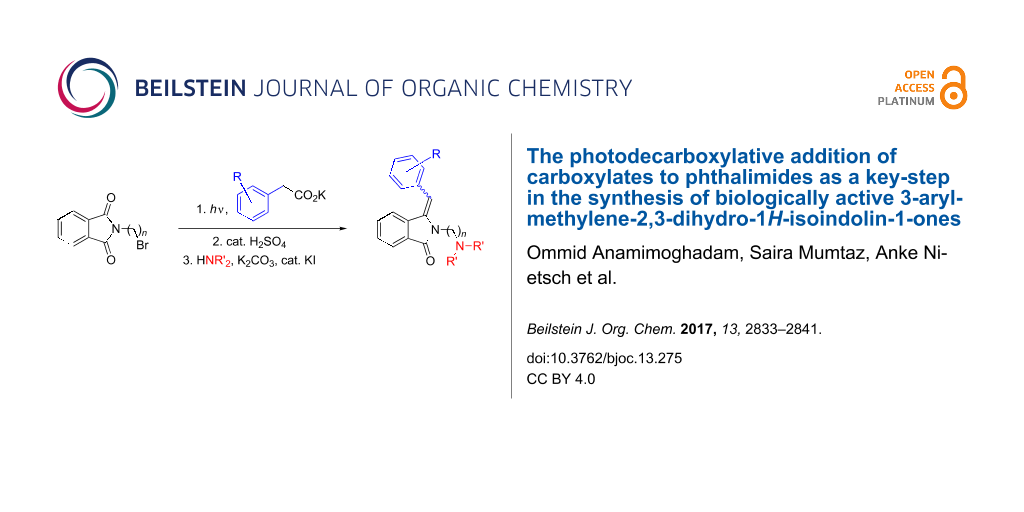
Abstract
The synthesis of various 3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones was realized following a simple three-step process. The protocol utilized the photodecarboxylative addition of readily available carboxylates to N-(bromoalkyl)phthalimides as a versatile and efficient key step. The initially obtained hydroxyphthalimidines were readily converted to the desired N-diaminoalkylated 3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones via acid-catalyzed dehydration and subsequent nucleophilic substitution with the corresponding secondary amines. The procedure was successfully applied to the synthesis of known local anesthetics (AL-12, AL-12B and AL-5) in their neutral forms.

Graphical Abstract
Introduction
Phthalimides and their related 3-alkyl- and 3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones play an important role in medicinal chemistry due to their biological activities for a wide range of therapeutic applications [1-7]. AL-12, AL-12B and AL-5 (Figure 1), for example, were described as highly active local anesthetics distinctly exceeding the efficiencies of common local anesthetics such as procaine, xylocaine/lidocaine and tetracaine [8]. Their molecular structures contain the three key elements of all local anesthetics: (a) a lipophilic aromatic ring, (b) an amide (or ester) linker, and (c) a terminal tertiary amine [9]. The original synthesis of these bioactive compounds involved a Perkin condensation followed by an amination reaction. An alternative pathway to AL-12B has been described by Couture and co-workers and incorporated an intramolecular Horner–Wadsworth–Emmons reaction as a crucial step [10].
![[1860-5397-13-275-1]](/bjoc/content/figures/1860-5397-13-275-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structures of AL-12, AL-12B and AL-5.
Figure 1: Molecular structures of AL-12, AL-12B and AL-5.
Due to their diverse biological activities, a variety of synthetic pathways to 3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones has been developed over the past two decades [10-22]. The photodecarboxylative addition of phenylacetates to phthalimides represents a mild alternative entrance to these target molecules [23-31]. The reaction utilizes phenylacetate salts as readily available alkylation agents [32,33]. Selected transformations have been furthermore realized on large multigram scales [25,34,35] and in continuous-flow mode [36-40]. The photodecarboxylation procedure was subsequently applied to the synthesis of 2-dialkylaminoalkyl-3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones.
The retrosynthetic analysis is depicted in Scheme 1. The initial step comprises the photodecarboxylative addition of phenylacetate to commercially available N-(bromoalkyl)phthalimides, yielding the corresponding benzylated hydroxyphthalimidine derivatives as key intermediates. Subsequent acid-catalyzed dehydration [41], followed by amination [42] furnishes the desired target compounds. The amino group is introduced in the final step as it would otherwise interfere with the desired photoreaction. In fact, amines are very potent electron donors and are easily oxidized by the excited phthalimide chromophore [43-46].
![[1860-5397-13-275-i1]](/bjoc/content/inline/1860-5397-13-275-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of AL-12, AL-12B and AL-5 (in their neutral forms) and their derivatives.
Scheme 1: Retrosynthetic analysis of AL-12, AL-12B and AL-5 (in their neutral forms) and their derivatives.
Results and Discussion
Initial irradiation experiments were conducted with N-(2-bromoethyl)phthalimide (1a) and potassium phenylacetate (2a) as a model system (Scheme 2 and Table 1). The salt 2a, generated from the corresponding phenylacetic acid and potassium carbonate, was used in excess amounts to suppress competing ‘simple’ decarboxylation (-CO2− ↔ -H exchange) reactions to the corresponding toluene derivatives [25].
![[1860-5397-13-275-i2]](/bjoc/content/inline/1860-5397-13-275-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Optimization study using phenylacetate 2a.
Scheme 2: Optimization study using phenylacetate 2a.
Table 1: Impact of the reaction medium on the photodecarboxylative addition.
| Entry | Co-solvent | T [°C] | Time [h] | Yield [%] |
| 1a | water | 30 | 4 | 45b (3a) |
| 2a | water | 20 | 4 | 49b (3a) |
| 3 | pH 7 buffer | 20 | 3 | 87 (3a) |
aCompound 4 formed as byproduct (25–30%). bIsolated yield after column chromatography.
Following the established protocol and utilizing a 1:1 acetone/water mixture as reaction medium [27,30], irradiations with UVB light (300 ± 30 nm) for 4 hours under nitrogen purging in Pyrex flasks (cutoff: <300 nm [47]) furnished the desired addition product 3a in yields of 45 and 49% (Table 1, entries 1 and 2). The pH raised from approx. 7.5 at the beginning to about 11.5 at the end of each irradiation experiment. In both cases, the tetracyclic oxazolidine compound 4, originating from intramolecular nucleophilic substitution, was obtained as a byproduct in 25–30%. Noteworthy, compound 4 is formed as the only product when 1a is treated with organometallic reagents [48-50]. When the photoreaction was repeated in a 1:1 mixture of acetone and pH 7 buffer at 20 °C [28], the formation of 4 was prevented and the desired benzylated hydroxyphthalimidine 3a precipitated during irradiation for 3 hours. Subsequently, 3a was obtained in an excellent yield of 87% by simple filtration and washing (Table 1, entry 3). The pH increased from about 7.8 at the start to ca. 8.7 at the end of the irradiation. Temperature control was crucial as thermal conversion of 3a into 4 was found to occur above 40 °C. Compound 1a could be converted quantitatively to the oxazolidine derivative 4 by treatment with either sodium carbonate or potassium tert-butoxide in acetonitrile.
