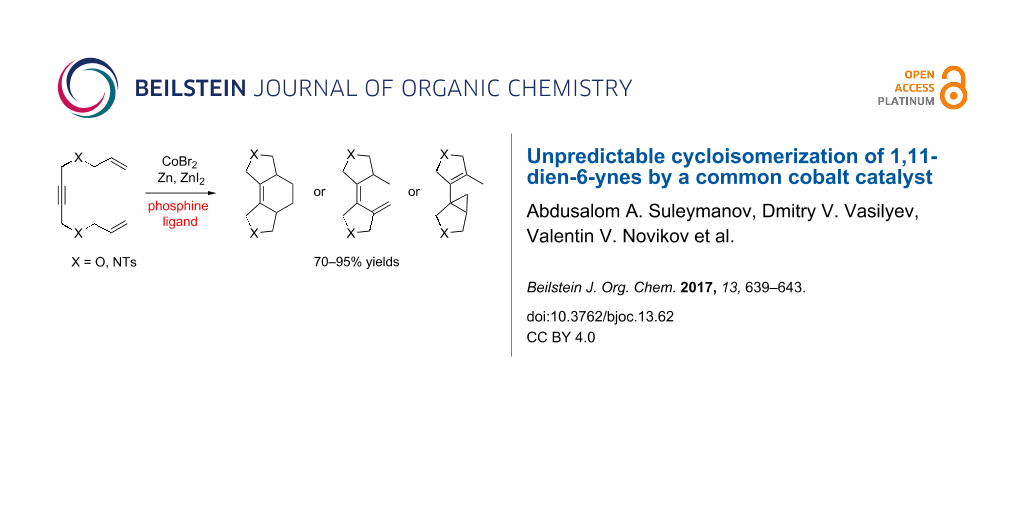
Abstract
1,11-Dien-6-ynes undergo cycloisomerization in the presence of the cobalt catalytic system CoBr2/phosphine ligand/Zn/ZnI2 giving cyclohexene, diene or cyclopropane structures depending on the type of the phosphine ligand. This unpredictable behaviour suggests that, although the availability of the cobalt catalytic system is appealing, the development of well-defined catalysts is desirable for further progress.

Graphical Abstract
Introduction
Metal-catalyzed reactions of enynes represent an atom- and step-economical route to complex organic molecules with a broad range of functionalities [1-4]. In particular, cycloisomerization of enynes allows one to prepare compounds with exocyclic double bonds and cyclopropanes in a highly selective manner [5-7]. While catalytic transformations of enynes have been investigated in detail, there are only a few examples of similar reactions of dienynes [8-14]. Among dienynes, 1,11-dien-6-ynes 1 are of particular interest, because they are readily available in 1–2 steps from the commercial precursors (e.g., allyl bromides and 1,4-dibromo-2-butyne). Transformations of 1,11-dien-6-ynes provide an access to highly substituted polycycles [8,11-13] (Scheme 1). However, all these transformations require expensive noble metal catalysts with rather sophisticated ligands. Therefore, the development of a cheap catalyst for such reactions is highly desirable.
![[1860-5397-13-62-i1]](/bjoc/content/inline/1860-5397-13-62-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Examples of metal-catalyzed transformations of 1,11-dien-6-ynes.
Scheme 1: Examples of metal-catalyzed transformations of 1,11-dien-6-ynes.
Over the last decade the application of the cobalt catalytic system CoBr2/phosphine ligand/Zn/ZnI2 for the carbon–carbon bond formation has become a subject of a growing interest (for reviews see [15-21]). This catalytic system has many advantages, including the availability of the cobalt salts and high tolerance to organic functional groups (for some recent examples see [22-27]). However, it has been noted that the direction of the catalytic reactions often depends on the structure of the phosphine ligand. Major research on these ligand-controlled reactions has been made by the groups of Hilt and Cheng, in particular, on the selective formation of meta/para-products of Diels–Alder reactions [28-32], linear/branched products of hydrovinylation reactions [33,34], [2 + 2] cycloadditions vs Alder-ene reactions [35], E/Z isomerizations of alkenes [36,37] and other processes [38-42].
Herein, we report the cycloisomerizations of 1,11-dien-6-ynes in the presence of a cobalt catalytic system. We have found that the regioselectivity of these reactions dramatically and unpredictably depends on the structure of the phosphine ligand and on the structure of the substrate.
