence of sodium methoxide. The one-pot multicomponent method provided two times higher yields than the stepwise method.
![[1860-5397-14-15-i70]](/bjoc/content/inline/1860-5397-14-15-i70.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 70: Microwave-assisted synthesis of 4-aminopyrazolo[1,5-a][1,3,5]triazine.
Scheme 70: Microwave-assisted synthesis of 4-aminopyrazolo[1,5-a][1,3,5]triazine.
Synthesis of pyrazolo[3,4-d][1,2,3]triazine
Pyrazolo[3,4-d][1,2,3]triazines are important fused pyrazole derivatives because of their biological activity and are valuable synthons in organic transformations. These are also structural analogues of adenosine and guanosine [157,158]. But surprisingly, only a few literature reports are available for synthesis and biological potential of this nucleus.
Pyrazolo[3,4-d][1,2,3]triazines 262 were synthesized by Rabie et al. [159] from the diazotization of 4-(N-arylcarboxamide)-3-(N-phenyl)-3,5-diaminopyrazole derivatives 260 with sodium nitrite which underwent in situ cyclization (Scheme 71).
![[1860-5397-14-15-i71]](/bjoc/content/inline/1860-5397-14-15-i71.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 71: Synthesis of pyrazolo[3,4-d][1,2,3]triazines from pyrazol-5-yl diazonium salts.
Scheme 71: Synthesis of pyrazolo[3,4-d][1,2,3]triazines from pyrazol-5-yl diazonium salts.
Synthesis of pyrazolo[3,4-e][1,2,4]triazines
Matar et al. [160] reported that 3-amino-4-phenylhydrazono-1-phenyl-2-pyrazolin-5-ones 263 undergo cyclization on refluxing in DMFDMA to afford 2,5-dihydropyrazolo[5,1-c][1,2,4]triazines 264 in good yields (Scheme 72).
![[1860-5397-14-15-i72]](/bjoc/content/inline/1860-5397-14-15-i72.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 72: Synthesis of 2,5-dihydropyrazolo[3,4-e][1,2,4]triazines.
Scheme 72: Synthesis of 2,5-dihydropyrazolo[3,4-e][1,2,4]triazines.
Synthesis of pyrazolo[5,1-c][1,2,4]triazines
The pyrazolo[5,1-c][1,2,4]triazine nucleus is present in compounds possessing a variety of pharmacological and biological activities [161-163]. The association of biological properties with this nucleus stimulated the interest of organic chemists in the development of novel synthetic approaches for the construction of the pyrazolo[5,1-c][1,2,4]triazine nucleus.
Shawali et al. [164] have reported the synthesis of 3-[(4,5-disubstituted-pyrazol-3-yl)carbonyl]-pyrazolo[5,1-c][1,2,4]triazines 269 by cyclization of diazopyrazolylenaminones 268. 3-Acetylpyrazoles 265 on condensation with DMFDMA provided enaminones 266 which were converted to diazopyrazolylenaminones 268 on coupling with 3-phenylpyrazolyldiazonium salt 267 in pyridine at 0–5 °C. Diazopyrazolylenaminones 268 underwent cyclization under reaction conditions to give pyrazolo[5,1-c][1,2,4]triazines 269 (Scheme 73).
![[1860-5397-14-15-i73]](/bjoc/content/inline/1860-5397-14-15-i73.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 73: Synthesis of pyrazolo[5,1-c][1,2,4]triazines via diazopyrazolylenaminones.
Scheme 73: Synthesis of pyrazolo[5,1-c][1,2,4]triazines via diazopyrazolylenaminones.
Abdelhamid et al. [165] in a similar report synthesized pyrazolo[5,1-c][1,2,4]triazines 272 from the reaction of enaminone 270 with pyrazol-3-yl diazonium salt 271 in ethanolic sodium acetate solution (Scheme 74).
![[1860-5397-14-15-i74]](/bjoc/content/inline/1860-5397-14-15-i74.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 74: Synthesis of pyrazolo[5,1-c][1,2,4]triazines in presence of sodium acetate.
Scheme 74: Synthesis of pyrazolo[5,1-c][1,2,4]triazines in presence of sodium acetate.
Metwally et al. [166] reported the synthesis of pyrazolo[5,1-c][1,2,4]triazines 275, 277, 280 and 283 from coupling of pyrazolyldiazonium salt 273 with various nitrile derivatives 94, β-diketones 58, 2-aminoprop-1-ene-1,1,3-tricarbonitrile (278) and 3-methyl-1H-pyrazol-5(4H)-one (281) involving the intermediacy of corresponding hydrazones 274, 276, 279 and 282 in acetic acid or POCl3/DMF (Scheme 75).
![[1860-5397-14-15-i75]](/bjoc/content/inline/1860-5397-14-15-i75.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 75: Synthesis of various 7-diazopyrazolo[5,1-c][1,2,4]triazine derivatives.
Scheme 75: Synthesis of various 7-diazopyrazolo[5,1-c][1,2,4]triazine derivatives.
Adapting a similar procedure Al-Adiwish et al. [167] reported the synthesis of pyrazolo[5,1-c][1,2,4]triazines 285 and 286. 5-Aminopyrazoles 284 were diazotized to the corresponding diazonium salts and subsequently condensed with active methylene 94 and 104 to give hydrazono intermediates which underwent cyclization in acetic acid to provide the desired pyrazolo[5,1-c][1,2,4]triazines 285 and 286, respectively (Scheme 76). Selected pyrazolo[5,1-c][1,2,4]triazines 285 and 286 were screened for antibacterial activity and cytotoxicity against Vero cells.
![[1860-5397-14-15-i76]](/bjoc/content/inline/1860-5397-14-15-i76.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 76: One pot synthesis of pyrazolo[5,1-c][1,2,4]triazines.
Scheme 76: One pot synthesis of pyrazolo[5,1-c][1,2,4]triazines.
Schulze et al. [168] reported that 3,4-dinitropyrazole (287) on treatment with trimethylhydrazinium iodide (TMHI) provided 5-amino-3,4-dinitropyrazole (288) in 54% yields. The subsequent diazotization of aminopyrazole 288 and its coupling with sodium salt of nitroacetonitrile provided with 56% of 4-amino-3,7,8-trinitropyrazolo-[5,1-c][1,2,4]triazine (290) which has promising explosive properties (Scheme 77).
![[1860-5397-14-15-i77]](/bjoc/content/inline/1860-5397-14-15-i77.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 77: Synthesis of 4-amino-3,7,8-trinitropyrazolo-[5,1-c][1,2,4]triazines.
Scheme 77: Synthesis of 4-amino-3,7,8-trinitropyrazolo-[5,1-c][1,2,4]triazines.
Ledenyova et al. [169] have reported the synthesis of tricyclic pyrazolo[5,1-c][1,2,4]triazines 294, 297, 300 and 301 from azocoupling reaction of pyrazolediazonium salts 291 with various heterocyclic components, e.g., barbituric acid and thiobarbituric acid 292. Attempts were made to cyclize azocoupled intermediates 293 by heating with polyphosphoric acid (PPA) but only the intermediate formed from barbituric acid (X = O) provided pyrazolo[5,1-c]pyrimido[4,5-e][1,2,4]triazine-4(3H)-one 294 while the intermediate (X = S) failed to cyclize with PPA and anhydrous sodium acetate in acetic acid (Scheme 78).
![[1860-5397-14-15-i78]](/bjoc/content/inline/1860-5397-14-15-i78.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 78: Synthesis of tricyclic pyrazolo[5,1-c][1,2,4]triazines by azocoupling reaction.
Scheme 78: Synthesis of tricyclic pyrazolo[5,1-c][1,2,4]triazines by azocoupling reaction.
The reaction of pyrazolediazonium salts 291 with 2-hetarylpyrimidine-4,6-diones 295 in the presence of sodium acetate in acetic provided coloured compounds 296 which underwent smooth cyclization on heating with PPA to give 8-methyl-7-phenyl-2-hetaryl-pyrazolo[5,1-c]pyrimido[4,5-e][1,2,4]triazin-4(3H)-ones 297 in 65–70% yields.
The coupling reaction of 4-hydroxy-6-methyl-1H-pyridine-2-one 4-hydroxy-6-methyl-2H-pyran-2-one (triacetic acid, 298) with pyrazolediazonium salts 291 provided pyrazolo[5,1-c][1,2,4]triazine derivatives 300 through the intermediacy of azo coupled product 299. While 4-hydroxy-6-methyl-2H-pyran-2-one (298, Y = O) underwent cyclization differently with formation of bicyclic carboxylic acid derivative of pyrazolo[5,1-c][1,2,4]triazine 301 probably due to lactone ring opening (Scheme 78).
Conclusion
In this review article, we have systematically summarized various synthetic methods developed in the last decade for the construction of various pyrazoloazines as a group of fused pyrazolo derivatives utilizing 5-aminopyrazole as a synthetic precursor. The 5-aminopyrazole nucleus possesses ubiquitous distinctive structural features and its coupling reactions with different types of electrophilic reagents like aldehydes, ketones, β-diketones, β-ketoesters, and α,β-unsaturated ketones truly justifies its synthetic potential to construct fused heterocycles. This review opens the scope for future developments in new methodologies which promise the synthesis of novel fused heterocyclic systems with a wide range of medicinal and synthetic applications.
References
-
Kumar, H.; Saini, D.; Jain, S.; Jain, N. Eur. J. Med. Chem. 2013, 70, 248–258. doi:10.1016/j.ejmech.2013.10.004
Return to citation in text: [1] -
Gomha, S. M.; Edrees, M. M.; Faty, R. A. M.; Muhammad, Z. A.; Mabkhot, Y. N. Chem. Cent. J. 2017, 11, No. 37. doi:10.1186/s13065-017-0266-4.