Mixtures of N-(bromoalkyl)phthalimides 1 and phenylacetates 2 in acetone/pH 7 buffer were subsequently irradiated with UVB light for 2–4 hours (Scheme 3 and Table 2). With the N-(2-bromoethyl)- and N-(3-bromopropyl)phthalimides 1a and 1b, the desired benzylated products 3a–q were obtained as colorless crystalline solids in good to excellent yields of 63–95% (Table 2, entries 1–17). In many cases, the photoproducts 3 simply precipitated during irradiation or after removal of the co-solvent acetone and could be isolated by filtration. In all other cases, the desired products 3 were obtained after extraction and subsequent column chromatography. All compounds 3 showed a characteristic pair of doublets between 3 and 4 ppm with a large geminal 2J coupling of 12–16 Hz for the benzylic methylene group (-CH2Ar) in their 1H NMR spectra and a singlet at 90 ± 3 ppm for the newly formed tertiary alcohol (C–OH) in their 13C NMR spectra, respectively. Small amounts (<10%) of non-volatile toluene derivatives were occasionally detected in the crude products by 1H NMR spectroscopic analysis but no attempts were made to isolate these compounds. The reaction was additionally studied with natural sunlight [51,52]. Solutions of 1a and 2a were exposed to direct sunlight in a solar float developed by Liu and co-workers [53,54]. After 6 hours of illumination, the reaction had reached a conversion of 47% and 3a was subsequently isolated by column chromatography in 26% yield (Table 2, entry 18).
![[1860-5397-13-275-i3]](/bjoc/content/inline/1860-5397-13-275-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photodecarboxylative additions to N-(bromoalkyl)phthalimides.
Scheme 3: Photodecarboxylative additions to N-(bromoalkyl)phthalimides.
Table 2: Experimental results for photodecarboxylative additions.
| Entry | n | R | Time [h] | Yield 3 [%] |
| 1 | 2 (1a) | H (2a) | 3 | 87 (3a) |
| 2 | 2 (1a) | 4-F (2b) | 3 | 83 (3b) |
| 3 | 2 (1a) | 4-Cl (2c) | 3 | 79 (3c) |
| 4 | 2 (1a) | 4-Br (2d) | 3 | 63 (3d) |
| 5 | 2 (1a) | 4-MeO (2e) | 3 | 85 (3e) |
| 6 | 2 (1a) | 4-Me (2f) | 3 | 88 (3f) |
| 7 | 2 (1a) | 3-Me (2g) | 3 | 66 (3g) |
| 8 | 2 (1a) | 2-Me (2h) | 3 | 71 (3h) |
| 9 | 2 (1a) | 4-AcO (2i) | 2 | 77 (3i) |
| 10 | 3 (1b) | H (2a) | 3 | 95 (3j) |
| 11 | 3 (1b) | 4-F (2b) | 3 | 75 (3k) |
| 12 | 3 (1b) | 4-Cl (2c) | 3 | 80 (3l) |
| 13 | 3 (1b) | 4-Br (2d) | 3 | 73 (3m) |
| 14 | 3 (1b) | 4-MeO (2e) | 3 | 76 (3n) |
| 15 | 3 (1b) | 4-Me (2f) | 3 | 89 (3o) |
| 16 | 3 (1b) | 3-Me (2g) | 3 | 91 (3p) |
| 17 | 3 (1b) | 2-Me (2h) | 3 | 73 (3q) |
| 18a | 2 (1a) | H (2a) | 6 | 26 (3a)b |
| 19 | 1 (1c) | H (2a) | 4 | 37 (5) |
aExposure to sunlight in a solar float. b47% conversion of 1a.
The structures of the photoaddition products 3a and 3b were unambiguously confirmed by X-ray crystallography (Figure 2). In the solid state, molecules of both compounds undergo hydrogen bonding between the newly formed hydroxy group and the intact carbonyl group, resulting in a one-dimensional network (see Supporting Information File 1).
![[1860-5397-13-275-2]](/bjoc/content/figures/1860-5397-13-275-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structures of photoaddition products 3a (left) and 3b (right).
Figure 2: Crystal structures of photoaddition products 3a (left) and 3b (right).
Notably, irradiation of N-(bromomethyl)phthalimide (1c) in the presence of phenylacetate (2a) did not furnish the desired addition product, but the benzylated ester 5 in 37% yield instead (Scheme 4). The electron-withdrawing character of the phthalimide group favored thermal nucleophilic substitution between 1c and phenylacetate (2a) to phthalimide 6. Ester 6 was indeed obtained in 37% yield by gently heating a mixture of phthalimide 1c and phenylacetate (2a) in acetone/water. Subsequently, compound 5 was independently prepared by photodecarboxylative benzylation of 6 for 4 h in 91% yield.
![[1860-5397-13-275-i4]](/bjoc/content/inline/1860-5397-13-275-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Formation of 5 by photodecarboxylative additions.
Scheme 4: Formation of 5 by photodecarboxylative additions.
Sulfuric acid-catalyzed dehydrations of the benzylated hydroxyphthalimidines 3a–q in dichloromethane at room temperature resulted in the corresponding olefins 7a–q in good to excellent yields of 66–95% (Scheme 5 and Table 3). The simple reaction protocol enabled parallel operations in a Radleys Carousel 6 Plus Reaction Station™. In line with DFT calculations by Kise et al. [12] and independently by Li and Janesko [21], the thermodynamically favored E-isomer was obtained as the main or sole product. The high E-selectivity was furthermore confirmed by 1H NMR analyses. The olefinic protons of the minor Z-isomers are shifted downfield by approx. 0.25 ppm due to the shielding effect of the adjacent isoindolin-1-one ring [31,41]. A similar but deshielding effect was found for the N-bromoalkyl protons and the arylmethylene group. No such shifts were observed for the corresponding major E-isomers.
![[1860-5397-13-275-i5]](/bjoc/content/inline/1860-5397-13-275-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Acid-catalyzed dehydration of 3a–q.
Scheme 5: Acid-catalyzed dehydration of 3a–q.
Table 3: Experimental results for acid-catalyzed dehydrations.
| Entry | n | R | E/Z ratioa | Yield 7 [%] |
| 1 | 2 | H | 9:1 | 91 (7a) |
| 2 | 2 | 4-F | >10:1 | 85 (7b) |
| 3 | 2 | 4-Cl | >10:1 | 88 (7c) |
| 4 | 2 | 4-Br | >10:1 | 83 (7d) |
| 5 | 2 | 4-MeO | >10:1 | 90 (7e) |
| 6 | 2 | 4-Me | >10:1 | 94 (7f) |
| 7 | 2 | 3-Me | >10:1 | 83 (7g) |
| 8 | 2 | 2-Me | >10:1 | 90 (7h) |
| 9 | 2 | 4-AcO | 8:1 | 66 (7i) |
| 10 | 3 | H | >10:1 | 83 (7j) |
| 11 | 3 | 4-F | >10:1 | 91 (7k) |
| 12 | 3 | 4-Cl | >10:1 | 90 (7l) |
| 13 | 3 | 4-Br | >10:1 | 84 (7m) |
| 14 | 3 | 4-MeO | >10:1 | 95 (7n) |
| 15 | 3 | 4-Me | >10:1 | 92 (7o) |
| 16 | 3 | 3-Me | >10:1 | 92 (7p) |
| 17 | 3 | 2-Me | >10:1 | 83 (7q) |
aDetermined by 1H NMR analysis.