Results and Discussion
First, we chose 1,4-bis(allyloxy)but-2-yne (1a) and 1,4-di(N-allyltosylamido)-2-butyne (1b) as test substrates for our investigation (Scheme 2). It was found that in the presence of CoBr2, Zn and ZnI2 (20 mol % each) as well as the triphenylphosphine ligand (40 mol %) in 1,2-dichloroethane (DCE) these substrates underwent clean [2 + 2 + 2]-cyclotrimerization to give cyclohexene derivatives 2a,b in 70–90% yields. The rate of the reaction notably varied from experiment to experiment, but full conversion of 1a,b into 2a,b was usually achieved after 24 h. Attempts to employ 10 mol % loading of the pre-catalysts often led to incomplete conversion. Apparently, because of the heterogeneous nature of the mixture, only a small fraction of the cobalt salt actually formed the catalytically active species. Moreover, we found that it was important to activate the Zn/ZnI2 mixture prior addition of the substrate either by heating or by generation of ZnI2 in situ from the excess of Zn and I2. Variation of the solvent (MeCN, THF, DCE) and the reaction temperature (20–80 °C) had no significant influence on the reaction outcome.
![[1860-5397-13-62-i2]](/bjoc/content/inline/1860-5397-13-62-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cobalt-catalyzed cycloisomerizations of 1,11-dien-6-ynes.
Scheme 2: Cobalt-catalyzed cycloisomerizations of 1,11-dien-6-ynes.
Although the yields of 2a,b were good, the compounds were formed as roughly 1:1 mixtures of cis- and trans-isomers (cis-isomers were reported previously [11] which helped us to assign the structures of 2a,b). In attempt to improve the selectivity we used 1,2-bis(diphenylphosphino)ethane (dppe) as ligand instead of PPh3. However, this ligand completely changed the direction of the cyclization, converting 1a,b into the diene compounds 3a,b in 90–95% yields. These compounds were also obtained using a 1,2-bis(diphenylphosphino)benzene (dppbz) ligand. Despite the extensive studies of enyne transformations, this type of cyclization has been achieved only recently, using a cyclobutadiene rhodium complex as a catalyst [43].
Next, we employed 1,3-bis(diphenylphosphino)propane (dppp) as a phosphine ligand, which surprisingly led to conversion of 1 into some new isomeric products. The 1H NMR spectra of these products contained a number of signals in the 0.5–0.8 ppm region, which is characteristic for cyclopropane protons. On the basis of 1H–1H and 1H–13C correlation NMR spectra as well as GC–MS analysis we had assigned the structures of these products as the cyclopropane derivatives with internal 4 and exocyclic 5 double bonds (Scheme 2). Our spectral data correlate well with that of similar compounds, which were recently prepared by the rhodium-catalyzed cyclopropanation [44]. Compounds 4 and 5 were obtained as mixtures with approximately 1:1 ratio and total 80–90% yields. Numerous attempts to convert 5 into the more stable isomer 4 using strong bases or transition metal catalysts were unsuccessful. The cobalt catalytic system with other phosphine ligands, such as dppm, dppf, or BINAP, did not catalyze cycloisomerization of 1 into any products.
In order to explain the formation of products 2–5 we proposed a possible mechanism (Scheme 3) in accordance with the generally accepted concepts [15,18]. The crucial step of this mechanism is the formation of metallacycle 6 from unsaturated cobalt(I) species [Co(PR3)2Br] and the double and the triple bond of dienyne 1 (see Supporting Information File 1 for the calculated structure of 6). Subsequent insertion of the double bond into the Co–C bond of 6 leads to the 7-membered metallacycle 7, which can undergo reductive elimination to give the cyclohexene 2, or β-hydride elimination followed by the reductive elimination to give the diene 3. It may be speculated that the steric crowding in the intermediate 7 determines the direction of the reaction. In contrast to the relatively small dppe ligand, two large Ph3P ligands hinder the β-elimination (which requires proximal syn-position of metal and hydrogen) and therefore push reaction towards direct reductive elimination to give 2.
![[1860-5397-13-62-i3]](/bjoc/content/inline/1860-5397-13-62-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Possible mechanism for formation of the compounds 2–5.
