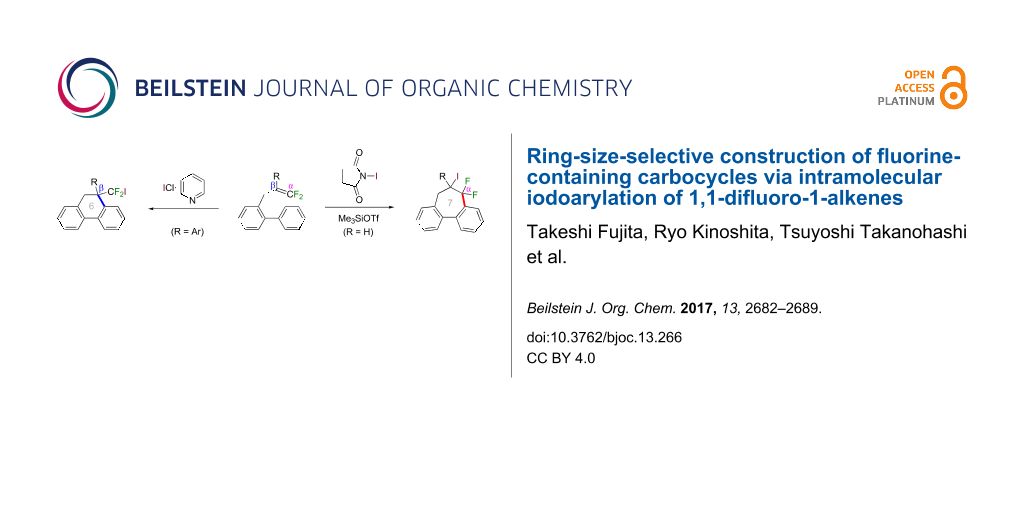
Abstract
1,1-Difluoro-1-alkenes bearing a biaryl-2-yl group effectively underwent site-selective intramolecular iodoarylation by the appropriate cationic iodine species. Iodoarylation of 2-(2-aryl-3,3-difluoroallyl)biaryls proceeded via regioselective carbon–carbon bond formation at the carbon atoms in β-position to the fluorine substituents, thereby constructing dibenzo-fused six-membered carbocycles bearing a difluoroiodomethyl group. In contrast, 2-(3,3-difluoroallyl)biaryls underwent a similar cyclization at the α-carbon atoms to afford ring-difluorinated seven-membered carbocycles.

Graphical Abstract
Introduction
As 1,1-difluoro-1-alkenes have an electron-deficient carbon–carbon double bond, they readily undergo intramolecular substitution of nucleophiles through an addition–elimination mechanism [1,2]. Thus, under basic conditions, they serve as useful precursors for ring-fluorinated heterocycles and carbocycles that are promising candidates for pharmaceuticals, agrochemicals, and functional materials. In contrast, the cationic cyclization of 1,1-difluoro-1-alkenes using electrophilic reagents (under acidic conditions) has been quite limited because of their low electron densities caused by fluorine substituents [3-5]. Despite the limitation, the cationic cyclization of difluoroalkenes possesses high potential for the synthesis of fluorine-containing cyclic compounds. Thus, the development of this type of cyclization is highly desirable to further expand the utility of difluoroalkenes in organic synthesis.
We have already achieved the metal-catalyzed and acid-mediated cationic cyclization of 1,1-difluoro-1-alkenes. In the former case, we reported the palladium [6-10], indium [10-13], and silver-catalyzed construction of ring-fluorinated carbocycles and heterocycles [14]. In the latter case, the domino-Friedel–Crafts-type cyclization proceeded via the cleavage of two carbon–fluorine bonds to afford polycyclic aromatic hydrocarbons [15-21]. Both types of cationic cyclization proceeded exclusively at the carbon atoms α to the fluorine substituents, because the β-selective metalation or protonation of difluoroalkenes generates α-fluorocarbocations, which were stabilized by the resonance effect of fluorine substituents.
In the course of our studies on the cationic cyclization of 1,1-difluoro-1-alkenes, we undertook an investigation of their iodine-mediated cyclization. Three-membered iodonium intermediates generated in the reaction course were expected to exhibit switchable regioselectivities [22]. This is because their cationic charge might be less localized on the carbon atoms α to the fluorine substituents, as compared to the aforementioned cationic intermediates [23-27]. Thus, we examined and eventually achieved complete control over the regioselectivity at the carbon atoms in β-position as well as those in α-position to the fluorine in the intramolecular Friedel–Crafts-type iodoarylation of 1,1-difluoro-1-alkenes bearing a biaryl group. Among the 1,1-difluoro-1-alkenes examined, 2-(2-aryl-3,3-difluoroallyl)biaryls underwent cyclization at the carbon atoms in β-position to the fluorine substituents to construct six-membered carbocycles bearing a difluoroiodomethyl group (Scheme 1a). On the other hand the cyclization of 2-(3,3-difluoroallyl)biaryls proceeded at the α-carbon atoms to give ring-fluorinated seven-membered carbocycles (Scheme 1b) [28].