The structure of the dehydration product (E)-7a was furthermore confirmed by X-ray crystallographic analysis (Figure 3). Remarkably, the phenyl group of the arylmethylene unit is positioned almost perpendicular to the isoindolinone ring. Compound (E)-7a forms dimers through CH–π interactions between the phenyl ring and the olefinic =CH group (see Supporting Information File 1).
![[1860-5397-13-275-3]](/bjoc/content/figures/1860-5397-13-275-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Crystal structure of (E)-7a. Side view and front view.
Figure 3: Crystal structure of (E)-7a. Side view and front view.
The final amination step was achieved following a modified procedure by Marzabadi and co-workers [42]. Catalytic amounts of potassium iodide were added for in situ halogen exchange. The dehydrated products 7a–q were heated in the presence of the respective secondary amine, K2CO3 and KI in DMF (Scheme 6 and Table 4). Subsequent work-up and isolation by column chromatography furnished the desired 2-dialkylaminoalkyl-3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones 8a–y in moderate yields of 49–61% (Table 4), among these the biologically active AL-12 (8e), AL-12A (8i) and AL-5 (8w) in their neutral forms. Interestingly, the Z-isomer of 8a–y was formed as the sole product in almost all cases as confirmed by 1H NMR analyses and by comparison with literature data (Table 4, entries 1–25) [10]. Subsequent investigations revealed that E-to-Z isomerization was caused during acidic work-up or purification. When products 8a and 8j were isolated by extraction under neutral conditions, the high E-selectivity of the dehydration step was retained. After column chromatographic purification on silica gel, partial isomerization to the Z-isomers was again observed (Table 4, entries 26 and 27).
![[1860-5397-13-275-i6]](/bjoc/content/inline/1860-5397-13-275-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Amination of dehydrated products 7a–q.
Scheme 6: Amination of dehydrated products 7a–q.
Table 4: Experimental results for amination reactions.
| Entry | n | R | R’ | E/Z ratioa | Yield 8 [%] |
| 1 (7a) | 2 | H | C2H5 | >1:10 | 55 (8a) |
| 2 (7b) | 2 | 4-F | C2H5 | >1:10 | 55 (8b) |
| 3 (7c) | 2 | 4-Cl | C2H5 | >1:10 | 53 (8c) |
| 4 (7d) | 2 | 4-Br | C2H5 | >1:10 | 51 (8d) |
| 5 (7e) | 2 | 4-MeO | C2H5 | >1:10 | 50 (8e) |
| 6 (7f) | 2 | 4-Me | C2H5 | >1:10 | 57 (8f) |
| 7 (7g) | 2 | 3-Me | C2H5 | >1:10 | 51 (8g) |
| 8 (7h) | 2 | 2-Me | C2H5 | >1:10 | 58 (8h) |
| 9 (7i) | 2 | 4-AcO | C2H5 | >1:10 | 57 (8i) |
| 10 (7j) | 3 | H | C2H5 | >1:10 | 53 (8j) |
| 11 (7k) | 3 | 4-F | C2H5 | >1:10 | 57 (8k) |
| 12 (7l) | 3 | 4-Cl | C2H5 | >1:10 | 60 (8l) |
| 13 (7m) | 3 | 4-Br | C2H5 | >1:10 | 59 (8m) |
| 14 (7n) | 3 | 4-MeO | C2H5 | >1:10 | 55 (8n) |
| 15 (7o) | 3 | 4-Me | C2H5 | >1:10 | 58 (8o) |
| 16 (7p) | 3 | 3-Me | C2H5 | >1:10 | 61 (8p) |
| 17 (7q) | 3 | 2-Me | C2H5 | >1:10 | 49 (8q) |
| 18 (7a) | 2 | H | CH3 | >1:10 | 58 (8r) |
| 19 (7b) | 2 | 4-F | CH3 | >1:10 | 57 (8s) |
| 20 (7c) | 2 | 4-Cl | CH3 | >1:10 | 55 (8t) |
| 21 (7e) | 2 | 4-MeO | CH3 | >1:10 | 49 (8u) |
| 22 (7f) | 2 | 4-Me | CH3 | >1:10 | 60 (8v) |
| 23 (7j) | 3 | H | CH3 | >1:10 | 58 (8w) |
| 24 (7k) | 3 | 4-F | CH3 | >1:10 | 55 (8x) |
| 25 (7q) | 3 | 2-Me | CH3 | >1:10 | 53 (8y) |
| 26 (7a) | 2 | H | C2H5 | >10:1b/1:1c | 65 (8a) |
| 27 (7j) | 3 | H | C2H5 | >10:1b/10:7c | 72 (8j) |
aAfter acidic work-up. Determined by 1H NMR analysis. bCrude product after neutral work-up. cPure product after neutral work-up followed by column chromatography on silica gel.
The solid state structure of the amination product (Z)-8a was furthermore established by X-ray crystal structure analysis (Figure 4). The phenyl group of the arylmethylene unit is positioned almost perpendicular to the isoindolinone ring on top of one of the N-ethyl groups of the side chain.
![[1860-5397-13-275-4]](/bjoc/content/figures/1860-5397-13-275-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Crystal structure of (Z)-8a. Side view and front view.
Figure 4: Crystal structure of (Z)-8a. Side view and front view.
The mechanism of the photodecarboxylation is well established (Scheme 7) and involves triplet sensitization by acetone and electron transfer between the phenylacetate and the excited phthalimide [55-57]. Subsequent decarboxylation, radical combination and protonation furnishes the observed benzylated hydroxyphthalimidine derivatives 3a–q.
![[1860-5397-13-275-i7]](/bjoc/content/inline/1860-5397-13-275-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Mechanistic scenario for the photodecarboxylative addition.
Scheme 7: Mechanistic scenario for the photodecarboxylative addition.
The nucleophilic cyclization to the oxazolidine derivative 4 may occur at different stages of the photodecarboxylation (Scheme 8). The phthalimide radical anion may undergo nucleophilic cyclization prior to C–C bond formation (path A), as postulated by Griesbeck et al. for the photocyclization of phthaloyl-L-methionine [58]. Alternatively, the alcoholate obtained after C–C bond formation may cyclize to the oxazolidine (path B), as known from reactions of 1a with organometallic reagents [48-50]. When compound 1a was treated with either sodium carbonate or potassium tert-butoxide it was indeed converted quantitatively to the oxazolidine derivative 4. Similarly, the final benzylated hydroxyphthalimidine may undergo cyclization instead (path C), as was found to occur at elevated temperature of >40 °C. Temperature control during the reaction, work-up and isolation was thus important to suppress this undesired transformation. The usage of pH 7 buffer as a co-solvent significantly improved yields and selectivity, possibly due to the avoidance of extreme basic conditions found for photodecarboxylations in acetone/water [34,59].