Return to citation in text: [1] -
Aggarwal, R.; Bansal, A.; Rozas, I.; Kelly, B.; Kaushik, P.; Kaushik, D. Eur. J. Med. Chem. 2013, 70, 350–357. doi:10.1016/j.ejmech.2013.09.052
Return to citation in text: [1] -
Aggarwal, R.; Kumar, S.; Kaushik, P.; Kaushik, D.; Gupta, G. K. Eur. J. Med. Chem. 2013, 62, 508–514. doi:10.1016/j.ejmech.2012.11.046
Return to citation in text: [1] -
Mukarram, S.; Bandgar, B. P.; Shaikh, R. U.; Ganapure, S. D.; Chavan, H. V. Med. Chem. Res. 2017, 26, 262–273. doi:10.1007/s00044-016-1744-2
Return to citation in text: [1] -
Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Eur. J. Med. Chem. 2005, 40, 922–927. doi:10.1016/j.ejmech.2005.03.021
Return to citation in text: [1] [2] -
Aggarwal, R.; Bansal, A.; Rozas, I.; Diez-Cecilia, E.; Kaur, A.; Mahajan, R.; Sharma, J. Med. Chem. Res. 2014, 23, 1454–1464. doi:10.1007/s00044-013-0751-9
Return to citation in text: [1] -
Aggarwal, R.; Kumar, R.; Kumar, S.; Garg, G.; Mahajan, R.; Sharma, J. J. Fluorine Chem. 2011, 132, 965–972. doi:10.1016/j.jfluchem.2011.07.029
Return to citation in text: [1] -
Saad, H. A.; Osman, N. A.; Moustafa, A. H. Molecules 2011, 16, 10187–10201. doi:10.3390/molecules161210187
Return to citation in text: [1] -
El-sabbhag, O. I.; Baraka, M. M.; Ibrahim, S. M.; Pannecouque, C.; Andrei, G.; Snoeck, R.; Balzarini, J.; Rashad, A. A. Eur. J. Med. Chem. 2009, 44, 3746–3753. doi:10.1016/j.ejmech.2009.03.038
Return to citation in text: [1] -
Ouyang, G.; Chen, Z.; Cai, X.-J.; Song, B.-A.; Bhadury, P. S.; Yang, S.; Jin, L.-H.; Xue, W.; Hu, D.-Y.; Zeng, S. Bioorg. Med. Chem. 2008, 16, 9699–9707. doi:10.1016/j.bmc.2008.09.070
Return to citation in text: [1] -
Aggarwal, R.; Kumar, V.; Gupta, G. K.; Kumar, V. Med. Chem. Res. 2013, 22, 3566–3573. doi:10.1007/s00044-012-0343-0
Return to citation in text: [1] -
Bekhit, A. A.; Ashour, H. M. A.; Ghany, Y. S. A.; Bekhit, A. E.-D. A.; Baraka, A. Eur. J. Med. Chem. 2008, 43, 456–463. doi:10.1016/j.ejmech.2007.03.030
Return to citation in text: [1] -
Bebernitz, G. R.; Argentieri, G.; Battle, B.; Brennan, C.; Balkan, B.; Burkey, B. F.; Eckhardt, M.; Gao, J.; Kapa, P.; Strohschein, R. J.; Schuster, H. F.; Wilson, M.; Xu, D. D. J. Med. Chem. 2001, 44, 2601–2611. doi:10.1021/jm010032c
Return to citation in text: [1] -
Yadava, U.; Shukla, B. K.; Roychoudhury, M.; Kumar, D. J. Mol. Model. 2015, 21, 96. doi:10.1007/s00894-015-2631-3
Return to citation in text: [1] -
Manikannan, R.; Venkatesan, R.; Muthusubramanian, S.; Yogeeswari, P.; Sriram, D. Bioorg. Med. Chem. Lett. 2010, 20, 6920–6924. doi:10.1016/j.bmcl.2010.09.137
Return to citation in text: [1] -
Özdemir, A.; Altıntop, M. D.; Kaplancıklı, Z. A.; Can, Ö. D.; Özkay, Ü. D.; Turan-Zitouni, G. Molecules 2015, 20, 2668–2684. doi:10.3390/molecules20022668
Return to citation in text: [1] -
Zhang, L.; Balan, G.; Barreiro, G.; Boscoe, B. P.; Chenard, L. K.; Cianfrogna, J.; Claffey, M. M.; Chen, L.; Coffman, K. J.; Drozda, S. E.; Dunetz, J. R.; Fonseca, K. R.; Galatsis, P.; Grimwood, S.; Lazzaro, J. T.; Mancuso, J. Y.; Miller, E. L.; Reese, M. R.; Rogers, B. N.; Sakurada, I.; Skaddan, M.; Smith, D. L.; Stepan, A. F.; Trapa, P.; Tuttle, J. B.; Verhoest, P. R.; Walker, D. P.; Wright, A. S.; Zaleska, M. M.; Zasadny, K.; Shaffer, C. L. J. Med. Chem. 2014, 57, 861–877. doi:10.1021/jm401622k
Return to citation in text: [1] -
Damont, A.; Médran-Navarrete, V.; Cacheux, F.; Kuhnast, B.; Pottier, G.; Bernards, N.; Marguet, F.; Puech, F.; Boisgard, R.; Dollé, F. J. Med. Chem. 2015, 58, 7449–7464. doi:10.1021/acs.jmedchem.5b00932
Return to citation in text: [1] [2] -
Kaplan, J.; Verheijen, J. C.; Brooijmans, N.; Toral-Barza, L.; Hollander, I.; Yu, K.; Zask, A. Bioorg. Med. Chem. Lett. 2010, 20, 640–643. doi:10.1016/j.bmcl.2009.11.050
Return to citation in text: [1] [2] -
El-Morsy, A. M.; El-Sayed, M. S.; Abulkhair, H. S. Open J. Med. Chem. 2017, 7, No. 1. doi:10.4236/ojmc.2017.71001
Return to citation in text: [1] -
Elnagdi, M. H.; Abdel-Galil, F. M.; Riad, B. Y.; Elgemeie, G. E. H. Heterocycles 1983, 20, 2437–2470. doi:10.3987/R-1983-12-2437
Return to citation in text: [1] -
Guerrini, G.; Ciciani, G.; Cambi, G.; Bruni, F.; Selleri, S.; Besnard, F.; Montali, M.; Martini, C.; Ghelardini, C.; Galeotti, N.; Costanzo, A. Bioorg. Med. Chem. 2007, 15, 2573–2586. doi:10.1016/j.bmc.2007.01.053
Return to citation in text: [1] -
Sun, L.; Bera, H.; Chui, W. K. Eur. J. Med. Chem. 2013, 65, 1–11. doi:10.1016/j.ejmech.2013.03.063
Return to citation in text: [1] [2] -
Elmaati, T. M. A.; El-Taweel, F. M. J. Heterocycl. Chem. 2004, 41, 109–134. doi:10.1002/jhet.5570410201
Return to citation in text: [1] [2] -
Anwar, H. F.; Elnagdi, M. H. ARKIVOC 2009, No. 1, 198–250. doi:10.3998/ark.5550190.0010.107
Return to citation in text: [1] -
Aggarwal, R.; Kumar, V.; Kumar, R.; Singh, S. P. Beilstein J. Org. Chem. 2011, 7, 179–197. doi:10.3762/bjoc.7.25
Return to citation in text: [1] -
Marinozzi, M.; Marcelli, G.; Carotti, A. Mini-Rev. Med. Chem. 2015, 15, 272–299. doi:10.2174/1389557515666150312154536
Return to citation in text: [1] -
Lee, S.; Park, S. B. Org. Lett. 2009, 22, 5214–5217. doi:10.1021/ol902147u
Return to citation in text: [1] -
Elnagdi, M. N.; Elmoghayar, M. R. H.; Elgemeie, G. E. H. Adv. Heterocycl. Chem. 1987, 41, 319–376. doi:10.1016/S0065-2725(08)60164-6
Return to citation in text: [1] -
Huang, S.; Lin, R.; Yu, Y.; Lu, Y.; Connolly, P. J.; Chiu, G.; Li, S.; Emanuel, S. L.; Middleton, S. A. Bioorg. Med. Chem. Lett. 2007, 17, 1243–1245. doi:10.1016/j.bmcl.2006.12.031
Return to citation in text: [1] -
Chiu, G.; Li, S.; Connolly, P. J.; Middleton, S. A.; Emanuel, S. L.; Huang, S.; Lin, R.; Lu, Y.; N. V. Janssen Pharmaceutica, Belgium. PCT Int. Appl. WO 2006130673, Dec 7, 2006.
Return to citation in text: [1] -
Gudmundsson, K. S.; Johns, B. A.; Wang, Z.; Turner, E. M.; Allen, S. H.; Freeman, G. A.; Boyd, F. L., Jr.; Sexton, C. J.; Selleseth, D. W.; Moniri, K. R.; Creech, K. L. Bioorg. Med. Chem. 2005, 13, 5346–5361. doi:10.1016/j.bmc.2005.05.043
Return to citation in text: [1] -
Abdel-Monem, Y. K.; El-Enein, S. A. A.; El-Sheikh-Amer, M. M. J. Mol. Struct. 2017, 1127, 386–396. doi:10.1016/j.molstruc.2016.07.110
Return to citation in text: [1] -
Straub, A.; Stasch, J.-P.; Alonso-Alija, C. Bioorg. Med. Chem. Lett. 2001, 11, 781–784. doi:10.1016/S0960-894X(01)00073-7
Return to citation in text: [1] -
Elnagdi, M. H.; Negm, A. M.; Sadek, K. U. Synlett 1994, 27–37. doi:10.1055/s-1994-22729
Return to citation in text: [1] -
Quiroga, J.; Alvarado, M.; Insuasty, B.; Moreno, R.; Raviña, E.; Estevez, I.; De Almeida S., R. H. J. Heterocycl. Chem. 1999, 36, 1311–1316. doi:10.1002/jhet.5570360533
Return to citation in text: [1] -
Quiroga, J.; Hormaza, A.; Insuasty, B.; Márquez, M. J. Heterocycl. Chem. 1998, 35, 409–412. doi:10.1002/jhet.5570350225
Return to citation in text: [1] -
Rao, H. S. P.; Adigopula, L. N.; Ramadas, K. ACS Comb. Sci. 2017, 19, 279–285. doi:10.1021/acscombsci.6b00156
Return to citation in text: [1] -
Lee, S.; Park, S. B. Org. Lett. 2009, 11, 5214–5217. doi:10.1021/ol902147u
Return to citation in text: [1] -
Bou-Petit, E.; Picas, E.; Puigjaner, C.; Font-Bardia, M.; Ferrer, N.; Sempere, J.; de la Bellacasa, R. P.; Batllori, X.; Teixidó, J.; Estrada-Tejedor, R.; y Cajal, S. R.; Borrell, J. I. ChemistrySelect 2017, 2, 3668–3672. doi:10.1002/slct.201700732
Return to citation in text: [1] -
Aggarwal, R.; Kumar, V.; Bansal, A.; Sanz, D.; Claramunt, R. M. J. Fluorine Chem. 2012, 140, 31–37. doi:10.1016/j.jfluchem.2012.04.007
Return to citation in text: [1] -
Ghaedi, A.; Bardajee, G. R.; Mirshokrayi, A.; Mahdavi, M.; Shafiee, A.; Akbarzadeh, T. RSC Adv. 2015, 5, 89652–89658. doi:10.1039/C5RA16769H
Return to citation in text: [1] -
Shi, D.-Q.; Zhou, Y.; Liu, H. Synth. Commun. 2010, 40, 3660–3668. doi:10.1080/00397910903471796
Return to citation in text: [1] -
Shi, D.-Q.; Yang, F. J. Chin. Chem. Soc. 2008, 55, 755–760. doi:10.1002/jccs.200800113
Return to citation in text: [1] -
Shi, D.-Q.; Ni, S.-N.; Yang, F.; Shi, J.-W.; Dou, G.-L.; Li, X.-Y.; Wang, X.-S. J. Heterocycl. Chem. 2008, 45, 653–660. doi:10.1002/jhet.5570450303
Return to citation in text: [1] -
Chen, J.; Liu, W.; Ma, J.; Xu, H.; Wu, J.; Tang, X.; Fan, Z.; Wang, P. J. Org. Chem. 2012, 77, 3475–3482. doi:10.1021/jo3002722
Return to citation in text: [1] -
Prakash, R.; Shekarrao, K.; Saikia, P.; Gogoi, S.; Boruah, R. C. RSC Adv. 2015, 5, 21099–21102. doi:10.1039/C4RA17190J
Return to citation in text: [1] -
Shekarrao, K.; Kaishap, P. P.; Saddanapu, V.; Addlagatta, A.; Gogoi, S.; Boruah, R. C. RSC Adv. 2014, 4, 24001–24006. doi:10.1039/C4RA02865A
Return to citation in text: [1] [2] -
Pandey, G.; Bhowmik, S.; Batra, S. Org. Lett. 2013, 15, 5044–5047. doi:10.1021/ol4023722
Return to citation in text: [1] -
Abdel-Aziz, H. A.; Saleh, T. S.; El-Zahabi, H. S. A. Arch. Pharm. 2010, 343, 24–30. doi:10.1002/ardp.200900082