![[1860-5397-13-266-i1]](/bjoc/content/inline/1860-5397-13-266-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Intramolecular site-selective iodoarylation of 1,1-difluoro-1-alkenes bearing a biaryl group.
Scheme 1: Intramolecular site-selective iodoarylation of 1,1-difluoro-1-alkenes bearing a biaryl group.
Results and Discussion
First, we sought an electrophilic iodine species suitable for the intramolecular iodoarylation of 2-(2-aryl-3,3-difluoroallyl)biaryls 1 using 2-(2-phenyl-3,3-difluoroallyl)biphenyl (1a) as a model substrate. To generate a highly reactive, cationic iodine species, several iodine sources were used with acid or metal activators (Table 1, entries 1–3). Upon treatment with N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) in a 1:1 mixed solvent of 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) and dichloromethane, 1a afforded the expected iodoarylation product 2a and its overreacted product, 9-benzoylphenanthrene (3a), in 34% and 15% yields, respectively (Table 1, entry 1). Ketone 3a was formed probably via the sequence consisting of iodide elimination from 2a, 1,2-migration of the phenyl group, and deprotonation, followed by hydrolysis of the resulting doubly activated benzylic difluoromethylene unit (Scheme 2). Neither a combination of bis(pyridine)iodonium (IPy2BF4) and trifluoromethanesulfonic acid nor a combination of I2 and silver(I) triflate improved the yield of 2a (Table 1, entries 2 and 3). Although iodine monochloride, which is known as a cationic iodine species, afforded only ketone 3a (Table 1, entry 4), its pyridine complex (PyICl) exclusively afforded the iodoarylation product 2a in 31% yield (Table 1, entry 5). The use of a 9:1 mixed solvent of HFIP and CH2Cl2 improved the yield of 2a to 85% (Table 1, entry 6). Lastly, increasing the concentration up to 0.075 M suppressed the formation of 3a and selectively afforded 2a in 91% yield (Table 1, entry 7). In this reaction, the nucleophilic benzene ring attacked the carbon atom β to the fluorine substituents of the cyclic iodonium intermediate, which was derived from 1a and PyICl. This indicates that the cationic charge in the cyclic iodonium intermediate might be localized at the β-carbon atom because of stabilization by the proximal phenyl group [28].
Table 1: Screening of conditions for the iodoarylation of 1a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-266-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | I+ (X equiv) | Y:Z | conditions | 2a (%)a | 3a (%)a |
|---|---|---|---|---|---|
| 1 | NIS (1.2), TMSOTf (1.2) | 1:1 | 0 °C, 40 min | 34 | 15 |
| 2 | IPy2BF4 (1.0), TfOH (2.0) | 1:1 | 0 °C, 1.5 h | N.D.b | N.D.b |
| 3 | I2 (1.2), AgOTf (1.2) | 1:1 | 0 °C, 1.5 h | 16 | 33 |
| 4 | ICl (2.0) | 1:1 | 0 °C, 20 min | N.D.b | 43 |
| 5 | PyICl (2.0) | 1:1 | 0 °C, 1 h | 31 | N.D.b |
| 6 | PyICl (2.0) | 9:1 | 0 °C, 1 h | 85 | 12 |
| 7c | PyICl (2.0) | 9:1 | 0 °C, 1 h | 91 | 1 |
aYield was determined by 19F NMR spectroscopy using PhCF3 as an internal standard. bN.D. = not detected. c0.075 M.
![[1860-5397-13-266-i2]](/bjoc/content/inline/1860-5397-13-266-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Under the optimal conditions obtained above, iodoarylation of several 2-(2-aryl-3,3-difluoroallyl)biaryls 1 was examined (Table 2). Difluoroiodomethylated dihydrophenanthrenes, 2a and 2b, bearing a phenyl and a biphenyl-4-yl group were obtained in 79% and 74% isolated yields, respectively. 2-(2-Aryl-3,3-difluoroallyl)biaryls 1c–e bearing electron-donating substituents (4-Me, 3-Me, and 4-MeO) on the benzene ring attached to the vinylic position successfully underwent iodoarylation to afford the corresponding dihydrophenanthrenes 2c–e. In contrast, electron-withdrawing substituents on similar positions hardly promoted the iodoarylation, which was presumably because of inefficient cyclic iodonium formation. Substrates 1f and 1g bearing electron-donating groups on the nucleophilic aryl groups also participated in the iodoarylation to afford the corresponding difluoroiodomethylated dihydrophenanthrenes 2f and 2g in 54% and 80% yields, respectively. However, with substrates bearing a strong electron-withdrawing group (e.g., CF3) on the nucleophilic benzene ring, the iodoarylation hardly proceeded. The unambiguous structural determination of the iodoarylation products 2 was accomplished by X-ray crystallographic analysis of 2a (Figure 1), which revealed that the iodoarylation products 2 have six-membered carbocycles bearing a difluoroiodomethyl group.