![[1860-5397-13-275-i8]](/bjoc/content/inline/1860-5397-13-275-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Possible scenarios for nucleophilic cyclization to 4.
Scheme 8: Possible scenarios for nucleophilic cyclization to 4.
The high stereoselectivity during the formation of compounds 7a–q is supported by the higher stability of the E-isomer, as shown for other simple N-substituted benzylideneisoindolin-1-ones [12,21]. Remarkably, acidic reaction conditions induced thermal isomerization of compounds 8a–y, possibly via an acylaminium cation intermediate (Scheme 9) [60]. The change in stability may be caused by a stabilizing ionic–π interaction for the Z-isomer during isomerization [61]. A similar thermal isomerization towards the more stable isomer induced by catalytic amounts of pyridinium p-toluenesulfonate has been recently reported by Kise and co-workers [12].
![[1860-5397-13-275-i9]](/bjoc/content/inline/1860-5397-13-275-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Possible E/Z isomerization for compounds 8a–y.
Scheme 9: Possible E/Z isomerization for compounds 8a–y.
Conclusion
Photodecarboxylation reactions are emerging as versatile transformations in organic synthesis [24,33,62-65]. The photodecarboxylative addition of phenylacetates to N-(bromoalkyl)phthalimides was used as a mild key step in the synthesis of 2-dialkylaminoalkyl-3-arylmethylene-2,3-dihydro-1H-isoindolin-1-ones, among these the potent local anesthetics AL-12, AL-12A and AL-5 (in their neutral forms). The simple procedures make this three-step process attractive for in-series continuous flow applications [40,66,67].
Supporting Information
| Supporting Information File 1: Experimental details, detailed spectroscopic and crystallographic data. | ||
| Format: PDF | Size: 757.2 KB | Download |
Acknowledgements
This work was financially supported by the Australian Research Council (ARC, DP130100794). SM thanks the Higher Education Commission Pakistan for providing a Scholarship under the International Research Support Initiative Program. MO thanks Professor Emeritus Robert (Bob) S. H. Liu from the University of Hawaii for the donation of the solar float reactor.
References
-
Kushwaha, N.; Kaushik, D. J. Appl. Pharm. Sci. 2016, 6, 159–171. doi:10.7324/JAPS.2016.60330
Return to citation in text: [1] -
Frlan, R.; Gobec, S. Expert Opin. Ther. Pat. 2017, 27, 637–641. doi:10.1080/13543776.2017.1322954
Return to citation in text: [1] -
Bhatia, R. K. Curr. Top. Med. Chem. 2017, 17, 189–207. doi:10.2174/1568026616666160530154100
Return to citation in text: [1] -
Sharma, U.; Kumar, P.; Kumar, N.; Singh, B. Mini-Rev. Med. Chem. 2010, 10, 678–704. doi:10.2174/138955710791572442
Return to citation in text: [1] -
Speck, K.; Magauer, T. Beilstein J. Org. Chem. 2013, 9, 2048–2078. doi:10.3762/bjoc.9.243
Return to citation in text: [1] -
Csende, F.; Stájer, G. Curr. Org. Chem. 2005, 9, 1261–1276. doi:10.2174/1385272054863961
Return to citation in text: [1] -
Csende, F.; Miklós, F.; Stájer, G. Curr. Org. Chem. 2012, 16, 1005–1050. doi:10.2174/138527212800194683
Return to citation in text: [1] -
Marsili, A. 2-(Omega-alkylaminoalkyl)-and (2-omega-dialkylaminoalkyl-3-(4-X-benxylidene)-phthalimidines. Eur. Pat. Appl. EP-0105131A1, April 11, 1984.
Chem. Abstr. 1984, 101, 54922.
Return to citation in text: [1] -
Becker, D. E.; Reed, K. L. Anesth. Prog. 2012, 59, 90–102. doi:10.2344/0003-3006-59.2.90
Return to citation in text: [1] -
Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7
Return to citation in text: [1] [2] [3] -
Li, L.; Wang, M.; Zhang, X.; Jiang, Y.; Ma, D. Org. Lett. 2009, 11, 1309–1312. doi:10.1021/ol9000922
Return to citation in text: [1] -
Kise, N.; Kawano, Y.; Sakurai, T. J. Org. Chem. 2013, 78, 12453–12459. doi:10.1021/jo402125u
Return to citation in text: [1] [2] [3] [4] -
Zhang, L.; Zhang, Y.; Wang, X.; Shen, J. Molecules 2013, 18, 654–665. doi:10.3390/molecules18010654
Return to citation in text: [1] -
Chatterjee, N.; Sarkar, S.; Pal, R.; Sen, A. K. Tetrahedron Lett. 2013, 54, 3748–3751. doi:10.1016/j.tetlet.2013.05.004
Return to citation in text: [1] -
Irudayanathan, F. M.; Noh, J.; Choi, J.; Lee, S. Adv. Synth. Catal. 2014, 356, 3433–3442. doi:10.1002/adsc.201400451
Return to citation in text: [1] -
Reyes-González, M. Á.; Zamudio-Medina, Á.; Ramírez-Marroquín, O. A.; Ordóñez, M. Monatsh. Chem. 2014, 145, 1001–1007. doi:10.1007/s00706-013-1146-9
Return to citation in text: [1] -
Gogoi, A.; Guin, S.; Rout, S. K.; Majji, G.; Patel, B. K. RSC Adv. 2014, 4, 59902–59907. doi:10.1039/C4RA12782J
Return to citation in text: [1] -
Zheng, X.-X.; Du, C.; Zhao, X.-M.; Zhu, X.; Suo, J.-F.; Hao, X.-Q.; Niu, J.-L.; Song, M.-P. J. Org. Chem. 2016, 81, 4002–4011. doi:10.1021/acs.joc.6b00129