Return to citation in text: [1] -
Yen, W.-P.; Liu, P.-L.; Uramaru, N.; Lin, H.-Y.; Wong, F. F. Tetrahedron 2015, 71, 8798–8803. doi:10.1016/j.tet.2015.09.042
Return to citation in text: [1] -
Nascimento-Júnior, N. M.; Mendes, T. C. F.; Leal, D. M.; Corrêa, C. M. N.; Sudo, R. T.; Zapata-Sudo, G.; Barreiro, E. J.; Fragaa, C. A. M. Bioorg. Med. Chem. Lett. 2010, 20, 74–77. doi:10.1016/j.bmcl.2009.11.038
Return to citation in text: [1] -
Jiang, B.; Liu, Y.-P.; Tu, S.-J. Eur. J. Org. Chem. 2011, 3026–3035. doi:10.1002/ejoc.201100127
Return to citation in text: [1] -
Liu, M.; Yin, G.; Zhu, C.; Yao, C. J. Heterocycl. Chem. 2015, 53, 1617–1625. doi:10.1002/jhet.2473
Return to citation in text: [1] -
Bazgir, A.; Ahadi, S.; Ghahremanzadeh, R.; Khavasi, H. R.; Mirzaei, P. Ultrason. Sonochem. 2010, 17, 447–452. doi:10.1016/j.ultsonch.2009.09.009
Return to citation in text: [1] -
Wang, Z.; Gao, L.; Xu, Z.; Ling, Z.; Qin, Y.; Rong, L.; Tu, S.-J. Tetrahedron 2017, 73, 385–394. doi:10.1016/j.tet.2016.12.015
Return to citation in text: [1] -
Quiroga, J.; Portillo, S.; Pérez, A.; Gálvez, J.; Abonia, R.; Insuasty, B. Tetrahedron Lett. 2011, 52, 2664–2666. doi:10.1016/j.tetlet.2011.03.067
Return to citation in text: [1] -
De, K.; Bhaumik, A.; Banerjee, B.; Mukhopadhyay, C. Tetrahedron Lett. 2015, 56, 1614–1618. doi:10.1016/j.tetlet.2015.01.163
Return to citation in text: [1] -
Dandia, A.; Laxkar, A. K.; Singh, R. Tetrahedron Lett. 2012, 53, 3012–3017. doi:10.1016/j.tetlet.2012.03.136
Return to citation in text: [1] -
Gao, X.; Yang, Z.; Hao, W.-J.; Jiang, B.; Tu, S.-J. J. Heterocycl. Chem. 2017, 54, 2434–2439. doi:10.1002/jhet.2840
Return to citation in text: [1] -
Hao, Y.; Xu, X.-P.; Chen, T.; Zhao, L.-L.; Ji, S.-J. Org. Biomol. Chem. 2012, 10, 724–728. doi:10.1039/C1OB06624B
Return to citation in text: [1] -
Anand, D.; Yadav, P. K.; Patel, O. P. S.; Parmar, N.; Maurya, R. K.; Vishwakarma, P.; Raju, K. S. R.; Taneja, I.; Wahajuddin, M.; Kar, S.; Yadav, P. P. J. Med. Chem. 2017, 60, 1041–1059. doi:10.1021/acs.jmedchem.6b01447
Return to citation in text: [1] -
Insuasty, D.; Abonia, R.; Insuasty, B.; Quiroga, J.; Laali, K. K.; Nogueras, M.; Cobo, J. ACS Comb. Sci. 2017, 19, 555–563. doi:10.1021/acscombsci.7b00091
Return to citation in text: [1] -
Quiroga, J.; Trilleras, J.; Pantoja, D.; Abonía, R.; Insuasty, B.; Nogueras, M.; Cobo, J. Tetrahedron Lett. 2010, 51, 4717–4719. doi:10.1016/j.tetlet.2010.07.009
Return to citation in text: [1] -
Nikpassand, M.; Mamaghani, M.; Shirini, F.; Tabatabaeian, K. Ultrason. Sonochem. 2010, 17, 301–305. doi:10.1016/j.ultsonch.2009.08.001
Return to citation in text: [1] -
Yao, C.-S.; Wang, C.-H.; Jiang, B.; Tu, S.-J. J. Comb. Chem. 2010, 12, 472–475. doi:10.1021/cc100017f
Return to citation in text: [1] -
Frolova, L. V.; Malik, I.; Uglinskii, P. Y.; Rogelj, S.; Kornienko, A.; Magedov, I. V. Tetrahedron Lett. 2011, 52, 6643–6645. doi:10.1016/j.tetlet.2011.10.012
Return to citation in text: [1] -
Fan, L.; Yao, C.; Shu, M. Heterocycl. Commun. 2016, 22, 63–67. doi:10.1515/hc-2015-0234
Return to citation in text: [1] -
Huang, Z.; Hu, Y.; Zhou, Y.; Shi, D. ACS Comb. Sci. 2011, 13, 45–49. doi:10.1021/co1000162
Return to citation in text: [1] -
El-borai, M. A.; Rizk, H. F.; Abd-Aal, M. F.; El-Deeb, I. Y. Eur. J. Med. Chem. 2012, 48, 92–96. doi:10.1016/j.ejmech.2011.11.038
Return to citation in text: [1] -
Hill, M. D.; Fang, H.; Brown, J. M.; Molski, T.; Easton, A.; Han, X.; Miller, R.; Hill-Drzewi, M.; Gallagher, L.; Matchett, M.; Gulianello, M.; Balakrishnan, A.; Bertekap, R. L.; Santone, K. S.; Whiterock, V. J.; Zhuo, X.; Bronson, J. J.; Macor, J. E.; Degnan, A. P. ACS Med. Chem. Lett. 2016, 7, 1082–1086. doi:10.1021/acsmedchemlett.6b00292
Return to citation in text: [1] -
Hill, M. D. Synthesis 2016, 48, 2201–2204. doi:10.1055/s-0035-1562230
Return to citation in text: [1] -
Aggarwal, R.; Singh, G.; Kumar, S.; McCabe, T.; Rozas, I. ChemistrySelect 2016, 1, 5990–5994. doi:10.1002/slct.201601201
Return to citation in text: [1] -
Rahmati, A. Tetrahedron Lett. 2010, 51, 2967–2970. doi:10.1016/j.tetlet.2010.03.109
Return to citation in text: [1] -
Dandia, A.; Gupta, S. L.; Singh, P.; Quraishi, M. A. ACS Sustainable Chem. Eng. 2013, 1, 1303–1310. doi:10.1021/sc400155u
Return to citation in text: [1] -
Gunasekaran, P.; Prasanna, P.; Perumal, S. Tetrahedron Lett. 2014, 55, 329–332. doi:10.1016/j.tetlet.2013.11.016
Return to citation in text: [1] -
Jiang, B.; Ye, Q.; Fan, W.; Wang, S.-L.; Tu, S.-J.; Li, G. Chem. Commun. 2014, 50, 6108–6111. doi:10.1039/C4CC00740A
Return to citation in text: [1] -
Wang, J.-J.; Feng, X.; Xun, Z.; Shi, D.-Q.; Huang, Z.-B. J. Org. Chem. 2015, 80, 8435–8442. doi:10.1021/acs.joc.5b01314
Return to citation in text: [1] -
Trapani, G.; Laquintana, V.; Denora, N.; Trapani, A.; Lopedota, A.; Latrofa, A.; Franco, M.; Serra, M.; Pisu, M. G.; Floris, I.; Sanna, E.; Biggio, G.; Liso, G. J. Med. Chem. 2005, 48, 292–305. doi:10.1021/jm049610q
Return to citation in text: [1] [2] -
Wu, Y.-C.; Zou, X.-M.; Hu, F.-Z.; Yang, H.-Z. J. Heterocycl. Chem. 2005, 42, 609–613. doi:10.1002/jhet.5570420421
Return to citation in text: [1] -
Aggarwal, R.; Sumran, G.; Garg, N.; Aggarwal, A. Eur. J. Med. Chem. 2011, 46, 3038–3048. doi:10.1016/j.ejmech.2011.04.041
Return to citation in text: [1] -
Hassan, A. S.; Mady, M. F.; Awad, H. M.; Hafez, T. S. Chin. Chem. Lett. 2017, 28, 388–393. doi:10.1016/j.cclet.2016.10.022
Return to citation in text: [1] -
Al-Mousawi, S. M.; Mohammad, M. A.; Elnagdi, M. H. J. Heterocycl. Chem. 2001, 38, 989–991. doi:10.1002/jhet.5570380430
Return to citation in text: [1] -
Jakše, R.; Svete, J.; Stanovnik, B.; Golobič, A. Tetrahedron 2004, 60, 4601–4608. doi:10.1016/j.tet.2004.03.075
Return to citation in text: [1] -
Lager, E.; Andersson, P.; Nilsson, J.; Pettersson, I.; Nielsen, E. Ø.; Nielsen, M.; Sterner, O.; Liljefors, T. J. Med. Chem. 2006, 49, 2526–2533. doi:10.1021/jm058057p
Return to citation in text: [1] -
Médran-Navarrete, V.; Damont, A.; Peyronneau, M. A.; Kuhnast, B.; Bernards, N.; Pottier, G.; Marguet, F.; Puech, F.; Boisgard, R.; Dollé, F. Bioorg. Med. Chem. Lett. 2014, 24, 1550–1556. doi:10.1016/j.bmcl.2014.01.080
Return to citation in text: [1] -
Marjani, A. P.; Khalafy, J.; Salami, F.; Ezati, M. ARKIVOC 2015, No. 5, 277–286. doi:10.3998/ark.5550190.p009.062
Return to citation in text: [1] -
Aggarwal, R.; Sumran, G.; Claramunt, R. M.; Sanz, D.; Elguero, J. J. Mol. Struct. 2009, 934, 96–102. doi:10.1016/j.molstruc.2009.06.021
Return to citation in text: [1] -
Aggarwal, R.; Masan, E.; Kaushik, P.; Kaushik, D.; Sharma, C.; Aneja, K. R. J. Fluorine Chem. 2014, 168, 16–24. doi:10.1016/j.jfluchem.2014.08.017
Return to citation in text: [1] -
Mulakayala, N.; Upendar Reddy, C. H.; Chaitanya, M.; Manzoor Hussain, M.; Kumar, C. S.; Golla, N. J. Korean Chem. Soc. 2011, 55, 719–722. doi:10.5012/jkcs.2011.55.4.719
Return to citation in text: [1] -
Buriol, L.; München, T. S.; Frizzo, C. P.; Marzari, M. R. B.; Zanatta, N.; Bonacorso, H. G.; Martins, M. A. P. Ultrason. Sonochem. 2013, 20, 1139–1143. doi:10.1016/j.ultsonch.2013.02.006
Return to citation in text: [1] -
Saikia, P.; Gogoi, S.; Boruah, R. C. J. Org. Chem. 2015, 80, 6885–6889. doi:10.1021/acs.joc.5b00933
Return to citation in text: [1] -
Kamal, A.; Tamboli, J. R.; Nayak, V. L.; Adil, S. F.; Vishnuvardhan, M. V. P. S.; Ramakrishna, S. Bioorg. Med. Chem. Lett. 2013, 23, 3208–3215. doi:10.1016/j.bmcl.2013.03.129
Return to citation in text: [1] -
Kamal, A.; Faazil, S.; Hussaini, S. M. A.; Ramaiah, M. J.; Balakrishna, M.; Patel, N.; Pushpavalli, S. N. C. V. L.; Pal-Bhadra, M. Bioorg. Med. Chem. Lett. 2016, 26, 2077–2083. doi:10.1016/j.bmcl.2016.02.072
Return to citation in text: [1] -
Griffith, D. A.; Hargrove, D. M.; Maurer, T. S.; Blum, C. A.; De Lombaert, S.; Inthavongsay, J. K.; Klade, L. E.; Mack, C. M.; Rose, C. R.; Sanders, M. J.; Carpino, P. A. Bioorg. Med. Chem. Lett. 2011, 21, 2641–2645. doi:10.1016/j.bmcl.2010.12.116
Return to citation in text: [1] -
Dwyer, M. P.; Paruch, K.; Labroli, M.; Alvarez, C.; Keertikar, K. M.; Poker, C.; Rossman, R.; Fischmann, T. O.; Duca, J. S.; Madison, V.; Parry, D.; Davis, N.; Seghezzi, W.; Wiswell, D.; Guzi, T. J. Bioorg. Med. Chem. Lett. 2011, 21, 467–470. doi:10.1016/j.bmcl.2010.10.113
Return to citation in text: [1] -
Azeredo, L. F. S. P.; Coutinho, J. P.; Jabor, V. A. P.; Feliciano, P. R.; Nonato, M. C.; Kaiser, C. R.; Menezes, C. M. S.; Hammes, A. S. O.; Caffarena, E. R.; Hoelz, L. V. B.; Souza, N. B.; Pereira, G. A. N.; Cerávolo, I. P.; Krettli, A. U.; Boechat, N. Eur. J. Med. Chem. 2017, 126, 72–83. doi:10.1016/j.ejmech.2016.09.073
Return to citation in text: [1] -
Hylsová, M.; Carbain, B.; Fanfrlík, J.; Musilová, L.; Haldar, S.; Köprülüoğlu, C.; Ajani, H.; Brahmkshatriya, P. S.; Jorda, R.; Kryštof, V.; Hobza, P.; Echalier, A.; Paruch, K.; Lepšík, M. Eur. J. Med. Chem. 2017, 126, 1118–1128. doi:10.1016/j.ejmech.2016.12.023