Scheme 3: Possible mechanism for formation of the compounds 2–5.
Alternatively, β-hydride elimination can proceed in the intermediate 6 to give the hydride species 8. Further, intramolecular 1,2- or 1,4-hydrometallation in 8 produces carbene complexes, which give cyclopropane derivatives 4 and 5.
In order to broaden the substrate scope, we studied similar reactions of the tetramethyl-substituted dienyne 1c (Scheme 4). As expected, it produced the cyclohexene derivative 2c in the presence of the triphenylphosphine ligand. More surprisingly, the same product was obtained when dppe or dppp were used as ligands, although the small amounts of isomeric by-products were detected in these cases. Noteworthy, according to the NMR spectra, product 2c was formed as a single diastereomer, however, it was not possible to unambiguously determine whether it was the cis- or the trans-isomer by NMR (including various correlation techniques) because of the high symmetry of both possible isomers. Attempts to grow crystals of 2c or its Br2 adduct for X-ray crystallography were also unsuccessful.
![[1860-5397-13-62-i4]](/bjoc/content/inline/1860-5397-13-62-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Cycloisomerization of the substituted dienyne 1c.
Scheme 4: Cycloisomerization of the substituted dienyne 1c.
Further experiments have shown that many other 1,11-dien-6-ynes 1d–l (Figure 1) did not undergo cycloisomerization in the presence of the cobalt catalytic system regardless of the employed ligand. In the case of the substrates 1d–h this may be explained by the increased steric crowding, which is evident from the 3D structure of the proposed intermediate 6 (see Supporting Information File 1). In the case of 1i–k the electron acceptor effects or the restricted rotation of conjugated enones could be the problem. The case of dienyne 1l was particularly surprising as it seemed to have a very similar structure to that of 1a,b. We can speculate that COOEt groups of 1l formed a chelate complex with ZnI2 and therefore strongly diminished its Lewis acidity, which is necessary for catalyst activation.
![[1860-5397-13-62-1]](/bjoc/content/figures/1860-5397-13-62-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The substituted 1,11-dien-6-ynes that did not undergo cycloisomerization in the presence of the cobalt catalytic system.
Figure 1: The substituted 1,11-dien-6-ynes that did not undergo cycloisomerization in the presence of the cob...
All the compounds obtained were characterized by 1H and 13C NMR spectroscopy as well as high-resolution mass-spectrometry (for ethers, which are oils) or elemental analysis (for tosylamides, which are crystalline solids). The structures of the starting dienyne 1h and the cyclohexene trans-2b were established by X-ray crystallography. The structures of cis-2b and 3b had been reported previously [11,43].
Conclusion
To conclude, we have shown that the behavior of the popular catalytic system CoBr2/phosphine ligand/Zn/ZnI2 is very sensitive to the structure of the phosphine ligand and to the structure of the substrate. On the one hand, this allows one to discover unusual transformations of the unsaturated substrates. On the other hand, such sensitivity hinders the development of general methods, which can be applied to a wide range of compounds. Overall, although the availability of the cobalt catalytic system is appealing, the development of a well-defined catalyst with precisely-controlled ligand environment is necessary for further progress.
Supporting Information
Experimental details and detailed spectroscopic data of all compounds are available as Supporting Information. Single crystal data for 1h and 2b (CCDC 1516680 and 1516681) has been deposited in the Cambridge Crystallographic Data Center.