Return to citation in text: [1] -
Pathare, R. S.; Sharma, S.; Elagandhula, S.; Saini, V.; Sawant, D. M.; Yadav, M.; Sharon, A.; Khan, S.; Pardasani, R. T. Eur. J. Org. Chem. 2016, 5579–5587. doi:10.1002/ejoc.201600999
Return to citation in text: [1] -
Pal, R.; Chatterjee, N.; Roy, M.; Nouh, E. S. A.; Sarkar, S.; Jaisankar, P.; Sarkar, S.; Sen, A. K. Tetrahedron Lett. 2016, 57, 43–47. doi:10.1016/j.tetlet.2015.11.059
Return to citation in text: [1] -
Li, L.; Janesko, B. G. J. Org. Chem. 2016, 81, 10802–10808. doi:10.1021/acs.joc.6b01904
Return to citation in text: [1] [2] [3] -
Munoz, S. B.; Aloia, A. N.; Moore, A. K.; Papp, A.; Mathew, T.; Fustero, S.; Olah, G. A.; Prakash, G. K. S. Org. Biomol. Chem. 2016, 14, 85–92. doi:10.1039/C5OB02187A
Return to citation in text: [1] -
Griesbeck, A. G.; Oelgemöller, M. Synlett 1999, 492–494. doi:10.1055/s-1999-2622
Return to citation in text: [1] -
Griesbeck, A. G.; Kramer, W.; Oelgemöller, M. Synlett 1999, 1169–1178. doi:10.1055/s-1999-3159
Return to citation in text: [1] [2] -
Oelgemöller, M.; Cygon, P.; Lex, J.; Griesbeck, A. G. Heterocycles 2003, 59, 669–684. doi:10.3987/COM-02-S77
Return to citation in text: [1] [2] [3] -
Griesbeck, A. G.; Warzecha, K.-D.; Neudörfl, J. M.; Görner, H. Synlett 2004, 2347–2350. doi:10.1055/s-2004-832850
Return to citation in text: [1] -
Hatoum, F.; Gallagher, S.; Baragwanath, L.; Lex, J.; Oelgemöller, M. Tetrahedron Lett. 2009, 50, 6335–6338. doi:10.1016/j.tetlet.2009.08.115
Return to citation in text: [1] [2] -
Belluau, V.; Noeureuil, P.; Ratzke, E.; Skvortsov, A.; Gallagher, S.; Motti, C. A.; Oelgemöller, M. Tetrahedron Lett. 2010, 51, 4738–4741. doi:10.1016/j.tetlet.2010.07.017
Return to citation in text: [1] [2] -
Griesbeck, A. G.; Nazarov, N.; Neudörfl, J. M.; Heffen, M. Green Chem. 2012, 14, 3004–3006. doi:10.1039/c2gc36089f
Return to citation in text: [1] -
Hatoum, F.; Engler, J.; Zelmer, C.; Wißen, J.; Motti, C. A.; Lex, J.; Oelgemöller, M. Tetrahedron Lett. 2012, 53, 5573–5577. doi:10.1016/j.tetlet.2012.07.142
Return to citation in text: [1] [2] -
Griesbeck, A. G.; Neudörfel, J.-M.; Goldfuss, B.; Molitor, S. ChemPhotoChem 2017, 1, 355–362. doi:10.1002/cptc.201700057
Return to citation in text: [1] [2] -
Capaldo, L.; Buzzetti, L.; Merli, D.; Fagnoni, M.; Ravelli, D. J. Org. Chem. 2016, 81, 7102–7109. doi:10.1021/acs.joc.6b00984
Return to citation in text: [1] -
Budac, D.; Wan, P. J. Photochem. Photobiol., A: Chem. 1992, 67, 135–166. doi:10.1016/1010-6030(92)85224-I
Return to citation in text: [1] [2] -
Griesbeck, A. G.; Kramer, W.; Oelgemöller, M. Green Chem. 1999, 1, 205–208. doi:10.1039/a905076k
Return to citation in text: [1] [2] -
Griesbeck, A. G.; Maptue, N.; Bondock, S.; Oelgemöller, M. Photochem. Photobiol. Sci. 2003, 2, 450–451. doi:10.1039/B212357F
Return to citation in text: [1] -
Shvydkiv, O.; Gallagher, S.; Nolan, K.; Oelgemöller, M. Org. Lett. 2010, 12, 5170–5173. doi:10.1021/ol102184u
Return to citation in text: [1] -
Shvydkiv, O.; Nolan, K.; Oelgemöller, M. Beilstein J. Org. Chem. 2011, 7, 1055–1063. doi:10.3762/bjoc.7.121
Return to citation in text: [1] -
Oelgemöller, M.; Gallagher, S.; McCarthy, K. Processes 2014, 2, 158–166. doi:10.3390/pr2010158
Return to citation in text: [1] -
Pordanjani, H. M.; Faderl, C.; Wang, J.; Motti, C. A.; Junk, P. C.; Oelgemöller, M. Aust. J. Chem. 2015, 68, 1662–1667. doi:10.1071/CH15356
Return to citation in text: [1] -
Josland, S.; Mumtaz, S.; Oelgemöller, M. Chem. Eng. Technol. 2016, 39, 81–87. doi:10.1002/ceat.201500285
Return to citation in text: [1] [2] -
Ang, W. S.; Halton, B. Aust. J. Chem. 1971, 24, 851–856. doi:10.1071/CH9710851
Return to citation in text: [1] [2] -
Jiang, Y.; Chen, C.-A.; Lu, K.; Daniewska, I.; De Leon, J.; Kong, R.; Forray, C.; Li, B.; Hegde, L. G.; Wolinsky, T. D.; Craig, D. A.; Wetzel, J. M.; Anderson, K.; Marzabadi, M. R. J. Med. Chem. 2007, 50, 3870–3882. doi:10.1021/jm060381c
Return to citation in text: [1] [2] -
Oelgemöller, M.; Griesbeck, A. G. J. Photochem. Photobiol., C: Photochem. Rev. 2002, 3, 109–127. doi:10.1016/S1389-5567(02)00022-9
Return to citation in text: [1] -
McDermott, G.; Yoo, D. J.; Oelgemöller, M. Heterocycles 2005, 65, 2221–2257. doi:10.3987/REV-05-601
Return to citation in text: [1] -
Coyle, J. D.; Smart, L. E.; Challiner, J. F.; Haws, E. J. J. Chem. Soc., Perkin Trans. 1 1985, 121–129. doi:10.1039/p19850000121
Return to citation in text: [1] -
Coyle, J. D.; Bryant, L. R. B.; Cragg, J. E.; Challiner, J. F.; Haws, E. J. J. Chem. Soc., Perkin Trans. 1 1985, 1177–1180. doi:10.1039/p19850001177
Return to citation in text: [1] -
Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M. T. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, United States, 2006; pp 599 ff.