Table 2: Construction of six-membered carbocycles via iodoarylation of 1.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-266-i7.svg?max-width=637&scale=1.0)
|
||||
| entry | 1 | 2 | time | yield (%)a |
|---|---|---|---|---|
| 1b |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-266-i8.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-266-i9.svg?max-width=637&scale=1.0)
2a |
1 h | 79 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-266-i10.svg?max-width=637&scale=1.0)
1b |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-266-i11.svg?max-width=637&scale=1.0)
2b |
2 h | 74 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-266-i12.svg?max-width=637&scale=1.0)
1c |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-266-i13.svg?max-width=637&scale=1.0)
2c |
35 min | 82 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-266-i14.svg?max-width=637&scale=1.0)
1d |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-266-i15.svg?max-width=637&scale=1.0)
2d |
1 h | 53 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-266-i16.svg?max-width=637&scale=1.0)
1e |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-266-i17.svg?max-width=637&scale=1.0)
2e |
25 min | 83 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-266-i18.svg?max-width=637&scale=1.0)
1f |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-266-i19.svg?max-width=637&scale=1.0)
2f |
1.5 h | 54 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-266-i20.svg?max-width=637&scale=1.0)
1g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-266-i21.svg?max-width=637&scale=1.0)
2g |
1 h | 80 |
aIsolated yield. bHFIP/CH2Cl2 9:1.
![[1860-5397-13-266-1]](/bjoc/content/figures/1860-5397-13-266-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: ORTEP diagram of 2a with 50% ellipsoid probability.
Figure 1: ORTEP diagram of 2a with 50% ellipsoid probability.
Further transformation of the difluoroiodomethyl group of 2a was achieved by heating (Scheme 3). Thus, refluxing a DMF solution of 2a for 15 h induced iodine–hydrogen exchange to afford difluoromethylated dihydrophenanthrene derivative 4a in almost quantitative yield [29,30]. A difluoromethyl group functions as a hydrogen-bond donor and a bioisostere of a hydroxy group, as a result of which difluoromethyl-bearing compounds attract much attention as bioactive materials [31,32]. This sequence provides ready access to these compounds.
![[1860-5397-13-266-i3]](/bjoc/content/inline/1860-5397-13-266-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Transformation of a CF2I group of 2a into a CHF2 group.
Scheme 3: Transformation of a CF2I group of 2a into a CHF2 group.
Next, 2-(3,3-difluoroallyl)biphenyl (5a), without an aryl group at its vinylic position, was subjected to the conditions examined in entries 1–3 of Table 1. In contrast to 1a, iodoarylation of 5a proceeded through C–C-bond formation exclusively at the carbon atoms α to the fluorine substituents to afford dibenzo-fused cycloheptane 6a with geminal fluorine substituents on the ring in 92%, 38%, 49% yields, respectively. The thus-obtained selectivity might be attributed to the localization of the cationic charge at the carbon atoms α to the fluorine substituents in the three-membered iodonium intermediates. Since the combination of NIS and TMSOTf was found to be the best for an iodoarylation of 5, the reactions of a couple of 2-(3,3-difluoroallyl)biaryls 5 were examined under the same conditions (Scheme 4). The iodoarylation of difluoroallylbiphenyl 5b, bearing an electron-donating methyl group on the nucleophilic aryl group, was completed in 15 min to afford 6b in 60% isolated yield. Brominated difluoroallylbiphenyl 5c successfully underwent the same cyclization to afford the corresponding product 6c in 62% isolated yield. The structural characterization of 6 was achieved by X-ray analysis using a single crystal of 6a (Figure 2), and it was found that the iodoarylation products 6 have a seven-membered carbocycle bearing adjacent difluoromethylene and iodomethylene units.
![[1860-5397-13-266-i4]](/bjoc/content/inline/1860-5397-13-266-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Construction of seven-membered carbocycles via iodoarylation of 5.
Scheme 4: Construction of seven-membered carbocycles via iodoarylation of 5.
![[1860-5397-13-266-2]](/bjoc/content/figures/1860-5397-13-266-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram of 6a with 50% ellipsoid probability.