| Supporting Information File 1: Experimental details and detailed spectroscopic data of all compounds. | ||
| Format: PDF | Size: 1.6 MB | Download |
| Supporting Information File 2: Single crystal data for 1h and 2b. | ||
| Format: ZIP | Size: 457.8 KB | Download |
References
-
Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268. doi:10.1002/anie.200701589
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319
Return to citation in text: [1] -
Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2011, 40, 3430. doi:10.1039/c1cs15029d
Return to citation in text: [1] -
Domínguez, G.; Pérez-Castells, J. Chem. – Eur. J. 2016, 22, 6720. doi:10.1002/chem.201504987
Return to citation in text: [1] -
Bruneau, C. Angew. Chem., Int. Ed. 2005, 44, 2328. doi:10.1002/anie.200462568
Return to citation in text: [1] -
Inglesby, P. A.; Evans, P. A. Chem. Soc. Rev. 2010, 39, 2791. doi:10.1039/b913110h
Return to citation in text: [1] -
Amatore, M.; Aubert, C. Eur. J. Org. Chem. 2015, 265. doi:10.1002/ejoc.201403012
Return to citation in text: [1] -
van Boxtel, L. J.; Körbe, S.; Noltemeyer, M.; de Meijere, A. Eur. J. Org. Chem. 2001, 2283. doi:10.1002/1099-0690(200106)2001:12<2283::AID-EJOC2283>3.0.CO;2-G
Return to citation in text: [1] [2] -
Himes, R. A.; Fanwick, P. E.; Rothwell, I. P. Chem. Commun. 2003, 18. doi:10.1039/B209889J
Return to citation in text: [1] -
Shibata, T.; Tahara, Y.-k. J. Am. Chem. Soc. 2006, 128, 11766. doi:10.1021/ja0639160
Return to citation in text: [1] -
Tanaka, D.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2007, 129, 7730. doi:10.1021/ja071954t
Return to citation in text: [1] [2] [3] [4] -
Sagae, H.; Noguchi, K.; Hirano, M.; Tanaka, K. Chem. Commun. 2008, 3804. doi:10.1039/b804294b
Return to citation in text: [1] [2] -
Clavier, H.; Correa, A.; Escudero-Adán, E. C.; Benet-Buchholz, J.; Cavallo, L.; Nolan, S. P. Chem. – Eur. J. 2009, 15, 10244. doi:10.1002/chem.200900976
Return to citation in text: [1] [2] -
Saito, N.; Tanaka, D.; Mori, M.; Sato, Y. Chem. Rec. 2011, 11, 186. doi:10.1002/tcr.201100003
Return to citation in text: [1] -
Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 10876. doi:10.1002/chem.200800904
Return to citation in text: [1] [2] -
Hess, W.; Treutwein, J.; Hilt, G. Synthesis 2008, 3537. doi:10.1055/s-0028-1083210
Return to citation in text: [1] -
Cahiez, G.; Moyeux, A. Chem. Rev. 2010, 110, 1435. doi:10.1021/cr9000786
Return to citation in text: [1] -
Gao, K.; Yoshikai, N. Acc. Chem. Res. 2014, 47, 1208. doi:10.1021/ar400270x
Return to citation in text: [1] [2] -
Pellissier, H.; Clavier, H. Chem. Rev. 2014, 114, 2775. doi:10.1021/cr4004055
Return to citation in text: [1] -
Gandeepan, P.; Cheng, C.-H. Acc. Chem. Res. 2015, 48, 1194. doi:10.1021/ar500463r
Return to citation in text: [1] -
Röse, P.; Hilt, G. Synthesis 2016, 463. doi:10.1055/s-0035-1560378
Return to citation in text: [1] -
Saino, N.; Amemiya, F.; Tanabe, E.; Kase, K.; Okamoto, S. Org. Lett. 2006, 8, 1439. doi:10.1021/ol0602295
Return to citation in text: [1] -