Return to citation in text: [1] -
Bousquet, T.; Fleury, J.-F.; Daïch, A.; Netchitaïlo, P. Tetrahedron 2006, 62, 706–715. doi:10.1016/j.tet.2005.10.017
Return to citation in text: [1] [2] -
Wharton, C. J.; Wrigglesworth, R. J. Chem. Soc., Perkin Trans. 1 1985, 809–813. doi:10.1039/p19850000809
Return to citation in text: [1] [2] -
Torian, B. E.; Braun, L. L. J. Heterocycl. Chem. 1984, 21, 293–295. doi:10.1002/jhet.5570210204
Return to citation in text: [1] [2] -
Yoshimi, Y.; Nishio, A.; Hayashi, M.; Morita, T. J. Photochem. Photobiol., A: Chem. 2016, 331, 17–21. doi:10.1016/j.jphotochem.2016.03.012
Return to citation in text: [1] -
Oelgemöller, M. Chem. Rev. 2016, 116, 9664–9682. doi:10.1021/acs.chemrev.5b00720
Return to citation in text: [1] -
Zhao, Y.-P.; Campbell, R. O.; Liu, R. S. H. Green Chem. 2008, 10, 1038–1042. doi:10.1039/b809007f
Return to citation in text: [1] -
Zhao, Y.-P.; Yang, L.-Y.; Liu, R. S. H. Green Chem. 2009, 11, 837–842. doi:10.1039/b819207c
Return to citation in text: [1] -
Warzecha, K.-D.; Görner, H.; Griesbeck, A. G. J. Phys. Chem. A 2006, 110, 3356–3363. doi:10.1021/jp055878x
Return to citation in text: [1] -
Görner, H.; Griesbeck, A. G.; Heinrich, T.; Kramer, W.; Oelgemöller, M. Chem. – Eur. J. 2001, 7, 1530–1538. doi:10.1002/1521-3765(20010401)7:7<1530::AID-CHEM1530>3.0.CO;2-L
Return to citation in text: [1] -
Görner, H.; Oelgemöller, M.; Griesbeck, A. G. J. Phys. Chem. A 2002, 106, 1458–1464. doi:10.1021/jp011090c
Return to citation in text: [1] -
Griesbeck, A. G.; Mauder, H.; Müller, I.; Peters, E.-M.; Peters, K.; von Schnering, H. G. Tetrahedron Lett. 1993, 34, 453–456. doi:10.1016/0040-4039(93)85100-B
Return to citation in text: [1] -
Griesbeck, A. G.; Heinrich, T.; Oelgemöller, M.; Molis, A.; Heidtmann, A. Helv. Chim. Acta 2002, 85, 4561–4578. doi:10.1002/hlca.200290027
Return to citation in text: [1] -
Maryanoff, B. E.; Zhang, H.-C.; Cohen, J. H.; Turchi, I. J.; Maryanoff, C. A. Chem. Rev. 2004, 104, 1431–1628. doi:10.1021/cr0306182
Return to citation in text: [1] -
Salonen, L. M.; Ellermann, M.; Diederich, F. Angew. Chem., Int. Ed. 2011, 50, 4808–4842. doi:10.1002/anie.201007560
Return to citation in text: [1] -
Jin, Y.; Fu, H. Asian J. Org. Chem. 2017, 6, 368–385. doi:10.1002/ajoc.201600513
Return to citation in text: [1] -
Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 15632–15641. doi:10.1002/anie.201505731
Return to citation in text: [1] -
Yoshimi, Y. J. Photochem. Photobiol., A: Chem. 2017, 342, 116–130. doi:10.1016/j.jphotochem.2017.04.007
Return to citation in text: [1] -
Yoshimi, Y. J. Synth. Org. Chem., Jpn. 2013, 71, 935–943. doi:10.5059/yukigoseikyokaishi.71.935
Return to citation in text: [1] -
Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183
Return to citation in text: [1] -
Webb, D.; Jamison, T. F. Chem. Sci. 2010, 1, 675–680. doi:10.1039/c0sc00381f
Return to citation in text: [1]
| 34. | Griesbeck, A. G.; Kramer, W.; Oelgemöller, M. Green Chem. 1999, 1, 205–208. doi:10.1039/a905076k |
| 59. | Griesbeck, A. G.; Heinrich, T.; Oelgemöller, M.; Molis, A.; Heidtmann, A. Helv. Chim. Acta 2002, 85, 4561–4578. doi:10.1002/hlca.200290027 |
| 12. | Kise, N.; Kawano, Y.; Sakurai, T. J. Org. Chem. 2013, 78, 12453–12459. doi:10.1021/jo402125u |
| 21. | Li, L.; Janesko, B. G. J. Org. Chem. 2016, 81, 10802–10808. doi:10.1021/acs.joc.6b01904 |
| 60. | Maryanoff, B. E.; Zhang, H.-C.; Cohen, J. H.; Turchi, I. J.; Maryanoff, C. A. Chem. Rev. 2004, 104, 1431–1628. doi:10.1021/cr0306182 |
| 1. | Kushwaha, N.; Kaushik, D. J. Appl. Pharm. Sci. 2016, 6, 159–171. doi:10.7324/JAPS.2016.60330 |
| 2. | Frlan, R.; Gobec, S. Expert Opin. Ther. Pat. 2017, 27, 637–641. doi:10.1080/13543776.2017.1322954 |
| 3. | Bhatia, R. K. Curr. Top. Med. Chem. 2017, 17, 189–207. doi:10.2174/1568026616666160530154100 |
| 4. | Sharma, U.; Kumar, P.; Kumar, N.; Singh, B. Mini-Rev. Med. Chem. 2010, 10, 678–704. doi:10.2174/138955710791572442 |
| 5. | Speck, K.; Magauer, T. Beilstein J. Org. Chem. 2013, 9, 2048–2078. doi:10.3762/bjoc.9.243 |
| 6. | Csende, F.; Stájer, G. Curr. Org. Chem. 2005, 9, 1261–1276. doi:10.2174/1385272054863961 |
| 7. | Csende, F.; Miklós, F.; Stájer, G. Curr. Org. Chem. 2012, 16, 1005–1050. doi:10.2174/138527212800194683 |
| 10. | Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7 |
| 11. | Li, L.; Wang, M.; Zhang, X.; Jiang, Y.; Ma, D. Org. Lett. 2009, 11, 1309–1312. doi:10.1021/ol9000922 |
| 12. | Kise, N.; Kawano, Y.; Sakurai, T. J. Org. Chem. 2013, 78, 12453–12459. doi:10.1021/jo402125u |
| 13. | Zhang, L.; Zhang, Y.; Wang, X.; Shen, J. Molecules 2013, 18, 654–665. doi:10.3390/molecules18010654 |
| 14. | Chatterjee, N.; Sarkar, S.; Pal, R.; Sen, A. K. Tetrahedron Lett. 2013, 54, 3748–3751. doi:10.1016/j.tetlet.2013.05.004 |