Return to citation in text: [1] -
Aggarwal, R.; Kumar, V.; Singh, G.; Sanz, D.; Claramunt, R. M.; Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. J. Heterocycl. Chem. 2015, 52, 336–345. doi:10.1002/jhet.1955
Return to citation in text: [1] -
Aggarwal, R.; Singh, G.; Kaushik, P.; Kaushik, D.; Paliwal, D.; Kumar, A. Eur. J. Med. Chem. 2015, 101, 326–333. doi:10.1016/j.ejmech.2015.06.011
Return to citation in text: [1] -
Tian, Y.; Du, D.; Rai, D.; Wang, L.; Liu, H.; Zhan, P.; De Clercq, E.; Pannecouque, C.; Liu, X. Bioorg. Med. Chem. 2014, 22, 2052–2059. doi:10.1016/j.bmc.2014.02.029
Return to citation in text: [1] -
Kaswan, P.; Pericherla, K.; Purohit, D.; Kumar, A. Tetrahedron Lett. 2015, 56, 549–553. doi:10.1016/j.tetlet.2014.11.121
Return to citation in text: [1] -
Chobe, S. S.; Dawane, B. S.; Tumbi, K. M.; Nandekar, P. P.; Sangamwar, A. T. Bioorg. Med. Chem. Lett. 2012, 22, 7566–7572. doi:10.1016/j.bmcl.2012.10.027
Return to citation in text: [1] -
Ahmetaj, S.; Velikanje, N.; Grošelj, U.; Šterbal, I.; Prek, B.; Golobič, A.; Kočar, D.; Dahmann, G.; Stanovnik, B.; Svete, J. Mol. Diversity 2013, 17, 731–743. doi:10.1007/s11030-013-9469-3
Return to citation in text: [1] -
Abdelhamid, A. O.; Gomha, S. M. J. Chem. 2013, No. 327095. doi:10.1155/2013/327095
Return to citation in text: [1] -
Mostafa, S. S.; Salem Nada, A. M. M. Eur. Chem. Bull. 2015, 4, 50–59. doi:10.17628/ecb.2015.4.50-59
Return to citation in text: [1] -
Ren, L.; Laird, E. R.; Buckmelter, A. J.; Dinkel, V.; Gloor, S. L.; Grina, J.; Newhouse, B.; Rasor, K.; Hastings, G.; Gradl, S. N.; Rudolph, J. Bioorg. Med. Chem. Lett. 2012, 22, 1165–1168. doi:10.1016/j.bmcl.2011.11.092
Return to citation in text: [1] -
Stepaniuk, O. O.; Matviienko, V. O.; Kontratov, I. S.; Vitruk, I. V.; Tolmachev, A. O. Synthesis 2013, 45, 925–930. doi:10.1055/s-0032-1318329
Return to citation in text: [1] -
Ma, Y.-Q.; Zhang, Z.-T.; He, Q.; Xue, D.; Zhang, P.-F. Synth. Commun. 2012, 42, 933–941. doi:10.1080/00397911.2010.521902
Return to citation in text: [1] -
El-Bendary, E. R.; Badria, F. A. Arch. Pharm. 2000, 333, 99–103. doi:10.1002/(SICI)1521-4184(20004)333:4<99::AID-ARDP99>3.0.CO;2-O
Return to citation in text: [1] -
Chern, J.-H.; Shia, K.-S.; Hsu, T.-A.; Tai, C.-L.; Lee, C.-C.; Lee, Y.-C.; Chang, C.-S.; Tseng, S.-N.; Shih, S.-R. Bioorg. Med. Chem. Lett. 2004, 14, 2519–2525. doi:10.1016/j.bmcl.2004.02.092
Return to citation in text: [1] -
Ram, V. J.; Pandey, H. N.; Mishra, L. Arch. Pharm. 1979, 312, 586–590. doi:10.1002/ardp.19793120705
Return to citation in text: [1] -
Amir, M.; Javed, S.; Kumar, H. Indian J. Pharm. Sci. 2007, 69, 337–343. doi:10.4103/0250-474X.34540
Return to citation in text: [1] [2] -
Bakavoli, M.; Bagherzadeh, G.; Vaseghifar, M.; Shiri, A.; Pordel, M.; Mashreghi, M.; Pordeli, P.; Araghi, M. Eur. J. Med. Chem. 2010, 45, 647–650. doi:10.1016/j.ejmech.2009.10.051
Return to citation in text: [1] [2] -
Abunada, N. M.; Hassaneen, H. M.; Kandile, N. G.; Miqdad, O. A. Molecules 2008, 13, 1501–1517. doi:10.3390/molecules13071501
Return to citation in text: [1] -
Raffa, D.; Maggio, B.; Plescia, F.; Cascioferro, S.; Raimondi, M. V.; Plescia, S.; Cusimano, M. G. Arch. Pharm. 2009, 342, 321–326. doi:10.1002/ardp.200800140
Return to citation in text: [1] -
Petrie, C. R.; Cottam, H. B.; McKernan, P. A.; Robins, R. K.; Revankar, G. R. J. Med. Chem. 1985, 28, 1010–1016. doi:10.1021/jm00146a007
Return to citation in text: [1] -
Celano, M.; Schenone, S.; Cosco, D.; Navarra, M.; Puxeddu, E.; Racanicchi, L.; Brullo, C.; Varano, E.; Alcaro, S.; Ferretti, E.; Botta, G.; Filetti, S.; Fresta, M.; Botta, M.; Russo, D. Endocr.-Relat. Cancer 2008, 15, 499–510. doi:10.1677/ERC-07-0243
Return to citation in text: [1] -
Fallacara, A. L.; Mancini, A.; Zamperini, C.; Dreassi, E.; Marianelli, S.; Chiariello, M.; Pozzi, G.; Santoro, F.; Botta, M.; Schenone, S. Bioorg. Med. Chem. Lett. 2017, 27, 3196–3200. doi:10.1016/j.bmcl.2017.05.015
Return to citation in text: [1] -
Fu, Y.; Wang, Y.; Wan, S.; Li, Z.; Wang, G.; Zhang, J.; Wu, X. Molecules 2017, 22, 542. doi:10.3390/molecules22040542
Return to citation in text: [1] -
Wang, C.; Liu, H.; Song, Z.; Ji, Y.; Xing, L.; Peng, X.; Wang, X.; Ai, J.; Geng, M.; Zhang, A. Bioorg. Med. Chem. Lett. 2017, 27, 2544–2548. doi:10.1016/j.bmcl.2017.03.088
Return to citation in text: [1] -
Hutson, P. H.; Finger, E. N.; Magliaro, B. C.; Smith, S. M.; Converso, A.; Sanderson, P. E.; Mullins, D.; Hyde, L. A.; Eschle, B. K.; Turnbull, Z.; Sloan, H.; Guzzi, M.; Zhang, X.; Wang, A.; Rindgen, D.; Mazzola, R.; Vivian, J. A.; Eddins, D.; Uslaner, J. M.; Bednar, R.; Gambone, C.; Le-Mair, W.; Marino, M. J.; Sachs, N.; Xu, G.; Parmentier-Batteur, S. Neuropharmacology 2011, 61, 665–676. doi:10.1016/j.neuropharm.2011.05.009
Return to citation in text: [1] -
Guccione, S.; Modica, M.; Longmore, J.; Shaw, D.; Barretta, G. U.; Santagati, A.; Santagati, M.; Russo, F. Bioorg. Med. Chem. Lett. 1996, 6, 59–64. doi:10.1016/0960-894X(95)00558-B
Return to citation in text: [1] -
Xia, Y.; Chackalamannil, S.; Czarniecki, M.; Tsai, H.; Vaccaro, H.; Cleven, R.; Cook, J.; Fawzi, A.; Watkins, R.; Zhang, H. J. Med. Chem. 1997, 40, 4372–4377. doi:10.1021/jm970495b
Return to citation in text: [1] -
Trivedi, A.; Dodiya, D.; Surani, J.; Jarsania, S.; Mathukiya, H.; Ravat, N.; Shah, V. Arch. Pharm. 2008, 341, 435–439. doi:10.1002/ardp.200800027
Return to citation in text: [1] -
Trivedi, A.; Vaghasiya, S.; Dholariya, B.; Dodiya, D.; Shah, V. J. Enzyme Inhib. Med. Chem. 2010, 25, 893–899. doi:10.3109/14756360903540276
Return to citation in text: [1] -
Ghozlan, S. A. S.; Abdelrazek, F. M.; Mohamed, M. H.; Azmy, K. E. J. Heterocycl. Chem. 2010, 47, 1379–1385. doi:10.1002/jhet.482
Return to citation in text: [1] -
El-Moghazy, S. M.; George, R. F.; Osman, E. E. A.; Elbatrawy, A. A.; Kissova, M.; Colombo, A.; Crespan, E.; Maga, G. Eur. J. Med. Chem. 2016, 123, 1–13. doi:10.1016/j.ejmech.2016.07.034
Return to citation in text: [1] -
Hassan, G. S.; Kadry, H. H.; Abou-Seri, S. M.; Ali, M. M.; Mahmoud, A. E. E.-D. Bioorg. Med. Chem. 2011, 19, 6808–6817. doi:10.1016/j.bmc.2011.09.036
Return to citation in text: [1] -
Singla, P.; Luxami, V.; Singh, R.; Tandon, V.; Paul, K. Eur. J. Med. Chem. 2017, 126, 24–35. doi:10.1016/j.ejmech.2016.09.093
Return to citation in text: [1] -
Venkatesan, G.; Paira, P.; Cheong, S. L.; Federico, S.; Klotz, K. N.; Spalluto, G.; Pastorin, G. Eur. J. Med. Chem. 2015, 92, 784–798. doi:10.1016/j.ejmech.2015.01.046
Return to citation in text: [1] -
Liu, J.; Zhang, X.-W.; Song, D.-Z.; Cai, Z.-q.; Zhou, Y.-p. J. Chem. Res. 2016, 40, 63–125. doi:10.3184/174751916X14531173426246
Return to citation in text: [1] -
Song, X. J.; Shao, Y.; Dong, X. G. Chin. Chem. Lett. 2011, 22, 1036–1038. doi:10.1016/j.cclet.2011.05.012
Return to citation in text: [1] -
Zhang, X.; Lin, Q.; Zhong, P. Molecules 2010, 15, 3079–3086. doi:10.3390/molecules15053079
Return to citation in text: [1] -
Todorovic, N.; Awuah, E.; Shakya, T.; Wright, G. D.; Capretta, A. Tetrahedron Lett. 2011, 52, 5761–5763. doi:10.1016/j.tetlet.2011.08.103
Return to citation in text: [1] -
Binch, H.; Robinson, D.; Miller, A.; Fraysse, D. Pyrrolopyrazines and pyrazolopyrazines useful as inhibitors of protein kinases. WO Patent WO 2006058074, June 1, 2006.
Return to citation in text: [1] -
Sado, T.; Inoue, A. Chem. Abstr. 1990, 113, 78422k.
Return to citation in text: [1] -
Imaizumi, K.; Sado, T. Chem. Abstr. 1994, 121, 91797w.
Return to citation in text: [1] -
El-Emary, T. I. J. Chin. Chem. Soc. 2006, 53, 391–401. doi:10.1002/jccs.200600050
Return to citation in text: [1] -
Foks, H.; Pancechowska-Ksepko, D.; Kędzia, A.; Zwolska, Z.; Janowiec, M.; Augustynowicz-Kopeć, E. Farmaco 2005, 60, 513–517. doi:10.1016/j.farmac.2005.05.002
Return to citation in text: [1] -
El-Kashef, H. S.; El-Emary, T. I.; Gasquet, M.; Timon-David, P.; Maldonado, J.; Vanelle, P. Pharmazie 2000, 55, 572–576.
Return to citation in text: [1] -
El-Emary, T. I.; Kamal El-Dean, A. M.; El-Kashef, H. S. Farmaco 1998, 53, 383–388. doi:10.1016/S0014-827X(98)00014-7
Return to citation in text: [1] -
Quiroga, J.; Sánchez, N. E.; Acosta, P.; Insuasty, B.; Abonia, R. Tetrahedron Lett. 2012, 53, 3181–3187. doi:10.1016/j.tetlet.2012.04.083
Return to citation in text: [1] -
El-Emary, T.; El-Kashef, H. Eur. J. Med. Chem. 2013, 62, 478–485. doi:10.1016/j.ejmech.2013.01.025
Return to citation in text: [1] -
Farghaly, A.-R.; Esmail, S.; Abdel-Zaher, A.; Abdel-Hafez, A.; El-Kashef, H. Bioorg. Med. Chem. 2014, 22, 2166–2175. doi:10.1016/j.bmc.2014.02.019
Return to citation in text: [1] -
Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Lu, J.; Averill, A.; Almassy, R.; Chu, S. Bioorg. Med. Chem. Lett. 2008, 18, 619–623. doi:10.1016/j.bmcl.2007.11.074
Return to citation in text: [1] -
El-Hawash, S. A.; El-Mallah, A. I. Pharmazie 1998, 53, 368–373.