Sakurada, T.; Sugiyama, Y.-k.; Okamoto, S. J. Org. Chem. 2013, 78, 3583. doi:10.1021/jo400064b
Return to citation in text: [1] -
Geny, A.; Gaudrel, S.; Slowinski, F.; Amatore, M.; Chouraqui, G.; Malacria, M.; Aubert, C.; Gandon, V. Adv. Synth. Catal. 2009, 351, 271. doi:10.1002/adsc.200800646
Return to citation in text: [1] -
Achard, M.; Tenaglia, A.; Buono, G. Org. Lett. 2005, 7, 2353. doi:10.1021/ol050618j
Return to citation in text: [1] -
Toselli, N.; Martin, D.; Achard, M.; Tenaglia, A.; Bürgi, T.; Buono, G. Adv. Synth. Catal. 2008, 350, 280. doi:10.1002/adsc.200700424
Return to citation in text: [1] -
Nishimura, A.; Tamai, E.; Ohashi, M.; Ogoshi, S. Chem. – Eur. J. 2014, 20, 6613. doi:10.1002/chem.201402218
Return to citation in text: [1] -
Hilt, G.; Korn, T. J. Tetrahedron Lett. 2001, 42, 2783. doi:10.1016/S0040-4039(01)00289-1
Return to citation in text: [1] -
Hilt, G.; Janikowski, J.; Hess, W. Angew. Chem., Int. Ed. 2006, 45, 5204. doi:10.1002/anie.200601974
Return to citation in text: [1] -
Kuttner, J. R.; Warratz, S.; Hilt, G. Synthesis 2012, 1293. doi:10.1055/s-0031-1289752
Return to citation in text: [1] -
Fiebig, L.; Kuttner, J.; Hilt, G.; Schwarzer, M. C.; Frenking, G.; Schmalz, H.-G.; Schäfer, M. J. Org. Chem. 2013, 78, 10485. doi:10.1021/jo402001g
Return to citation in text: [1] -
Hilt, G.; Janikowski, J.; Schwarzer, M.; Burghaus, O.; Sakow, D.; Bröring, M.; Drüschler, M.; Huber, B.; Roling, B.; Harms, K.; Frenking, G. J. Organomet. Chem. 2014, 749, 219. doi:10.1016/j.jorganchem.2013.09.020
Return to citation in text: [1] -
Arndt, M.; Dindaroğlu, M.; Schmalz, H.-G.; Hilt, G. Org. Lett. 2011, 13, 6236. doi:10.1021/ol202696n
Return to citation in text: [1] -
Mannathan, S.; Cheng, C.-H. Chem. – Eur. J. 2012, 18, 11771. doi:10.1002/chem.201200779
Return to citation in text: [1] -
Hilt, G.; Paul, A.; Treutwein, J. Org. Lett. 2010, 12, 1536. doi:10.1021/ol100266u
Return to citation in text: [1] -
Pünner, F.; Schmidt, A.; Hilt, G. Angew. Chem., Int. Ed. 2012, 51, 1270. doi:10.1002/anie.201107512
Return to citation in text: [1] -
Schmidt, A.; Nödling, A. R.; Hilt, G. Angew. Chem., Int. Ed. 2015, 54, 801. doi:10.1002/anie.201409902
Return to citation in text: [1] -
Chang, H.-T.; Thiruvellore, T. J.; Wang, C.-C.; Cheng, C.-H. J. Am. Chem. Soc. 2007, 129, 12032. doi:10.1021/ja073604c
Return to citation in text: [1] -
Wei, C.-H.; Mannathan, S.; Cheng, C.-H. J. Am. Chem. Soc. 2011, 133, 6942. doi:10.1021/ja201827j
Return to citation in text: [1] -
Wei, C.-H.; Mannathan, S.; Cheng, C.-H. Angew. Chem., Int. Ed. 2012, 51, 10592. doi:10.1002/anie.201205115
Return to citation in text: [1] -
Santhoshkumar, R.; Mannathan, S.; Cheng, C.-H. Org. Lett. 2014, 16, 4208. doi:10.1021/ol501904e
Return to citation in text: [1] -
Santhoshkumar, R.; Mannathan, S.; Cheng, C.-H. J. Am. Chem. Soc. 2015, 137, 16116. doi:10.1021/jacs.5b10447
Return to citation in text: [1] -
Perekalin, D. S.; Shvydkiy, N. V.; Nelyubina, Y. V.; Kudinov, A. R. Chem. – Eur. J. 2015, 21, 16344. doi:10.1002/chem.201503270
Return to citation in text: [1] [2] -