Figure 2: ORTEP diagram of 6a with 50% ellipsoid probability.
In addition, a selective HI elimination from 6a could be achieved by the choice of base, leading to the construction of a [7]annulene system (Scheme 5). The use of lithium bases, such as lithium diisopropylamide and lithium hexamethyldisilazide, induced HF eliminations as well as substantial HI elimination. However, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) exclusively promoted HI elimination to afford ring-difluorinated dibenzo[a,c][7]annulene 7a in an almost quantitative yield. Since dibenzo[a,c][7]annulenes serve as bioactive agents, this method would be of value in the research directed toward pharmaceutical and materials chemistry [33-35].
![[1860-5397-13-266-i5]](/bjoc/content/inline/1860-5397-13-266-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Selective HI elimination from 6a.
Scheme 5: Selective HI elimination from 6a.
Conclusion
In summary, we demonstrated selective constructions of six and seven-membered carbocyclic rings through the intramolecular iodoarylation of 3,3-difluoroallylic biaryls. The size selectivity in the cyclization was drastically controlled by the presence or absence of an aryl group in the 2-position of the 3,3-difluoroallylic moiety, which might perturb cationic charge distribution in the corresponding cyclic iodonium intermediates. The aryl group in the 2-position (at the carbon atom in β-position to the fluorine substituents) promoted a six-membered-ring closure, most likely because of the localization of cationic charge stabilized by the aromatic ring. In contrast, seven-membered carbocycles were constructed probably as a result of the cationic charge localized at the 3-position of difluoroallylic moiety (at the α-carbon atom of the fluorine substituents) due to the α-cation-stabilizing effect of fluorine.
Experimental
General: 1H NMR, 13C NMR, and 19F NMR spectra were recorded on a Bruker Avance 500 or a JEOL ECS-400 spectrometer. Chemical shift values are given in ppm relative to internal Me4Si (for 1H NMR: δ = 0.00 ppm), CDCl3 (for 13C NMR: δ = 77.0 ppm), C6F6 (for 19F NMR: δ = 0.0 ppm), and (4-MeC6H4)2C(CF3)2 (for 19F NMR: δ = 97.9 ppm). IR spectra were recorded on a Horiba FT-300S spectrometer using the attenuated total reflectance (ATR) method. Mass spectra were measured on a JEOL JMS-T100GCV spectrometer. X-ray diffraction studies were performed on a Bruker APEXII ULTRA instrument equipped with a CCD diffractometer using Mo Kα (graphite monochromated, λ = 0.71069 Å) radiation. The CCDC deposition numbers of compounds 2a and 6a are 1556804 and 1556803, respectively. All the reactions were conducted under argon or nitrogen atmosphere.
Materials: Column chromatography and preparative thin-layer chromatography (PTLC) were conducted on silica gel (Silica Gel 60 N, Kanto Chemical Co., Inc. for column chromatography and Wakogel B-5F, Wako Pure Chemical Industries, Ltd. for PTLC). Tetrahydrofuran (THF), dichloromethane, and N,N-dimethylformamide (DMF) were purified by a solvent-purification system (GlassContour) equipped with columns of activated alumina and supported-copper catalyst (Q-5) before use. 1,1,1,3,3,3-Hexafluoropropan-2-ol (HFIP) was distilled from CaH2 and stored over activated 4 Å molecular sieves. Unless otherwise noted, materials were obtained from commercial sources and used directly without further purifications.
Typical procedure for the iodoarylation of 2-(2-aryl-3,3-difluoroallyl)biaryls 1: To a HFIP (1.20 mL) and dichloromethane (0.13 mL) solution of 2-(2-phenyl-3,3-difluoroallyl)biphenyl (1a, 31 mg, 0.10 mmol) was added pyridine iodine monochloride (PyICl, 49 mg, 0.20 mmol) at 0 °C. After stirring at the same temperature for 1 h, the reaction was quenched with an aqueous NaHCO3 solution. The organic materials were extracted with CHCl3 three times. The combined extracts were washed with an aqueous Na2S2O3 solution and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by PTLC (hexane/ethyl acetate 10:1) to give 9-(difluoroiodomethyl)-9-phenyl-9,10-dihydrophenanthrene (2a, 34 mg, 79%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 3.68 (d, J = 15.8 Hz, 1H), 3.71 (d, J = 15.8 Hz, 1H), 7.07–7.08 (m, 3H), 7.15–7.24 (m, 5H), 7.42–7.49 (m, 2H), 7.52–7.54 (m, 1H), 7.79 (d, J = 7.5 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 38.7, 59.5 (t, JCF = 17 Hz), 110.6 (t, JCF = 316 Hz), 123.6, 125.1, 127.2, 127.4, 127.50, 127.52, 128.0, 128.46, 128.50, 128.6 (t, JCF = 4 Hz), 130.1, 132.7, 133.64, 133.64, 134.6, 136.8; 19F NMR (470 MHz, CDCl3) δ 124.7 (br s); IR (neat): 3068, 1489, 1454, 1126, 1147, 1097, 964, 850, 742, 696, 592 cm−1; HRMS–EI (m/z): [M]+ calcd for C21H15F2I, 432.0186; found: 432.0166.