Return to citation in text: [1] -
Trincavelli, M. L.; Da Pozzo, E.; Daniele, S.; Martini, C. Curr. Top. Med. Chem. 2012, 12, 254–269. doi:10.2174/1568026799078787
Return to citation in text: [1] -
Youdim, M. B. H.; Bakhle, Y. S. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S287–S296. doi:10.1038/sj.bjp.0706464
Return to citation in text: [1] -
Kulkarni, S. K.; Dhir, A. Expert Opin. Invest. Drugs 2009, 18, 767–788. doi:10.1517/13543780902880850
Return to citation in text: [1] -
Bera, H.; Ojha, P. k.; Tan, B. J.; Sun, L.; Dolzhenko, A. V.; Chui, W.-K.; Chiu, G. N. C. Eur. J. Med. Chem. 2014, 78, 294–303. doi:10.1016/j.ejmech.2014.03.063
Return to citation in text: [1] -
Abbas-Temirek, H. H.; Abo-Bakr, A. M. Eur. J. Chem. 2016, 7, 107–114. doi:10.5155/eurjchem.7.1.107-114.1369
Return to citation in text: [1] -
Insuasty, H.; Insuasty, B.; Castro, E.; Quiroga, J.; Abonía, R.; Nogueras, M.; Cobo, J. Tetrahedron 2012, 68, 9384–9390. doi:10.1016/j.tet.2012.09.029
Return to citation in text: [1] -
Insuasty, H.; Castro, E.; Escobar, J. C.; Murillo, V.; Rodríguez, J.; Cuca, L. E.; Estrada, M.; Insuasty, B.; Guerrero, M. F. Rev. Colomb. Cienc. Quim.-Farm. 2014, 43, 22–38. doi:10.15446/rcciquifa.v43n1.45462
Return to citation in text: [1] -
Lim, F. P. L.; Luna, G.; Dolzhenko, A. V. Tetrahedron Lett. 2015, 56, 7016–7019. doi:10.1016/j.tetlet.2015.11.006
Return to citation in text: [1] -
Seela, F.; Lindner, M.; Glaçon, V.; Lin, W. J. Org. Chem. 2004, 69, 4695–4700. doi:10.1021/jo040150i
Return to citation in text: [1] -
Migawa, M.; Drach, J.; Townsend, L. J. Med. Chem. 2005, 48, 3840–3851. doi:10.1021/jm0402014
Return to citation in text: [1] -
Fadda, A. A.; Rabie, R.; Etman, H. A. J. Heterocycl. Chem. 2017, 54, 1015–1023. doi:10.1002/jhet.2669
Return to citation in text: [1] -
Al-Matar, H. M.; Khalil, K. D.; Al-Dorri, D. M.; Elnagdi, M. H. Molecules 2010, 15, 3302–3310. doi:10.3390/molecules15053302
Return to citation in text: [1] -
Rusinov, V. L.; Ulomskii, E. N.; Chupakhin, O. N.; Charushin, V. N. Russ. Chem. Bull. 2008, 57, 985–1014. doi:10.1007/s11172-008-0130-8
Return to citation in text: [1] -
Berger, D. M.; Dutia, M. D.; Hopper, D.; Torres, N. Int. Appl. PCT WO 2009/039387 A1, March 26, 2009.
Return to citation in text: [1] -
Guerrini, G.; Costanzo, A.; Ciciani, G.; Bruni, F.; Selleri, S.; Costagli, C.; Besnard, F.; Costa, B.; Martini, C.; De Siena, G.; Malmberg-Aiello, P. Bioorg. Med. Chem. 2006, 14, 758–775. doi:10.1016/j.bmc.2005.08.058
Return to citation in text: [1] -
Shawali, A. S. J. Adv. Res. 2012, 3, 185–188. doi:10.1016/j.jare.2011.07.004
Return to citation in text: [1] -
Abdelhamid, A. O.; Fahmi, A. A.; Halim, K. N. M. Synth. Commun. 2013, 43, 1101–1126. doi:10.1080/00397911.2011.616639
Return to citation in text: [1] -
Metwally, M. A.; Gouda, M. A.; Harmal, A. N.; Khalil, A. M. Eur. J. Med. Chem. 2012, 56, 254–262. doi:10.1016/j.ejmech.2012.08.034
Return to citation in text: [1] -
Al-Adiwish, W. M.; Tahir, M. I. M.; Siti-Noor-Adnalizawati, A.; Hashim, S. F.; Ibrahim, N.; Yaacob, W. A. Eur. J. Med. Chem. 2013, 64, 464–476. doi:10.1016/j.ejmech.2013.04.029
Return to citation in text: [1] -
Schulze, M. C.; Scott, B. L.; Chavez, D. E. J. Mater. Chem. A 2015, 3, 17963–17965. doi:10.1039/C5TA05291B
Return to citation in text: [1] -
Ledenyova, I. V.; Didenko, V. V.; Shestakov, A. S.; Shikhaliev, K. S. J. Heterocycl. Chem. 2013, 50, 573–578. doi:10.1002/jhet.1533
Return to citation in text: [1]
| 91. | Mulakayala, N.; Upendar Reddy, C. H.; Chaitanya, M.; Manzoor Hussain, M.; Kumar, C. S.; Golla, N. J. Korean Chem. Soc. 2011, 55, 719–722. doi:10.5012/jkcs.2011.55.4.719 |
| 92. | Buriol, L.; München, T. S.; Frizzo, C. P.; Marzari, M. R. B.; Zanatta, N.; Bonacorso, H. G.; Martins, M. A. P. Ultrason. Sonochem. 2013, 20, 1139–1143. doi:10.1016/j.ultsonch.2013.02.006 |
| 80. | Trapani, G.; Laquintana, V.; Denora, N.; Trapani, A.; Lopedota, A.; Latrofa, A.; Franco, M.; Serra, M.; Pisu, M. G.; Floris, I.; Sanna, E.; Biggio, G.; Liso, G. J. Med. Chem. 2005, 48, 292–305. doi:10.1021/jm049610q |
| 89. | Aggarwal, R.; Sumran, G.; Claramunt, R. M.; Sanz, D.; Elguero, J. J. Mol. Struct. 2009, 934, 96–102. doi:10.1016/j.molstruc.2009.06.021 |
| 90. | Aggarwal, R.; Masan, E.; Kaushik, P.; Kaushik, D.; Sharma, C.; Aneja, K. R. J. Fluorine Chem. 2014, 168, 16–24. doi:10.1016/j.jfluchem.2014.08.017 |
| 88. | Marjani, A. P.; Khalafy, J.; Salami, F.; Ezati, M. ARKIVOC 2015, No. 5, 277–286. doi:10.3998/ark.5550190.p009.062 |
| 97. | Dwyer, M. P.; Paruch, K.; Labroli, M.; Alvarez, C.; Keertikar, K. M.; Poker, C.; Rossman, R.; Fischmann, T. O.; Duca, J. S.; Madison, V.; Parry, D.; Davis, N.; Seghezzi, W.; Wiswell, D.; Guzi, T. J. Bioorg. Med. Chem. Lett. 2011, 21, 467–470. doi:10.1016/j.bmcl.2010.10.113 |
| 95. | Kamal, A.; Faazil, S.; Hussaini, S. M. A.; Ramaiah, M. J.; Balakrishna, M.; Patel, N.; Pushpavalli, S. N. C. V. L.; Pal-Bhadra, M. Bioorg. Med. Chem. Lett. 2016, 26, 2077–2083. doi:10.1016/j.bmcl.2016.02.072 |
| 96. | Griffith, D. A.; Hargrove, D. M.; Maurer, T. S.; Blum, C. A.; De Lombaert, S.; Inthavongsay, J. K.; Klade, L. E.; Mack, C. M.; Rose, C. R.; Sanders, M. J.; Carpino, P. A. Bioorg. Med. Chem. Lett. 2011, 21, 2641–2645. doi:10.1016/j.bmcl.2010.12.116 |
| 93. | Saikia, P.; Gogoi, S.; Boruah, R. C. J. Org. Chem. 2015, 80, 6885–6889. doi:10.1021/acs.joc.5b00933 |
| 94. | Kamal, A.; Tamboli, J. R.; Nayak, V. L.; Adil, S. F.; Vishnuvardhan, M. V. P. S.; Ramakrishna, S. Bioorg. Med. Chem. Lett. 2013, 23, 3208–3215. doi:10.1016/j.bmcl.2013.03.129 |
| 102. | Tian, Y.; Du, D.; Rai, D.; Wang, L.; Liu, H.; Zhan, P.; De Clercq, E.; Pannecouque, C.; Liu, X. Bioorg. Med. Chem. 2014, 22, 2052–2059. doi:10.1016/j.bmc.2014.02.029 |
| 130. | Hassan, G. S.; Kadry, H. H.; Abou-Seri, S. M.; Ali, M. M.; Mahmoud, A. E. E.-D. Bioorg. Med. Chem. 2011, 19, 6808–6817. doi:10.1016/j.bmc.2011.09.036 |
| 103. | Kaswan, P.; Pericherla, K.; Purohit, D.; Kumar, A. Tetrahedron Lett. 2015, 56, 549–553. doi:10.1016/j.tetlet.2014.11.121 |
| 129. | El-Moghazy, S. M.; George, R. F.; Osman, E. E. A.; Elbatrawy, A. A.; Kissova, M.; Colombo, A.; Crespan, E.; Maga, G. Eur. J. Med. Chem. 2016, 123, 1–13. doi:10.1016/j.ejmech.2016.07.034 |
| 100. | Aggarwal, R.; Kumar, V.; Singh, G.; Sanz, D.; Claramunt, R. M.; Alkorta, I.; Sánchez-Sanz, G.; Elguero, J. J. Heterocycl. Chem. 2015, 52, 336–345. doi:10.1002/jhet.1955 |
| 115. | Bakavoli, M.; Bagherzadeh, G.; Vaseghifar, M.; Shiri, A.; Pordel, M.; Mashreghi, M.; Pordeli, P.; Araghi, M. Eur. J. Med. Chem. 2010, 45, 647–650. doi:10.1016/j.ejmech.2009.10.051 |
| 101. | Aggarwal, R.; Singh, G.; Kaushik, P.; Kaushik, D.; Paliwal, D.; Kumar, A. Eur. J. Med. Chem. 2015, 101, 326–333. doi:10.1016/j.ejmech.2015.06.011 |
| 131. | Singla, P.; Luxami, V.; Singh, R.; Tandon, V.; Paul, K. Eur. J. Med. Chem. 2017, 126, 24–35. doi:10.1016/j.ejmech.2016.09.093 |
| 98. | Azeredo, L. F. S. P.; Coutinho, J. P.; Jabor, V. A. P.; Feliciano, P. R.; Nonato, M. C.; Kaiser, C. R.; Menezes, C. M. S.; Hammes, A. S. O.; Caffarena, E. R.; Hoelz, L. V. B.; Souza, N. B.; Pereira, G. A. N.; Cerávolo, I. P.; Krettli, A. U.; Boechat, N. Eur. J. Med. Chem. 2017, 126, 72–83. doi:10.1016/j.ejmech.2016.09.073 |
| 124. | Guccione, S.; Modica, M.; Longmore, J.; Shaw, D.; Barretta, G. U.; Santagati, A.; Santagati, M.; Russo, F. Bioorg. Med. Chem. Lett. 1996, 6, 59–64. doi:10.1016/0960-894X(95)00558-B |
| 125. | Xia, Y.; Chackalamannil, S.; Czarniecki, M.; Tsai, H.; Vaccaro, H.; Cleven, R.; Cook, J.; Fawzi, A.; Watkins, R.; Zhang, H. J. Med. Chem. 1997, 40, 4372–4377. doi:10.1021/jm970495b |
| 99. | Hylsová, M.; Carbain, B.; Fanfrlík, J.; Musilová, L.; Haldar, S.; Köprülüoğlu, C.; Ajani, H.; Brahmkshatriya, P. S.; Jorda, R.; Kryštof, V.; Hobza, P.; Echalier, A.; Paruch, K.; Lepšík, M. Eur. J. Med. Chem. 2017, 126, 1118–1128. doi:10.1016/j.ejmech.2016.12.023 |