Torres, O.; Roglans, A.; Pla-Quintana, A. Adv. Synth. Catal. 2016, 358, 3512. doi:10.1002/adsc.201600789
Return to citation in text: [1]
| 1. | Michelet, V.; Toullec, P. Y.; Genêt, J.-P. Angew. Chem., Int. Ed. 2008, 47, 4268. doi:10.1002/anie.200701589 |
| 2. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326. doi:10.1021/cr0684319 |
| 3. | Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2011, 40, 3430. doi:10.1039/c1cs15029d |
| 4. | Domínguez, G.; Pérez-Castells, J. Chem. – Eur. J. 2016, 22, 6720. doi:10.1002/chem.201504987 |
| 15. | Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 10876. doi:10.1002/chem.200800904 |
| 16. | Hess, W.; Treutwein, J.; Hilt, G. Synthesis 2008, 3537. doi:10.1055/s-0028-1083210 |
| 17. | Cahiez, G.; Moyeux, A. Chem. Rev. 2010, 110, 1435. doi:10.1021/cr9000786 |
| 18. | Gao, K.; Yoshikai, N. Acc. Chem. Res. 2014, 47, 1208. doi:10.1021/ar400270x |
| 19. | Pellissier, H.; Clavier, H. Chem. Rev. 2014, 114, 2775. doi:10.1021/cr4004055 |
| 20. | Gandeepan, P.; Cheng, C.-H. Acc. Chem. Res. 2015, 48, 1194. doi:10.1021/ar500463r |
| 21. | Röse, P.; Hilt, G. Synthesis 2016, 463. doi:10.1055/s-0035-1560378 |
| 15. | Jeganmohan, M.; Cheng, C.-H. Chem. – Eur. J. 2008, 14, 10876. doi:10.1002/chem.200800904 |
| 18. | Gao, K.; Yoshikai, N. Acc. Chem. Res. 2014, 47, 1208. doi:10.1021/ar400270x |
| 8. | van Boxtel, L. J.; Körbe, S.; Noltemeyer, M.; de Meijere, A. Eur. J. Org. Chem. 2001, 2283. doi:10.1002/1099-0690(200106)2001:12<2283::AID-EJOC2283>3.0.CO;2-G |
| 11. | Tanaka, D.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2007, 129, 7730. doi:10.1021/ja071954t |
| 12. | Sagae, H.; Noguchi, K.; Hirano, M.; Tanaka, K. Chem. Commun. 2008, 3804. doi:10.1039/b804294b |
| 13. | Clavier, H.; Correa, A.; Escudero-Adán, E. C.; Benet-Buchholz, J.; Cavallo, L.; Nolan, S. P. Chem. – Eur. J. 2009, 15, 10244. doi:10.1002/chem.200900976 |
| 11. | Tanaka, D.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2007, 129, 7730. doi:10.1021/ja071954t |
| 43. | Perekalin, D. S.; Shvydkiy, N. V.; Nelyubina, Y. V.; Kudinov, A. R. Chem. – Eur. J. 2015, 21, 16344. doi:10.1002/chem.201503270 |
| 8. | van Boxtel, L. J.; Körbe, S.; Noltemeyer, M.; de Meijere, A. Eur. J. Org. Chem. 2001, 2283. doi:10.1002/1099-0690(200106)2001:12<2283::AID-EJOC2283>3.0.CO;2-G |
| 9. | Himes, R. A.; Fanwick, P. E.; Rothwell, I. P. Chem. Commun. 2003, 18. doi:10.1039/B209889J |
| 10. | Shibata, T.; Tahara, Y.-k. J. Am. Chem. Soc. 2006, 128, 11766. doi:10.1021/ja0639160 |
| 11. | Tanaka, D.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2007, 129, 7730. doi:10.1021/ja071954t |
| 12. | Sagae, H.; Noguchi, K.; Hirano, M.; Tanaka, K. Chem. Commun. 2008, 3804. doi:10.1039/b804294b |
| 13. | Clavier, H.; Correa, A.; Escudero-Adán, E. C.; Benet-Buchholz, J.; Cavallo, L.; Nolan, S. P. Chem. – Eur. J. 2009, 15, 10244. doi:10.1002/chem.200900976 |
| 14. | Saito, N.; Tanaka, D.; Mori, M.; Sato, Y. Chem. Rec. 2011, 11, 186. doi:10.1002/tcr.201100003 |