Typical procedure for the iodoarylation of 2-(3,3-difluoroallyl)biaryls 5: To a HFIP (2.5 mL) and dichloromethane (1.5 mL) solution of N-iodosuccinimide (NIS, 27 mg, 0.12 mmol) was added trimethylsilyl trifluoromethanesulfonate (22 μL, 0.12 mmol) at 0 °C. After stirring at the same temperature for 10 min, a dichloromethane (1.0 mL) solution of 2-(3,3-difluoroallyl)biphenyl (5a, 23 mg, 0.10 mmol) was added to the reaction mixture. After stirring at 0 °C for 40 min, the reaction was quenched with an aqueous NaHCO3 solution. The organic materials were extracted with dichloromethane three times. The combined extracts were washed with an aqueous Na2S2O3 solution and brine, and dried over anhydrous Na2SO4. After removal of the solvent under reduced pressure, the residue was purified by PTLC (hexane/ethyl acetate 10:1) to give 5,5-difluoro-6-iodo-6,7-dihydro-5H-dibenzo[a,c][7]annulene (6a, 33 mg, 92%) as a colorless liquid. 1H NMR (500 MHz, CDCl3) δ 3.06 (dd, J = 14.8, 4.9, 1H), 3.38 (dd, J = 14.8, 6.0 Hz, 1H), 4.91–4.98 (m, 1H), 7.28–7.35 (m, 2H), 7.41–7.44 (m, 3H), 7.47 (d, J = 7.8 Hz, 1H), 7.55–7.59 (m, 1H), 7.70 (d, J = 7.4 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 35.1 (dd, JCF = 27 Hz), 41.7, 118.9 (dd, JCF = 247 Hz), 125.2, 127.5, 128.0, 128.20, 128.23, 129.2, 129.7, 131.0, 131.4 (dd, JCF = 24 Hz), 134.6, 138.6 (dd, JCF = 5 Hz), 140.3; 19F NMR (470 MHz, DMSO-d6, 120 °C) δ 72.3 (d, JFF = 236 Hz, 1F), 86.5 (d, JFF = 236 Hz, 1F); IR (neat): 3068, 3030, 1450, 1149, 1055, 989, 752, 598 cm–1; HRMS–EI (m/z): [M]+ calcd for C15H11F2I, 355.9873; found: 355.9866.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and spectral data. | ||
| Format: PDF | Size: 5.3 MB | Download |
Acknowledgements
This work was financially supported by JSPS KAKENHI Grant Number JP16H04105 in Grant-in-Aid for Scientific Research (B) (J.I.), JSPS KAKENHI Grant Number JP16H01002 in Precisely Designed Catalysts with Customized Scaffolding (J.I.), and JSPS KAKENHI Grant Number JP16K20939 in Grant-in-Aid for Young Scientists (B) (T.F.). We acknowledge the generous gifts of 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP, Central Glass Co., Ltd.), 2-bromo-3,3,3-trifluoroprop-1-ene, and dibromodifluoromethane (Tosoh Finechem Co.).
References
-
Ichikawa, J. Chim. Oggi 2007, 25, 54–57.