| 123. | Hutson, P. H.; Finger, E. N.; Magliaro, B. C.; Smith, S. M.; Converso, A.; Sanderson, P. E.; Mullins, D.; Hyde, L. A.; Eschle, B. K.; Turnbull, Z.; Sloan, H.; Guzzi, M.; Zhang, X.; Wang, A.; Rindgen, D.; Mazzola, R.; Vivian, J. A.; Eddins, D.; Uslaner, J. M.; Bednar, R.; Gambone, C.; Le-Mair, W.; Marino, M. J.; Sachs, N.; Xu, G.; Parmentier-Batteur, S. Neuropharmacology 2011, 61, 665–676. doi:10.1016/j.neuropharm.2011.05.009 |
| 128. | Ghozlan, S. A. S.; Abdelrazek, F. M.; Mohamed, M. H.; Azmy, K. E. J. Heterocycl. Chem. 2010, 47, 1379–1385. doi:10.1002/jhet.482 |
| 126. | Trivedi, A.; Dodiya, D.; Surani, J.; Jarsania, S.; Mathukiya, H.; Ravat, N.; Shah, V. Arch. Pharm. 2008, 341, 435–439. doi:10.1002/ardp.200800027 |
| 127. | Trivedi, A.; Vaghasiya, S.; Dholariya, B.; Dodiya, D.; Shah, V. J. Enzyme Inhib. Med. Chem. 2010, 25, 893–899. doi:10.3109/14756360903540276 |
| 122. | Wang, C.; Liu, H.; Song, Z.; Ji, Y.; Xing, L.; Peng, X.; Wang, X.; Ai, J.; Geng, M.; Zhang, A. Bioorg. Med. Chem. Lett. 2017, 27, 2544–2548. doi:10.1016/j.bmcl.2017.03.088 |
| 106. | Abdelhamid, A. O.; Gomha, S. M. J. Chem. 2013, No. 327095. doi:10.1155/2013/327095 |
| 107. | Mostafa, S. S.; Salem Nada, A. M. M. Eur. Chem. Bull. 2015, 4, 50–59. doi:10.17628/ecb.2015.4.50-59 |
| 104. | Chobe, S. S.; Dawane, B. S.; Tumbi, K. M.; Nandekar, P. P.; Sangamwar, A. T. Bioorg. Med. Chem. Lett. 2012, 22, 7566–7572. doi:10.1016/j.bmcl.2012.10.027 |
| 105. | Ahmetaj, S.; Velikanje, N.; Grošelj, U.; Šterbal, I.; Prek, B.; Golobič, A.; Kočar, D.; Dahmann, G.; Stanovnik, B.; Svete, J. Mol. Diversity 2013, 17, 731–743. doi:10.1007/s11030-013-9469-3 |
| 132. | Venkatesan, G.; Paira, P.; Cheong, S. L.; Federico, S.; Klotz, K. N.; Spalluto, G.; Pastorin, G. Eur. J. Med. Chem. 2015, 92, 784–798. doi:10.1016/j.ejmech.2015.01.046 |
| 64. | Insuasty, D.; Abonia, R.; Insuasty, B.; Quiroga, J.; Laali, K. K.; Nogueras, M.; Cobo, J. ACS Comb. Sci. 2017, 19, 555–563. doi:10.1021/acscombsci.7b00091 |
| 65. | Quiroga, J.; Trilleras, J.; Pantoja, D.; Abonía, R.; Insuasty, B.; Nogueras, M.; Cobo, J. Tetrahedron Lett. 2010, 51, 4717–4719. doi:10.1016/j.tetlet.2010.07.009 |
| 141. | Foks, H.; Pancechowska-Ksepko, D.; Kędzia, A.; Zwolska, Z.; Janowiec, M.; Augustynowicz-Kopeć, E. Farmaco 2005, 60, 513–517. doi:10.1016/j.farmac.2005.05.002 |
| 63. | Anand, D.; Yadav, P. K.; Patel, O. P. S.; Parmar, N.; Maurya, R. K.; Vishwakarma, P.; Raju, K. S. R.; Taneja, I.; Wahajuddin, M.; Kar, S.; Yadav, P. P. J. Med. Chem. 2017, 60, 1041–1059. doi:10.1021/acs.jmedchem.6b01447 |
| 140. | El-Emary, T. I. J. Chin. Chem. Soc. 2006, 53, 391–401. doi:10.1002/jccs.200600050 |
| 1. | Kumar, H.; Saini, D.; Jain, S.; Jain, N. Eur. J. Med. Chem. 2013, 70, 248–258. doi:10.1016/j.ejmech.2013.10.004 |
| 2. | Gomha, S. M.; Edrees, M. M.; Faty, R. A. M.; Muhammad, Z. A.; Mabkhot, Y. N. Chem. Cent. J. 2017, 11, No. 37. doi:10.1186/s13065-017-0266-4. |
| 135. | Zhang, X.; Lin, Q.; Zhong, P. Molecules 2010, 15, 3079–3086. doi:10.3390/molecules15053079 |
| 134. | Song, X. J.; Shao, Y.; Dong, X. G. Chin. Chem. Lett. 2011, 22, 1036–1038. doi:10.1016/j.cclet.2011.05.012 |
| 137. | Binch, H.; Robinson, D.; Miller, A.; Fraysse, D. Pyrrolopyrazines and pyrazolopyrazines useful as inhibitors of protein kinases. WO Patent WO 2006058074, June 1, 2006. |
| 136. | Todorovic, N.; Awuah, E.; Shakya, T.; Wright, G. D.; Capretta, A. Tetrahedron Lett. 2011, 52, 5761–5763. doi:10.1016/j.tetlet.2011.08.103 |
| 9. | Saad, H. A.; Osman, N. A.; Moustafa, A. H. Molecules 2011, 16, 10187–10201. doi:10.3390/molecules161210187 |
| 72. | Hill, M. D.; Fang, H.; Brown, J. M.; Molski, T.; Easton, A.; Han, X.; Miller, R.; Hill-Drzewi, M.; Gallagher, L.; Matchett, M.; Gulianello, M.; Balakrishnan, A.; Bertekap, R. L.; Santone, K. S.; Whiterock, V. J.; Zhuo, X.; Bronson, J. J.; Macor, J. E.; Degnan, A. P. ACS Med. Chem. Lett. 2016, 7, 1082–1086. doi:10.1021/acsmedchemlett.6b00292 |
| 73. | Hill, M. D. Synthesis 2016, 48, 2201–2204. doi:10.1055/s-0035-1562230 |
| 6. | Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Eur. J. Med. Chem. 2005, 40, 922–927. doi:10.1016/j.ejmech.2005.03.021 |
| 7. | Aggarwal, R.; Bansal, A.; Rozas, I.; Diez-Cecilia, E.; Kaur, A.; Mahajan, R.; Sharma, J. Med. Chem. Res. 2014, 23, 1454–1464. doi:10.1007/s00044-013-0751-9 |
| 8. | Aggarwal, R.; Kumar, R.; Kumar, S.; Garg, G.; Mahajan, R.; Sharma, J. J. Fluorine Chem. 2011, 132, 965–972. doi:10.1016/j.jfluchem.2011.07.029 |
| 5. | Mukarram, S.; Bandgar, B. P.; Shaikh, R. U.; Ganapure, S. D.; Chavan, H. V. Med. Chem. Res. 2017, 26, 262–273. doi:10.1007/s00044-016-1744-2 |
| 70. | Huang, Z.; Hu, Y.; Zhou, Y.; Shi, D. ACS Comb. Sci. 2011, 13, 45–49. doi:10.1021/co1000162 |
| 133. | Liu, J.; Zhang, X.-W.; Song, D.-Z.; Cai, Z.-q.; Zhou, Y.-p. J. Chem. Res. 2016, 40, 63–125. doi:10.3184/174751916X14531173426246 |
| 3. | Aggarwal, R.; Bansal, A.; Rozas, I.; Kelly, B.; Kaushik, P.; Kaushik, D. Eur. J. Med. Chem. 2013, 70, 350–357. doi:10.1016/j.ejmech.2013.09.052 |
| 4. | Aggarwal, R.; Kumar, S.; Kaushik, P.; Kaushik, D.; Gupta, G. K. Eur. J. Med. Chem. 2013, 62, 508–514. doi:10.1016/j.ejmech.2012.11.046 |
| 71. | El-borai, M. A.; Rizk, H. F.; Abd-Aal, M. F.; El-Deeb, I. Y. Eur. J. Med. Chem. 2012, 48, 92–96. doi:10.1016/j.ejmech.2011.11.038 |
| 20. | Kaplan, J.; Verheijen, J. C.; Brooijmans, N.; Toral-Barza, L.; Hollander, I.; Yu, K.; Zask, A. Bioorg. Med. Chem. Lett. 2010, 20, 640–643. doi:10.1016/j.bmcl.2009.11.050 |
| 14. | Bebernitz, G. R.; Argentieri, G.; Battle, B.; Brennan, C.; Balkan, B.; Burkey, B. F.; Eckhardt, M.; Gao, J.; Kapa, P.; Strohschein, R. J.; Schuster, H. F.; Wilson, M.; Xu, D. D. J. Med. Chem. 2001, 44, 2601–2611. doi:10.1021/jm010032c |
| 68. | Frolova, L. V.; Malik, I.; Uglinskii, P. Y.; Rogelj, S.; Kornienko, A.; Magedov, I. V. Tetrahedron Lett. 2011, 52, 6643–6645. doi:10.1016/j.tetlet.2011.10.012 |
| 6. | Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Eur. J. Med. Chem. 2005, 40, 922–927. doi:10.1016/j.ejmech.2005.03.021 |
| 69. | Fan, L.; Yao, C.; Shu, M. Heterocycl. Commun. 2016, 22, 63–67. doi:10.1515/hc-2015-0234 |
| 12. | Aggarwal, R.; Kumar, V.; Gupta, G. K.; Kumar, V. Med. Chem. Res. 2013, 22, 3566–3573. doi:10.1007/s00044-012-0343-0 |
| 13. | Bekhit, A. A.; Ashour, H. M. A.; Ghany, Y. S. A.; Bekhit, A. E.-D. A.; Baraka, A. Eur. J. Med. Chem. 2008, 43, 456–463. doi:10.1016/j.ejmech.2007.03.030 |
| 66. | Nikpassand, M.; Mamaghani, M.; Shirini, F.; Tabatabaeian, K. Ultrason. Sonochem. 2010, 17, 301–305. doi:10.1016/j.ultsonch.2009.08.001 |
| 10. | El-sabbhag, O. I.; Baraka, M. M.; Ibrahim, S. M.; Pannecouque, C.; Andrei, G.; Snoeck, R.; Balzarini, J.; Rashad, A. A. Eur. J. Med. Chem. 2009, 44, 3746–3753. doi:10.1016/j.ejmech.2009.03.038 |
| 11. | Ouyang, G.; Chen, Z.; Cai, X.-J.; Song, B.-A.; Bhadury, P. S.; Yang, S.; Jin, L.-H.; Xue, W.; Hu, D.-Y.; Zeng, S. Bioorg. Med. Chem. 2008, 16, 9699–9707. doi:10.1016/j.bmc.2008.09.070 |
| 67. | Yao, C.-S.; Wang, C.-H.; Jiang, B.; Tu, S.-J. J. Comb. Chem. 2010, 12, 472–475. doi:10.1021/cc100017f |
| 76. | Dandia, A.; Gupta, S. L.; Singh, P.; Quraishi, M. A. ACS Sustainable Chem. Eng. 2013, 1, 1303–1310. doi:10.1021/sc400155u |
| 150. | Youdim, M. B. H.; Bakhle, Y. S. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S287–S296. doi:10.1038/sj.bjp.0706464 |
| 77. | Gunasekaran, P.; Prasanna, P.; Perumal, S. Tetrahedron Lett. 2014, 55, 329–332. doi:10.1016/j.tetlet.2013.11.016 |
| 149. | Trincavelli, M. L.; Da Pozzo, E.; Daniele, S.; Martini, C. Curr. Top. Med. Chem. 2012, 12, 254–269. doi:10.2174/1568026799078787 |