| 15. | Irudayanathan, F. M.; Noh, J.; Choi, J.; Lee, S. Adv. Synth. Catal. 2014, 356, 3433–3442. doi:10.1002/adsc.201400451 |
| 16. | Reyes-González, M. Á.; Zamudio-Medina, Á.; Ramírez-Marroquín, O. A.; Ordóñez, M. Monatsh. Chem. 2014, 145, 1001–1007. doi:10.1007/s00706-013-1146-9 |
| 17. | Gogoi, A.; Guin, S.; Rout, S. K.; Majji, G.; Patel, B. K. RSC Adv. 2014, 4, 59902–59907. doi:10.1039/C4RA12782J |
| 18. | Zheng, X.-X.; Du, C.; Zhao, X.-M.; Zhu, X.; Suo, J.-F.; Hao, X.-Q.; Niu, J.-L.; Song, M.-P. J. Org. Chem. 2016, 81, 4002–4011. doi:10.1021/acs.joc.6b00129 |
| 19. | Pathare, R. S.; Sharma, S.; Elagandhula, S.; Saini, V.; Sawant, D. M.; Yadav, M.; Sharon, A.; Khan, S.; Pardasani, R. T. Eur. J. Org. Chem. 2016, 5579–5587. doi:10.1002/ejoc.201600999 |
| 20. | Pal, R.; Chatterjee, N.; Roy, M.; Nouh, E. S. A.; Sarkar, S.; Jaisankar, P.; Sarkar, S.; Sen, A. K. Tetrahedron Lett. 2016, 57, 43–47. doi:10.1016/j.tetlet.2015.11.059 |
| 21. | Li, L.; Janesko, B. G. J. Org. Chem. 2016, 81, 10802–10808. doi:10.1021/acs.joc.6b01904 |
| 22. | Munoz, S. B.; Aloia, A. N.; Moore, A. K.; Papp, A.; Mathew, T.; Fustero, S.; Olah, G. A.; Prakash, G. K. S. Org. Biomol. Chem. 2016, 14, 85–92. doi:10.1039/C5OB02187A |
| 47. | Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M. T. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, United States, 2006; pp 599 ff. |
| 10. | Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7 |
| 48. | Bousquet, T.; Fleury, J.-F.; Daïch, A.; Netchitaïlo, P. Tetrahedron 2006, 62, 706–715. doi:10.1016/j.tet.2005.10.017 |
| 49. | Wharton, C. J.; Wrigglesworth, R. J. Chem. Soc., Perkin Trans. 1 1985, 809–813. doi:10.1039/p19850000809 |
| 50. | Torian, B. E.; Braun, L. L. J. Heterocycl. Chem. 1984, 21, 293–295. doi:10.1002/jhet.5570210204 |
| 9. | Becker, D. E.; Reed, K. L. Anesth. Prog. 2012, 59, 90–102. doi:10.2344/0003-3006-59.2.90 |
| 25. | Oelgemöller, M.; Cygon, P.; Lex, J.; Griesbeck, A. G. Heterocycles 2003, 59, 669–684. doi:10.3987/COM-02-S77 |
| 8. |
Marsili, A. 2-(Omega-alkylaminoalkyl)-and (2-omega-dialkylaminoalkyl-3-(4-X-benxylidene)-phthalimidines. Eur. Pat. Appl. EP-0105131A1, April 11, 1984.
Chem. Abstr. 1984, 101, 54922. |
| 27. | Hatoum, F.; Gallagher, S.; Baragwanath, L.; Lex, J.; Oelgemöller, M. Tetrahedron Lett. 2009, 50, 6335–6338. doi:10.1016/j.tetlet.2009.08.115 |
| 30. | Hatoum, F.; Engler, J.; Zelmer, C.; Wißen, J.; Motti, C. A.; Lex, J.; Oelgemöller, M. Tetrahedron Lett. 2012, 53, 5573–5577. doi:10.1016/j.tetlet.2012.07.142 |
| 36. | Shvydkiv, O.; Gallagher, S.; Nolan, K.; Oelgemöller, M. Org. Lett. 2010, 12, 5170–5173. doi:10.1021/ol102184u |
| 37. | Shvydkiv, O.; Nolan, K.; Oelgemöller, M. Beilstein J. Org. Chem. 2011, 7, 1055–1063. doi:10.3762/bjoc.7.121 |
| 38. | Oelgemöller, M.; Gallagher, S.; McCarthy, K. Processes 2014, 2, 158–166. doi:10.3390/pr2010158 |
| 39. | Pordanjani, H. M.; Faderl, C.; Wang, J.; Motti, C. A.; Junk, P. C.; Oelgemöller, M. Aust. J. Chem. 2015, 68, 1662–1667. doi:10.1071/CH15356 |
| 40. | Josland, S.; Mumtaz, S.; Oelgemöller, M. Chem. Eng. Technol. 2016, 39, 81–87. doi:10.1002/ceat.201500285 |
| 42. | Jiang, Y.; Chen, C.-A.; Lu, K.; Daniewska, I.; De Leon, J.; Kong, R.; Forray, C.; Li, B.; Hegde, L. G.; Wolinsky, T. D.; Craig, D. A.; Wetzel, J. M.; Anderson, K.; Marzabadi, M. R. J. Med. Chem. 2007, 50, 3870–3882. doi:10.1021/jm060381c |
| 24. | Griesbeck, A. G.; Kramer, W.; Oelgemöller, M. Synlett 1999, 1169–1178. doi:10.1055/s-1999-3159 |
| 33. | Budac, D.; Wan, P. J. Photochem. Photobiol., A: Chem. 1992, 67, 135–166. doi:10.1016/1010-6030(92)85224-I |
| 62. | Jin, Y.; Fu, H. Asian J. Org. Chem. 2017, 6, 368–385. doi:10.1002/ajoc.201600513 |
| 63. | Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 15632–15641. doi:10.1002/anie.201505731 |
| 64. | Yoshimi, Y. J. Photochem. Photobiol., A: Chem. 2017, 342, 116–130. doi:10.1016/j.jphotochem.2017.04.007 |
| 65. | Yoshimi, Y. J. Synth. Org. Chem., Jpn. 2013, 71, 935–943. doi:10.5059/yukigoseikyokaishi.71.935 |
| 25. | Oelgemöller, M.; Cygon, P.; Lex, J.; Griesbeck, A. G. Heterocycles 2003, 59, 669–684. doi:10.3987/COM-02-S77 |
| 34. | Griesbeck, A. G.; Kramer, W.; Oelgemöller, M. Green Chem. 1999, 1, 205–208. doi:10.1039/a905076k |
| 35. | Griesbeck, A. G.; Maptue, N.; Bondock, S.; Oelgemöller, M. Photochem. Photobiol. Sci. 2003, 2, 450–451. doi:10.1039/B212357F |
| 43. | Oelgemöller, M.; Griesbeck, A. G. J. Photochem. Photobiol., C: Photochem. Rev. 2002, 3, 109–127. doi:10.1016/S1389-5567(02)00022-9 |
| 44. | McDermott, G.; Yoo, D. J.; Oelgemöller, M. Heterocycles 2005, 65, 2221–2257. doi:10.3987/REV-05-601 |