Return to citation in text: [1] -
Zhang, X.; Cao, S. Tetrahedron Lett. 2017, 58, 375–392. doi:10.1016/j.tetlet.2016.12.054
Return to citation in text: [1] -
Suda, M. Tetrahedron Lett. 1980, 21, 2555–2556. doi:10.1016/0040-4039(80)80127-4
Return to citation in text: [1] -
Morikawa, T.; Kumadaki, I.; Shiro, M. Chem. Pharm. Bull. 1985, 33, 5144–5146. doi:10.1248/cpb.33.5144
Return to citation in text: [1] -
Kendrick, D. A.; Kolb, M. J. Fluorine Chem. 1989, 45, 273–276. doi:10.1016/S0022-1139(00)84152-4
Return to citation in text: [1] -
Yokota, M.; Fujita, D.; Ichikawa, J. Org. Lett. 2007, 9, 4639–4642. doi:10.1021/ol702279w
Return to citation in text: [1] -
Tanabe, H.; Ichikawa, J. Chem. Lett. 2010, 39, 248–249. doi:10.1246/cl.2010.248
Return to citation in text: [1] -
Fuchibe, K.; Morikawa, T.; Shigeno, K.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 1126–1129. doi:10.1021/ol503759d
Return to citation in text: [1] -
Fuchibe, K.; Morikawa, T.; Ueda, R.; Okauchi, T.; Ichikawa, J. J. Fluorine Chem. 2015, 179, 106–115. doi:10.1016/j.jfluchem.2015.06.008
Return to citation in text: [1] -
Fuchibe, K.; Shigeno, K.; Zhao, N.; Aihara, H.; Akisaka, R.; Morikawa, T.; Fujita, T.; Yamakawa, K.; Shimada, T.; Ichikawa, J. J. Fluorine Chem. 2017, 203, 173–184. doi:10.1016/j.jfluchem.2017.09.002
Return to citation in text: [1] [2] -
Fuchibe, K.; Mayumi, Y.; Zhao, N.; Watanabe, S.; Yokota, M.; Ichikawa, J. Angew. Chem., Int. Ed. 2013, 52, 7825–7828. doi:10.1002/anie.201302740
Return to citation in text: [1] -
Fuchibe, K.; Mayumi, Y.; Yokota, M.; Aihara, H.; Ichikawa, J. Bull. Chem. Soc. Jpn. 2014, 87, 942–949. doi:10.1246/bcsj.20140128
Return to citation in text: [1] -
Fuchibe, K.; Imaoka, H.; Ichikawa, J. Chem. – Asian J. 2017, 12, 2359–2363. doi:10.1002/asia.201700870
Return to citation in text: [1] -
Fujita, T.; Watabe, Y.; Yamashita, S.; Tanabe, H.; Nojima, T.; Ichikawa, J. Chem. Lett. 2016, 45, 964–966. doi:10.1246/cl.160427
Return to citation in text: [1] -
Ichikawa, J. Pure Appl. Chem. 2000, 72, 1685–1689. doi:10.1351/pac200072091685
Return to citation in text: [1] -
Ichikawa, J.; Jyono, H.; Kudo, T.; Fujiwara, M.; Yokota, M. Synthesis 2005, 39–46. doi:10.1055/s-2004-834910
Return to citation in text: [1] -
Ichikawa, J.; Kaneko, M.; Yokota, M.; Itonaga, M.; Yokoyama, T. Org. Lett. 2006, 8, 3167–3170. doi:10.1021/ol060912r
Return to citation in text: [1] -
Ichikawa, J.; Yokota, M.; Kudo, T.; Umezaki, S. Angew. Chem., Int. Ed. 2008, 47, 4870–4873. doi:10.1002/anie.200801396
Return to citation in text: [1] -
Isobe, H.; Hitosugi, S.; Matsuno, T.; Iwamoto, T.; Ichikawa, J. Org. Lett. 2009, 11, 4026–4028. doi:10.1021/ol901693y
Return to citation in text: [1] -
Fuchibe, K.; Jyono, H.; Fujiwara, M.; Kudo, T.; Yokota, M.; Ichikawa, J. Chem. – Eur. J. 2011, 17, 12175–12185. doi:10.1002/chem.201100618
Return to citation in text: [1] -
Suzuki, N.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 4984–4987. doi:10.1021/acs.orglett.5b02426
Return to citation in text: [1] -
Saito, A.; Okada, M.; Nakamura, Y.; Kitagawa, O.; Horikawa, H.; Taguchi, T. J. Fluorine Chem. 2003, 123, 75–80. doi:10.1016/S0022-1139(03)00138-6
Return to citation in text: [1] -
Freeman, F. Chem. Rev. 1975, 75, 439–490. doi:10.1021/cr60296a003
Return to citation in text: [1] -
Rodriguez, J.; Dulcère, J.-P. Synthesis 1993, 1177–1205. doi:10.1055/s-1993-26022
Return to citation in text: [1] -
French, A. N.; Bissmire, S.; Wirth, T. Chem. Soc. Rev. 2004, 33, 354–362. doi:10.1039/b310389g
Return to citation in text: [1] -
Denmark, S. E.; Kuester, W. E.; Burk, M. T. Angew. Chem., Int. Ed. 2012, 51, 10938–10953. doi:10.1002/anie.201204347
Return to citation in text: [1] -
Nolsøe, J. M. J.; Hansen, T. V. Eur. J. Org. Chem. 2014, 3051–3065. doi:10.1002/ejoc.201301400
Return to citation in text: [1] -
See reference [16] for an α-cation-stabilizing effect of fluorine exceeding that of an aryl group.