| 74. | Aggarwal, R.; Singh, G.; Kumar, S.; McCabe, T.; Rozas, I. ChemistrySelect 2016, 1, 5990–5994. doi:10.1002/slct.201601201 |
| 75. | Rahmati, A. Tetrahedron Lett. 2010, 51, 2967–2970. doi:10.1016/j.tetlet.2010.03.109 |
| 151. | Kulkarni, S. K.; Dhir, A. Expert Opin. Invest. Drugs 2009, 18, 767–788. doi:10.1517/13543780902880850 |
| 146. | Farghaly, A.-R.; Esmail, S.; Abdel-Zaher, A.; Abdel-Hafez, A.; El-Kashef, H. Bioorg. Med. Chem. 2014, 22, 2166–2175. doi:10.1016/j.bmc.2014.02.019 |
| 145. | El-Emary, T.; El-Kashef, H. Eur. J. Med. Chem. 2013, 62, 478–485. doi:10.1016/j.ejmech.2013.01.025 |
| 147. | Nie, Z.; Perretta, C.; Erickson, P.; Margosiak, S.; Lu, J.; Averill, A.; Almassy, R.; Chu, S. Bioorg. Med. Chem. Lett. 2008, 18, 619–623. doi:10.1016/j.bmcl.2007.11.074 |
| 142. | El-Kashef, H. S.; El-Emary, T. I.; Gasquet, M.; Timon-David, P.; Maldonado, J.; Vanelle, P. Pharmazie 2000, 55, 572–576. |
| 87. | Médran-Navarrete, V.; Damont, A.; Peyronneau, M. A.; Kuhnast, B.; Bernards, N.; Pottier, G.; Marguet, F.; Puech, F.; Boisgard, R.; Dollé, F. Bioorg. Med. Chem. Lett. 2014, 24, 1550–1556. doi:10.1016/j.bmcl.2014.01.080 |
| 144. | Quiroga, J.; Sánchez, N. E.; Acosta, P.; Insuasty, B.; Abonia, R. Tetrahedron Lett. 2012, 53, 3181–3187. doi:10.1016/j.tetlet.2012.04.083 |
| 19. | Damont, A.; Médran-Navarrete, V.; Cacheux, F.; Kuhnast, B.; Pottier, G.; Bernards, N.; Marguet, F.; Puech, F.; Boisgard, R.; Dollé, F. J. Med. Chem. 2015, 58, 7449–7464. doi:10.1021/acs.jmedchem.5b00932 |
| 143. | El-Emary, T. I.; Kamal El-Dean, A. M.; El-Kashef, H. S. Farmaco 1998, 53, 383–388. doi:10.1016/S0014-827X(98)00014-7 |
| 80. | Trapani, G.; Laquintana, V.; Denora, N.; Trapani, A.; Lopedota, A.; Latrofa, A.; Franco, M.; Serra, M.; Pisu, M. G.; Floris, I.; Sanna, E.; Biggio, G.; Liso, G. J. Med. Chem. 2005, 48, 292–305. doi:10.1021/jm049610q |
| 81. | Wu, Y.-C.; Zou, X.-M.; Hu, F.-Z.; Yang, H.-Z. J. Heterocycl. Chem. 2005, 42, 609–613. doi:10.1002/jhet.5570420421 |
| 82. | Aggarwal, R.; Sumran, G.; Garg, N.; Aggarwal, A. Eur. J. Med. Chem. 2011, 46, 3038–3048. doi:10.1016/j.ejmech.2011.04.041 |
| 83. | Hassan, A. S.; Mady, M. F.; Awad, H. M.; Hafez, T. S. Chin. Chem. Lett. 2017, 28, 388–393. doi:10.1016/j.cclet.2016.10.022 |
| 84. | Al-Mousawi, S. M.; Mohammad, M. A.; Elnagdi, M. H. J. Heterocycl. Chem. 2001, 38, 989–991. doi:10.1002/jhet.5570380430 |
| 85. | Jakše, R.; Svete, J.; Stanovnik, B.; Golobič, A. Tetrahedron 2004, 60, 4601–4608. doi:10.1016/j.tet.2004.03.075 |
| 86. | Lager, E.; Andersson, P.; Nilsson, J.; Pettersson, I.; Nielsen, E. Ø.; Nielsen, M.; Sterner, O.; Liljefors, T. J. Med. Chem. 2006, 49, 2526–2533. doi:10.1021/jm058057p |
| 78. | Jiang, B.; Ye, Q.; Fan, W.; Wang, S.-L.; Tu, S.-J.; Li, G. Chem. Commun. 2014, 50, 6108–6111. doi:10.1039/C4CC00740A |
| 79. | Wang, J.-J.; Feng, X.; Xun, Z.; Shi, D.-Q.; Huang, Z.-B. J. Org. Chem. 2015, 80, 8435–8442. doi:10.1021/acs.joc.5b01314 |
| 36. | Elnagdi, M. H.; Negm, A. M.; Sadek, K. U. Synlett 1994, 27–37. doi:10.1055/s-1994-22729 |
| 37. | Quiroga, J.; Alvarado, M.; Insuasty, B.; Moreno, R.; Raviña, E.; Estevez, I.; De Almeida S., R. H. J. Heterocycl. Chem. 1999, 36, 1311–1316. doi:10.1002/jhet.5570360533 |
| 38. | Quiroga, J.; Hormaza, A.; Insuasty, B.; Márquez, M. J. Heterocycl. Chem. 1998, 35, 409–412. doi:10.1002/jhet.5570350225 |
| 39. | Rao, H. S. P.; Adigopula, L. N.; Ramadas, K. ACS Comb. Sci. 2017, 19, 279–285. doi:10.1021/acscombsci.6b00156 |
| 40. | Lee, S.; Park, S. B. Org. Lett. 2009, 11, 5214–5217. doi:10.1021/ol902147u |
| 41. | Bou-Petit, E.; Picas, E.; Puigjaner, C.; Font-Bardia, M.; Ferrer, N.; Sempere, J.; de la Bellacasa, R. P.; Batllori, X.; Teixidó, J.; Estrada-Tejedor, R.; y Cajal, S. R.; Borrell, J. I. ChemistrySelect 2017, 2, 3668–3672. doi:10.1002/slct.201700732 |
| 161. | Rusinov, V. L.; Ulomskii, E. N.; Chupakhin, O. N.; Charushin, V. N. Russ. Chem. Bull. 2008, 57, 985–1014. doi:10.1007/s11172-008-0130-8 |
| 162. | Berger, D. M.; Dutia, M. D.; Hopper, D.; Torres, N. Int. Appl. PCT WO 2009/039387 A1, March 26, 2009. |
| 163. | Guerrini, G.; Costanzo, A.; Ciciani, G.; Bruni, F.; Selleri, S.; Costagli, C.; Besnard, F.; Costa, B.; Martini, C.; De Siena, G.; Malmberg-Aiello, P. Bioorg. Med. Chem. 2006, 14, 758–775. doi:10.1016/j.bmc.2005.08.058 |
| 42. | Aggarwal, R.; Kumar, V.; Bansal, A.; Sanz, D.; Claramunt, R. M. J. Fluorine Chem. 2012, 140, 31–37. doi:10.1016/j.jfluchem.2012.04.007 |
| 160. | Al-Matar, H. M.; Khalil, K. D.; Al-Dorri, D. M.; Elnagdi, M. H. Molecules 2010, 15, 3302–3310. doi:10.3390/molecules15053302 |
| 156. | Lim, F. P. L.; Luna, G.; Dolzhenko, A. V. Tetrahedron Lett. 2015, 56, 7016–7019. doi:10.1016/j.tetlet.2015.11.006 |
| 155. | Insuasty, H.; Castro, E.; Escobar, J. C.; Murillo, V.; Rodríguez, J.; Cuca, L. E.; Estrada, M.; Insuasty, B.; Guerrero, M. F. Rev. Colomb. Cienc. Quim.-Farm. 2014, 43, 22–38. doi:10.15446/rcciquifa.v43n1.45462 |
| 159. | Fadda, A. A.; Rabie, R.; Etman, H. A. J. Heterocycl. Chem. 2017, 54, 1015–1023. doi:10.1002/jhet.2669 |
| 157. | Seela, F.; Lindner, M.; Glaçon, V.; Lin, W. J. Org. Chem. 2004, 69, 4695–4700. doi:10.1021/jo040150i |
| 158. | Migawa, M.; Drach, J.; Townsend, L. J. Med. Chem. 2005, 48, 3840–3851. doi:10.1021/jm0402014 |
| 50. | Pandey, G.; Bhowmik, S.; Batra, S. Org. Lett. 2013, 15, 5044–5047. doi:10.1021/ol4023722 |
| 24. | Sun, L.; Bera, H.; Chui, W. K. Eur. J. Med. Chem. 2013, 65, 1–11. doi:10.1016/j.ejmech.2013.03.063 |
| 51. | Abdel-Aziz, H. A.; Saleh, T. S.; El-Zahabi, H. S. A. Arch. Pharm. 2010, 343, 24–30. doi:10.1002/ardp.200900082 |
| 152. | Bera, H.; Ojha, P. k.; Tan, B. J.; Sun, L.; Dolzhenko, A. V.; Chui, W.-K.; Chiu, G. N. C. Eur. J. Med. Chem. 2014, 78, 294–303. doi:10.1016/j.ejmech.2014.03.063 |
| 48. | Prakash, R.; Shekarrao, K.; Saikia, P.; Gogoi, S.; Boruah, R. C. RSC Adv. 2015, 5, 21099–21102. doi:10.1039/C4RA17190J |
| 49. | Shekarrao, K.; Kaishap, P. P.; Saddanapu, V.; Addlagatta, A.; Gogoi, S.; Boruah, R. C. RSC Adv. 2014, 4, 24001–24006. doi:10.1039/C4RA02865A |
| 154. | Insuasty, H.; Insuasty, B.; Castro, E.; Quiroga, J.; Abonía, R.; Nogueras, M.; Cobo, J. Tetrahedron 2012, 68, 9384–9390. doi:10.1016/j.tet.2012.09.029 |
| 49. | Shekarrao, K.; Kaishap, P. P.; Saddanapu, V.; Addlagatta, A.; Gogoi, S.; Boruah, R. C. RSC Adv. 2014, 4, 24001–24006. doi:10.1039/C4RA02865A |
| 153. | Abbas-Temirek, H. H.; Abo-Bakr, A. M. Eur. J. Chem. 2016, 7, 107–114. doi:10.5155/eurjchem.7.1.107-114.1369 |
| 45. | Shi, D.-Q.; Yang, F. J. Chin. Chem. Soc. 2008, 55, 755–760. doi:10.1002/jccs.200800113 |
| 46. | Shi, D.-Q.; Ni, S.-N.; Yang, F.; Shi, J.-W.; Dou, G.-L.; Li, X.-Y.; Wang, X.-S. J. Heterocycl. Chem. 2008, 45, 653–660. doi:10.1002/jhet.5570450303 |
| 47. | Chen, J.; Liu, W.; Ma, J.; Xu, H.; Wu, J.; Tang, X.; Fan, Z.; Wang, P. J. Org. Chem. 2012, 77, 3475–3482. doi:10.1021/jo3002722 |
| 43. | Ghaedi, A.; Bardajee, G. R.; Mirshokrayi, A.; Mahdavi, M.; Shafiee, A.; Akbarzadeh, T. RSC Adv. 2015, 5, 89652–89658. doi:10.1039/C5RA16769H |
| 44. | Shi, D.-Q.; Zhou, Y.; Liu, H. Synth. Commun. 2010, 40, 3660–3668. doi:10.1080/00397910903471796 |
| 52. | Yen, W.-P.; Liu, P.-L.; Uramaru, N.; Lin, H.-Y.; Wong, F. F. Tetrahedron 2015, 71, 8798–8803. doi:10.1016/j.tet.2015.09.042 |
| 53. | Nascimento-Júnior, N. M.; Mendes, T. C. F.; Leal, D. M.; Corrêa, C. M. N.; Sudo, R. T.; Zapata-Sudo, G.; Barreiro, E. J.; Fragaa, C. A. M. Bioorg. Med. Chem. Lett. 2010, 20, 74–77. doi:10.1016/j.bmcl.2009.11.038 |
| 54. | Jiang, B.; Liu, Y.-P.; Tu, S.-J. Eur. J. Org. Chem. 2011, 3026–3035. doi:10.1002/ejoc.201100127 |
| 169. | Ledenyova, I. V.; Didenko, V. V.; Shestakov, A. S.; Shikhaliev, K. S. J. Heterocycl. Chem. 2013, 50, 573–578. doi:10.1002/jhet.1533 |
| 61. | Gao, X.; Yang, Z.; Hao, W.-J.; Jiang, B.; Tu, S.-J. J. Heterocycl. Chem. 2017, 54, 2434–2439. doi:10.1002/jhet.2840 |