| 45. | Coyle, J. D.; Smart, L. E.; Challiner, J. F.; Haws, E. J. J. Chem. Soc., Perkin Trans. 1 1985, 121–129. doi:10.1039/p19850000121 |
| 46. | Coyle, J. D.; Bryant, L. R. B.; Cragg, J. E.; Challiner, J. F.; Haws, E. J. J. Chem. Soc., Perkin Trans. 1 1985, 1177–1180. doi:10.1039/p19850001177 |
| 40. | Josland, S.; Mumtaz, S.; Oelgemöller, M. Chem. Eng. Technol. 2016, 39, 81–87. doi:10.1002/ceat.201500285 |
| 66. | Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183 |
| 67. | Webb, D.; Jamison, T. F. Chem. Sci. 2010, 1, 675–680. doi:10.1039/c0sc00381f |
| 32. | Capaldo, L.; Buzzetti, L.; Merli, D.; Fagnoni, M.; Ravelli, D. J. Org. Chem. 2016, 81, 7102–7109. doi:10.1021/acs.joc.6b00984 |
| 33. | Budac, D.; Wan, P. J. Photochem. Photobiol., A: Chem. 1992, 67, 135–166. doi:10.1016/1010-6030(92)85224-I |
| 61. | Salonen, L. M.; Ellermann, M.; Diederich, F. Angew. Chem., Int. Ed. 2011, 50, 4808–4842. doi:10.1002/anie.201007560 |
| 23. | Griesbeck, A. G.; Oelgemöller, M. Synlett 1999, 492–494. doi:10.1055/s-1999-2622 |
| 24. | Griesbeck, A. G.; Kramer, W.; Oelgemöller, M. Synlett 1999, 1169–1178. doi:10.1055/s-1999-3159 |
| 25. | Oelgemöller, M.; Cygon, P.; Lex, J.; Griesbeck, A. G. Heterocycles 2003, 59, 669–684. doi:10.3987/COM-02-S77 |
| 26. | Griesbeck, A. G.; Warzecha, K.-D.; Neudörfl, J. M.; Görner, H. Synlett 2004, 2347–2350. doi:10.1055/s-2004-832850 |
| 27. | Hatoum, F.; Gallagher, S.; Baragwanath, L.; Lex, J.; Oelgemöller, M. Tetrahedron Lett. 2009, 50, 6335–6338. doi:10.1016/j.tetlet.2009.08.115 |
| 28. | Belluau, V.; Noeureuil, P.; Ratzke, E.; Skvortsov, A.; Gallagher, S.; Motti, C. A.; Oelgemöller, M. Tetrahedron Lett. 2010, 51, 4738–4741. doi:10.1016/j.tetlet.2010.07.017 |
| 29. | Griesbeck, A. G.; Nazarov, N.; Neudörfl, J. M.; Heffen, M. Green Chem. 2012, 14, 3004–3006. doi:10.1039/c2gc36089f |
| 30. | Hatoum, F.; Engler, J.; Zelmer, C.; Wißen, J.; Motti, C. A.; Lex, J.; Oelgemöller, M. Tetrahedron Lett. 2012, 53, 5573–5577. doi:10.1016/j.tetlet.2012.07.142 |
| 31. | Griesbeck, A. G.; Neudörfel, J.-M.; Goldfuss, B.; Molitor, S. ChemPhotoChem 2017, 1, 355–362. doi:10.1002/cptc.201700057 |
| 41. | Ang, W. S.; Halton, B. Aust. J. Chem. 1971, 24, 851–856. doi:10.1071/CH9710851 |
| 12. | Kise, N.; Kawano, Y.; Sakurai, T. J. Org. Chem. 2013, 78, 12453–12459. doi:10.1021/jo402125u |
| 53. | Zhao, Y.-P.; Campbell, R. O.; Liu, R. S. H. Green Chem. 2008, 10, 1038–1042. doi:10.1039/b809007f |
| 54. | Zhao, Y.-P.; Yang, L.-Y.; Liu, R. S. H. Green Chem. 2009, 11, 837–842. doi:10.1039/b819207c |
| 28. | Belluau, V.; Noeureuil, P.; Ratzke, E.; Skvortsov, A.; Gallagher, S.; Motti, C. A.; Oelgemöller, M. Tetrahedron Lett. 2010, 51, 4738–4741. doi:10.1016/j.tetlet.2010.07.017 |
| 51. | Yoshimi, Y.; Nishio, A.; Hayashi, M.; Morita, T. J. Photochem. Photobiol., A: Chem. 2016, 331, 17–21. doi:10.1016/j.jphotochem.2016.03.012 |
| 52. | Oelgemöller, M. Chem. Rev. 2016, 116, 9664–9682. doi:10.1021/acs.chemrev.5b00720 |
| 58. | Griesbeck, A. G.; Mauder, H.; Müller, I.; Peters, E.-M.; Peters, K.; von Schnering, H. G. Tetrahedron Lett. 1993, 34, 453–456. doi:10.1016/0040-4039(93)85100-B |
| 48. | Bousquet, T.; Fleury, J.-F.; Daïch, A.; Netchitaïlo, P. Tetrahedron 2006, 62, 706–715. doi:10.1016/j.tet.2005.10.017 |
| 49. | Wharton, C. J.; Wrigglesworth, R. J. Chem. Soc., Perkin Trans. 1 1985, 809–813. doi:10.1039/p19850000809 |
| 50. | Torian, B. E.; Braun, L. L. J. Heterocycl. Chem. 1984, 21, 293–295. doi:10.1002/jhet.5570210204 |
| 10. | Couture, A.; Deniau, E.; Grandclaudon, P. Tetrahedron 1997, 53, 10313–10330. doi:10.1016/S0040-4020(97)00680-7 |
| 55. | Warzecha, K.-D.; Görner, H.; Griesbeck, A. G. J. Phys. Chem. A 2006, 110, 3356–3363. doi:10.1021/jp055878x |
| 56. | Görner, H.; Griesbeck, A. G.; Heinrich, T.; Kramer, W.; Oelgemöller, M. Chem. – Eur. J. 2001, 7, 1530–1538. doi:10.1002/1521-3765(20010401)7:7<1530::AID-CHEM1530>3.0.CO;2-L |
| 57. | Görner, H.; Oelgemöller, M.; Griesbeck, A. G. J. Phys. Chem. A 2002, 106, 1458–1464. doi:10.1021/jp011090c |
| 31. | Griesbeck, A. G.; Neudörfel, J.-M.; Goldfuss, B.; Molitor, S. ChemPhotoChem 2017, 1, 355–362. doi:10.1002/cptc.201700057 |
| 41. | Ang, W. S.; Halton, B. Aust. J. Chem. 1971, 24, 851–856. doi:10.1071/CH9710851 |
| 42. | Jiang, Y.; Chen, C.-A.; Lu, K.; Daniewska, I.; De Leon, J.; Kong, R.; Forray, C.; Li, B.; Hegde, L. G.; Wolinsky, T. D.; Craig, D. A.; Wetzel, J. M.; Anderson, K.; Marzabadi, M. R. J. Med. Chem. 2007, 50, 3870–3882. doi:10.1021/jm060381c |
| 12. | Kise, N.; Kawano, Y.; Sakurai, T. J. Org. Chem. 2013, 78, 12453–12459. doi:10.1021/jo402125u |
| 21. | Li, L.; Janesko, B. G. J. Org. Chem. 2016, 81, 10802–10808. doi:10.1021/acs.joc.6b01904 |
© 2017 Anamimoghadam et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)