Return to citation in text: [1] [2] -
Zhang, L.; Cradlebaugh, J.; Litwinienko, G.; Smart, B. E.; Ingold, K. U.; Dolbier, W. R., Jr. Org. Biomol. Chem. 2004, 2, 689–694. doi:10.1039/b313757k
Return to citation in text: [1] -
Zhang, C.-P.; Chen, Q.-Y.; Xiao, J.-C.; Gu, Y.-C. J. Fluorine Chem. 2009, 130, 671–673. doi:10.1016/j.jfluchem.2009.05.005
Return to citation in text: [1] -
Hu, J. J. Fluorine Chem. 2009, 130, 1130–1139. doi:10.1016/j.jfluchem.2009.05.016
Return to citation in text: [1] -
Ichikawa, J. J. Synth. Org. Chem., Jpn. 2010, 68, 1175–1184. doi:10.5059/yukigoseikyokaishi.68.1175
Return to citation in text: [1] -
Bergemann, S.; Brecht, R.; Büttner, F.; Guénard, D.; Gust, R.; Seitz, G.; Stubbs, M. T.; Thoret, S. Bioorg. Med. Chem. 2003, 11, 1269–1281. doi:10.1016/S0968-0896(02)00639-9
Return to citation in text: [1] -
Büttner, F.; Bergemann, S.; Guénard, D.; Gust, R.; Seitz, G.; Thoret, S. Bioorg. Med. Chem. 2005, 13, 3497–3511. doi:10.1016/j.bmc.2005.02.059
Return to citation in text: [1] -
Lin, H.-C.; Lee, S.-S. J. Nat. Prod. 2012, 75, 1735–1743. doi:10.1021/np300402k
Return to citation in text: [1]
| 1. | Ichikawa, J. Chim. Oggi 2007, 25, 54–57. |
| 2. | Zhang, X.; Cao, S. Tetrahedron Lett. 2017, 58, 375–392. doi:10.1016/j.tetlet.2016.12.054 |
| 14. | Fujita, T.; Watabe, Y.; Yamashita, S.; Tanabe, H.; Nojima, T.; Ichikawa, J. Chem. Lett. 2016, 45, 964–966. doi:10.1246/cl.160427 |
| 10. | Fuchibe, K.; Shigeno, K.; Zhao, N.; Aihara, H.; Akisaka, R.; Morikawa, T.; Fujita, T.; Yamakawa, K.; Shimada, T.; Ichikawa, J. J. Fluorine Chem. 2017, 203, 173–184. doi:10.1016/j.jfluchem.2017.09.002 |
| 11. | Fuchibe, K.; Mayumi, Y.; Zhao, N.; Watanabe, S.; Yokota, M.; Ichikawa, J. Angew. Chem., Int. Ed. 2013, 52, 7825–7828. doi:10.1002/anie.201302740 |
| 12. | Fuchibe, K.; Mayumi, Y.; Yokota, M.; Aihara, H.; Ichikawa, J. Bull. Chem. Soc. Jpn. 2014, 87, 942–949. doi:10.1246/bcsj.20140128 |
| 13. | Fuchibe, K.; Imaoka, H.; Ichikawa, J. Chem. – Asian J. 2017, 12, 2359–2363. doi:10.1002/asia.201700870 |
| 6. | Yokota, M.; Fujita, D.; Ichikawa, J. Org. Lett. 2007, 9, 4639–4642. doi:10.1021/ol702279w |
| 7. | Tanabe, H.; Ichikawa, J. Chem. Lett. 2010, 39, 248–249. doi:10.1246/cl.2010.248 |
| 8. | Fuchibe, K.; Morikawa, T.; Shigeno, K.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 1126–1129. doi:10.1021/ol503759d |
| 9. | Fuchibe, K.; Morikawa, T.; Ueda, R.; Okauchi, T.; Ichikawa, J. J. Fluorine Chem. 2015, 179, 106–115. doi:10.1016/j.jfluchem.2015.06.008 |
| 10. | Fuchibe, K.; Shigeno, K.; Zhao, N.; Aihara, H.; Akisaka, R.; Morikawa, T.; Fujita, T.; Yamakawa, K.; Shimada, T.; Ichikawa, J. J. Fluorine Chem. 2017, 203, 173–184. doi:10.1016/j.jfluchem.2017.09.002 |
| 33. | Bergemann, S.; Brecht, R.; Büttner, F.; Guénard, D.; Gust, R.; Seitz, G.; Stubbs, M. T.; Thoret, S. Bioorg. Med. Chem. 2003, 11, 1269–1281. doi:10.1016/S0968-0896(02)00639-9 |