| 166. | Metwally, M. A.; Gouda, M. A.; Harmal, A. N.; Khalil, A. M. Eur. J. Med. Chem. 2012, 56, 254–262. doi:10.1016/j.ejmech.2012.08.034 |
| 62. | Hao, Y.; Xu, X.-P.; Chen, T.; Zhao, L.-L.; Ji, S.-J. Org. Biomol. Chem. 2012, 10, 724–728. doi:10.1039/C1OB06624B |
| 165. | Abdelhamid, A. O.; Fahmi, A. A.; Halim, K. N. M. Synth. Commun. 2013, 43, 1101–1126. doi:10.1080/00397911.2011.616639 |
| 59. | De, K.; Bhaumik, A.; Banerjee, B.; Mukhopadhyay, C. Tetrahedron Lett. 2015, 56, 1614–1618. doi:10.1016/j.tetlet.2015.01.163 |
| 168. | Schulze, M. C.; Scott, B. L.; Chavez, D. E. J. Mater. Chem. A 2015, 3, 17963–17965. doi:10.1039/C5TA05291B |
| 60. | Dandia, A.; Laxkar, A. K.; Singh, R. Tetrahedron Lett. 2012, 53, 3012–3017. doi:10.1016/j.tetlet.2012.03.136 |
| 167. | Al-Adiwish, W. M.; Tahir, M. I. M.; Siti-Noor-Adnalizawati, A.; Hashim, S. F.; Ibrahim, N.; Yaacob, W. A. Eur. J. Med. Chem. 2013, 64, 464–476. doi:10.1016/j.ejmech.2013.04.029 |
| 57. | Wang, Z.; Gao, L.; Xu, Z.; Ling, Z.; Qin, Y.; Rong, L.; Tu, S.-J. Tetrahedron 2017, 73, 385–394. doi:10.1016/j.tet.2016.12.015 |
| 58. | Quiroga, J.; Portillo, S.; Pérez, A.; Gálvez, J.; Abonia, R.; Insuasty, B. Tetrahedron Lett. 2011, 52, 2664–2666. doi:10.1016/j.tetlet.2011.03.067 |
| 55. | Liu, M.; Yin, G.; Zhu, C.; Yao, C. J. Heterocycl. Chem. 2015, 53, 1617–1625. doi:10.1002/jhet.2473 |
| 164. | Shawali, A. S. J. Adv. Res. 2012, 3, 185–188. doi:10.1016/j.jare.2011.07.004 |
| 56. | Bazgir, A.; Ahadi, S.; Ghahremanzadeh, R.; Khavasi, H. R.; Mirzaei, P. Ultrason. Sonochem. 2010, 17, 447–452. doi:10.1016/j.ultsonch.2009.09.009 |
| 114. | Amir, M.; Javed, S.; Kumar, H. Indian J. Pharm. Sci. 2007, 69, 337–343. doi:10.4103/0250-474X.34540 |
| 115. | Bakavoli, M.; Bagherzadeh, G.; Vaseghifar, M.; Shiri, A.; Pordel, M.; Mashreghi, M.; Pordeli, P.; Araghi, M. Eur. J. Med. Chem. 2010, 45, 647–650. doi:10.1016/j.ejmech.2009.10.051 |
| 116. | Abunada, N. M.; Hassaneen, H. M.; Kandile, N. G.; Miqdad, O. A. Molecules 2008, 13, 1501–1517. doi:10.3390/molecules13071501 |
| 117. | Raffa, D.; Maggio, B.; Plescia, F.; Cascioferro, S.; Raimondi, M. V.; Plescia, S.; Cusimano, M. G. Arch. Pharm. 2009, 342, 321–326. doi:10.1002/ardp.200800140 |
| 111. | El-Bendary, E. R.; Badria, F. A. Arch. Pharm. 2000, 333, 99–103. doi:10.1002/(SICI)1521-4184(20004)333:4<99::AID-ARDP99>3.0.CO;2-O |
| 112. | Chern, J.-H.; Shia, K.-S.; Hsu, T.-A.; Tai, C.-L.; Lee, C.-C.; Lee, Y.-C.; Chang, C.-S.; Tseng, S.-N.; Shih, S.-R. Bioorg. Med. Chem. Lett. 2004, 14, 2519–2525. doi:10.1016/j.bmcl.2004.02.092 |
| 113. | Ram, V. J.; Pandey, H. N.; Mishra, L. Arch. Pharm. 1979, 312, 586–590. doi:10.1002/ardp.19793120705 |
| 109. | Stepaniuk, O. O.; Matviienko, V. O.; Kontratov, I. S.; Vitruk, I. V.; Tolmachev, A. O. Synthesis 2013, 45, 925–930. doi:10.1055/s-0032-1318329 |
| 110. | Ma, Y.-Q.; Zhang, Z.-T.; He, Q.; Xue, D.; Zhang, P.-F. Synth. Commun. 2012, 42, 933–941. doi:10.1080/00397911.2010.521902 |
| 108. | Ren, L.; Laird, E. R.; Buckmelter, A. J.; Dinkel, V.; Gloor, S. L.; Grina, J.; Newhouse, B.; Rasor, K.; Hastings, G.; Gradl, S. N.; Rudolph, J. Bioorg. Med. Chem. Lett. 2012, 22, 1165–1168. doi:10.1016/j.bmcl.2011.11.092 |
| 22. | Elnagdi, M. H.; Abdel-Galil, F. M.; Riad, B. Y.; Elgemeie, G. E. H. Heterocycles 1983, 20, 2437–2470. doi:10.3987/R-1983-12-2437 |
| 23. | Guerrini, G.; Ciciani, G.; Cambi, G.; Bruni, F.; Selleri, S.; Besnard, F.; Montali, M.; Martini, C.; Ghelardini, C.; Galeotti, N.; Costanzo, A. Bioorg. Med. Chem. 2007, 15, 2573–2586. doi:10.1016/j.bmc.2007.01.053 |
| 19. | Damont, A.; Médran-Navarrete, V.; Cacheux, F.; Kuhnast, B.; Pottier, G.; Bernards, N.; Marguet, F.; Puech, F.; Boisgard, R.; Dollé, F. J. Med. Chem. 2015, 58, 7449–7464. doi:10.1021/acs.jmedchem.5b00932 |
| 20. | Kaplan, J.; Verheijen, J. C.; Brooijmans, N.; Toral-Barza, L.; Hollander, I.; Yu, K.; Zask, A. Bioorg. Med. Chem. Lett. 2010, 20, 640–643. doi:10.1016/j.bmcl.2009.11.050 |
| 21. | El-Morsy, A. M.; El-Sayed, M. S.; Abulkhair, H. S. Open J. Med. Chem. 2017, 7, No. 1. doi:10.4236/ojmc.2017.71001 |
| 16. | Manikannan, R.; Venkatesan, R.; Muthusubramanian, S.; Yogeeswari, P.; Sriram, D. Bioorg. Med. Chem. Lett. 2010, 20, 6920–6924. doi:10.1016/j.bmcl.2010.09.137 |
| 17. | Özdemir, A.; Altıntop, M. D.; Kaplancıklı, Z. A.; Can, Ö. D.; Özkay, Ü. D.; Turan-Zitouni, G. Molecules 2015, 20, 2668–2684. doi:10.3390/molecules20022668 |
| 121. | Fu, Y.; Wang, Y.; Wan, S.; Li, Z.; Wang, G.; Zhang, J.; Wu, X. Molecules 2017, 22, 542. doi:10.3390/molecules22040542 |
| 18. | Zhang, L.; Balan, G.; Barreiro, G.; Boscoe, B. P.; Chenard, L. K.; Cianfrogna, J.; Claffey, M. M.; Chen, L.; Coffman, K. J.; Drozda, S. E.; Dunetz, J. R.; Fonseca, K. R.; Galatsis, P.; Grimwood, S.; Lazzaro, J. T.; Mancuso, J. Y.; Miller, E. L.; Reese, M. R.; Rogers, B. N.; Sakurada, I.; Skaddan, M.; Smith, D. L.; Stepan, A. F.; Trapa, P.; Tuttle, J. B.; Verhoest, P. R.; Walker, D. P.; Wright, A. S.; Zaleska, M. M.; Zasadny, K.; Shaffer, C. L. J. Med. Chem. 2014, 57, 861–877. doi:10.1021/jm401622k |
| 118. | Petrie, C. R.; Cottam, H. B.; McKernan, P. A.; Robins, R. K.; Revankar, G. R. J. Med. Chem. 1985, 28, 1010–1016. doi:10.1021/jm00146a007 |
| 15. | Yadava, U.; Shukla, B. K.; Roychoudhury, M.; Kumar, D. J. Mol. Model. 2015, 21, 96. doi:10.1007/s00894-015-2631-3 |
| 114. | Amir, M.; Javed, S.; Kumar, H. Indian J. Pharm. Sci. 2007, 69, 337–343. doi:10.4103/0250-474X.34540 |
| 119. | Celano, M.; Schenone, S.; Cosco, D.; Navarra, M.; Puxeddu, E.; Racanicchi, L.; Brullo, C.; Varano, E.; Alcaro, S.; Ferretti, E.; Botta, G.; Filetti, S.; Fresta, M.; Botta, M.; Russo, D. Endocr.-Relat. Cancer 2008, 15, 499–510. doi:10.1677/ERC-07-0243 |
| 120. | Fallacara, A. L.; Mancini, A.; Zamperini, C.; Dreassi, E.; Marianelli, S.; Chiariello, M.; Pozzi, G.; Santoro, F.; Botta, M.; Schenone, S. Bioorg. Med. Chem. Lett. 2017, 27, 3196–3200. doi:10.1016/j.bmcl.2017.05.015 |
| 26. | Anwar, H. F.; Elnagdi, M. H. ARKIVOC 2009, No. 1, 198–250. doi:10.3998/ark.5550190.0010.107 |
| 27. | Aggarwal, R.; Kumar, V.; Kumar, R.; Singh, S. P. Beilstein J. Org. Chem. 2011, 7, 179–197. doi:10.3762/bjoc.7.25 |
| 28. | Marinozzi, M.; Marcelli, G.; Carotti, A. Mini-Rev. Med. Chem. 2015, 15, 272–299. doi:10.2174/1389557515666150312154536 |
| 24. | Sun, L.; Bera, H.; Chui, W. K. Eur. J. Med. Chem. 2013, 65, 1–11. doi:10.1016/j.ejmech.2013.03.063 |
| 25. | Elmaati, T. M. A.; El-Taweel, F. M. J. Heterocycl. Chem. 2004, 41, 109–134. doi:10.1002/jhet.5570410201 |
| 34. | Abdel-Monem, Y. K.; El-Enein, S. A. A.; El-Sheikh-Amer, M. M. J. Mol. Struct. 2017, 1127, 386–396. doi:10.1016/j.molstruc.2016.07.110 |
| 35. | Straub, A.; Stasch, J.-P.; Alonso-Alija, C. Bioorg. Med. Chem. Lett. 2001, 11, 781–784. doi:10.1016/S0960-894X(01)00073-7 |
| 32. | Chiu, G.; Li, S.; Connolly, P. J.; Middleton, S. A.; Emanuel, S. L.; Huang, S.; Lin, R.; Lu, Y.; N. V. Janssen Pharmaceutica, Belgium. PCT Int. Appl. WO 2006130673, Dec 7, 2006. |
| 33. | Gudmundsson, K. S.; Johns, B. A.; Wang, Z.; Turner, E. M.; Allen, S. H.; Freeman, G. A.; Boyd, F. L., Jr.; Sexton, C. J.; Selleseth, D. W.; Moniri, K. R.; Creech, K. L. Bioorg. Med. Chem. 2005, 13, 5346–5361. doi:10.1016/j.bmc.2005.05.043 |
| 30. | Elnagdi, M. N.; Elmoghayar, M. R. H.; Elgemeie, G. E. H. Adv. Heterocycl. Chem. 1987, 41, 319–376. doi:10.1016/S0065-2725(08)60164-6 |
| 31. | Huang, S.; Lin, R.; Yu, Y.; Lu, Y.; Connolly, P. J.; Chiu, G.; Li, S.; Emanuel, S. L.; Middleton, S. A. Bioorg. Med. Chem. Lett. 2007, 17, 1243–1245. doi:10.1016/j.bmcl.2006.12.031 |
| 25. | Elmaati, T. M. A.; El-Taweel, F. M. J. Heterocycl. Chem. 2004, 41, 109–134. doi:10.1002/jhet.5570410201 |
© 2018 Aggarwal and Kumar; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)