| 34. | Büttner, F.; Bergemann, S.; Guénard, D.; Gust, R.; Seitz, G.; Thoret, S. Bioorg. Med. Chem. 2005, 13, 3497–3511. doi:10.1016/j.bmc.2005.02.059 |
| 35. | Lin, H.-C.; Lee, S.-S. J. Nat. Prod. 2012, 75, 1735–1743. doi:10.1021/np300402k |
| 3. | Suda, M. Tetrahedron Lett. 1980, 21, 2555–2556. doi:10.1016/0040-4039(80)80127-4 |
| 4. | Morikawa, T.; Kumadaki, I.; Shiro, M. Chem. Pharm. Bull. 1985, 33, 5144–5146. doi:10.1248/cpb.33.5144 |
| 5. | Kendrick, D. A.; Kolb, M. J. Fluorine Chem. 1989, 45, 273–276. doi:10.1016/S0022-1139(00)84152-4 |
| 16. | Ichikawa, J.; Jyono, H.; Kudo, T.; Fujiwara, M.; Yokota, M. Synthesis 2005, 39–46. doi:10.1055/s-2004-834910 |
| 28. | See reference [16] for an α-cation-stabilizing effect of fluorine exceeding that of an aryl group. |
| 29. | Zhang, L.; Cradlebaugh, J.; Litwinienko, G.; Smart, B. E.; Ingold, K. U.; Dolbier, W. R., Jr. Org. Biomol. Chem. 2004, 2, 689–694. doi:10.1039/b313757k |
| 30. | Zhang, C.-P.; Chen, Q.-Y.; Xiao, J.-C.; Gu, Y.-C. J. Fluorine Chem. 2009, 130, 671–673. doi:10.1016/j.jfluchem.2009.05.005 |
| 23. | Freeman, F. Chem. Rev. 1975, 75, 439–490. doi:10.1021/cr60296a003 |
| 24. | Rodriguez, J.; Dulcère, J.-P. Synthesis 1993, 1177–1205. doi:10.1055/s-1993-26022 |
| 25. | French, A. N.; Bissmire, S.; Wirth, T. Chem. Soc. Rev. 2004, 33, 354–362. doi:10.1039/b310389g |
| 26. | Denmark, S. E.; Kuester, W. E.; Burk, M. T. Angew. Chem., Int. Ed. 2012, 51, 10938–10953. doi:10.1002/anie.201204347 |
| 27. | Nolsøe, J. M. J.; Hansen, T. V. Eur. J. Org. Chem. 2014, 3051–3065. doi:10.1002/ejoc.201301400 |
| 31. | Hu, J. J. Fluorine Chem. 2009, 130, 1130–1139. doi:10.1016/j.jfluchem.2009.05.016 |
| 32. | Ichikawa, J. J. Synth. Org. Chem., Jpn. 2010, 68, 1175–1184. doi:10.5059/yukigoseikyokaishi.68.1175 |
| 22. | Saito, A.; Okada, M.; Nakamura, Y.; Kitagawa, O.; Horikawa, H.; Taguchi, T. J. Fluorine Chem. 2003, 123, 75–80. doi:10.1016/S0022-1139(03)00138-6 |
| 15. | Ichikawa, J. Pure Appl. Chem. 2000, 72, 1685–1689. doi:10.1351/pac200072091685 |
| 16. | Ichikawa, J.; Jyono, H.; Kudo, T.; Fujiwara, M.; Yokota, M. Synthesis 2005, 39–46. doi:10.1055/s-2004-834910 |
| 17. | Ichikawa, J.; Kaneko, M.; Yokota, M.; Itonaga, M.; Yokoyama, T. Org. Lett. 2006, 8, 3167–3170. doi:10.1021/ol060912r |
| 18. | Ichikawa, J.; Yokota, M.; Kudo, T.; Umezaki, S. Angew. Chem., Int. Ed. 2008, 47, 4870–4873. doi:10.1002/anie.200801396 |
| 19. | Isobe, H.; Hitosugi, S.; Matsuno, T.; Iwamoto, T.; Ichikawa, J. Org. Lett. 2009, 11, 4026–4028. doi:10.1021/ol901693y |
| 20. | Fuchibe, K.; Jyono, H.; Fujiwara, M.; Kudo, T.; Yokota, M.; Ichikawa, J. Chem. – Eur. J. 2011, 17, 12175–12185. doi:10.1002/chem.201100618 |
| 21. | Suzuki, N.; Fujita, T.; Ichikawa, J. Org. Lett. 2015, 17, 4984–4987. doi:10.1021/acs.orglett.5b02426 |
| 28. | See reference [16] for an α-cation-stabilizing effect of fluorine exceeding that of an aryl group. |
© 2017 Fujita et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)