| 43. | Perekalin, D. S.; Shvydkiy, N. V.; Nelyubina, Y. V.; Kudinov, A. R. Chem. – Eur. J. 2015, 21, 16344. doi:10.1002/chem.201503270 |
| 5. | Bruneau, C. Angew. Chem., Int. Ed. 2005, 44, 2328. doi:10.1002/anie.200462568 |
| 6. | Inglesby, P. A.; Evans, P. A. Chem. Soc. Rev. 2010, 39, 2791. doi:10.1039/b913110h |
| 7. | Amatore, M.; Aubert, C. Eur. J. Org. Chem. 2015, 265. doi:10.1002/ejoc.201403012 |
| 44. | Torres, O.; Roglans, A.; Pla-Quintana, A. Adv. Synth. Catal. 2016, 358, 3512. doi:10.1002/adsc.201600789 |
| 35. | Hilt, G.; Paul, A.; Treutwein, J. Org. Lett. 2010, 12, 1536. doi:10.1021/ol100266u |
| 38. | Chang, H.-T.; Thiruvellore, T. J.; Wang, C.-C.; Cheng, C.-H. J. Am. Chem. Soc. 2007, 129, 12032. doi:10.1021/ja073604c |
| 39. | Wei, C.-H.; Mannathan, S.; Cheng, C.-H. J. Am. Chem. Soc. 2011, 133, 6942. doi:10.1021/ja201827j |
| 40. | Wei, C.-H.; Mannathan, S.; Cheng, C.-H. Angew. Chem., Int. Ed. 2012, 51, 10592. doi:10.1002/anie.201205115 |
| 41. | Santhoshkumar, R.; Mannathan, S.; Cheng, C.-H. Org. Lett. 2014, 16, 4208. doi:10.1021/ol501904e |
| 42. | Santhoshkumar, R.; Mannathan, S.; Cheng, C.-H. J. Am. Chem. Soc. 2015, 137, 16116. doi:10.1021/jacs.5b10447 |
| 33. | Arndt, M.; Dindaroğlu, M.; Schmalz, H.-G.; Hilt, G. Org. Lett. 2011, 13, 6236. doi:10.1021/ol202696n |
| 34. | Mannathan, S.; Cheng, C.-H. Chem. – Eur. J. 2012, 18, 11771. doi:10.1002/chem.201200779 |
| 11. | Tanaka, D.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2007, 129, 7730. doi:10.1021/ja071954t |
| 28. | Hilt, G.; Korn, T. J. Tetrahedron Lett. 2001, 42, 2783. doi:10.1016/S0040-4039(01)00289-1 |
| 29. | Hilt, G.; Janikowski, J.; Hess, W. Angew. Chem., Int. Ed. 2006, 45, 5204. doi:10.1002/anie.200601974 |
| 30. | Kuttner, J. R.; Warratz, S.; Hilt, G. Synthesis 2012, 1293. doi:10.1055/s-0031-1289752 |
| 31. | Fiebig, L.; Kuttner, J.; Hilt, G.; Schwarzer, M. C.; Frenking, G.; Schmalz, H.-G.; Schäfer, M. J. Org. Chem. 2013, 78, 10485. doi:10.1021/jo402001g |
| 32. | Hilt, G.; Janikowski, J.; Schwarzer, M.; Burghaus, O.; Sakow, D.; Bröring, M.; Drüschler, M.; Huber, B.; Roling, B.; Harms, K.; Frenking, G. J. Organomet. Chem. 2014, 749, 219. doi:10.1016/j.jorganchem.2013.09.020 |
| 22. | Saino, N.; Amemiya, F.; Tanabe, E.; Kase, K.; Okamoto, S. Org. Lett. 2006, 8, 1439. doi:10.1021/ol0602295 |
| 23. | Sakurada, T.; Sugiyama, Y.-k.; Okamoto, S. J. Org. Chem. 2013, 78, 3583. doi:10.1021/jo400064b |
| 24. | Geny, A.; Gaudrel, S.; Slowinski, F.; Amatore, M.; Chouraqui, G.; Malacria, M.; Aubert, C.; Gandon, V. Adv. Synth. Catal. 2009, 351, 271. doi:10.1002/adsc.200800646 |
| 25. | Achard, M.; Tenaglia, A.; Buono, G. Org. Lett. 2005, 7, 2353. doi:10.1021/ol050618j |
| 26. | Toselli, N.; Martin, D.; Achard, M.; Tenaglia, A.; Bürgi, T.; Buono, G. Adv. Synth. Catal. 2008, 350, 280. doi:10.1002/adsc.200700424 |
| 27. | Nishimura, A.; Tamai, E.; Ohashi, M.; Ogoshi, S. Chem. – Eur. J. 2014, 20, 6613. doi:10.1002/chem.201402218 |
| 36. | Pünner, F.; Schmidt, A.; Hilt, G. Angew. Chem., Int. Ed. 2012, 51, 1270. doi:10.1002/anie.201107512 |
| 37. | Schmidt, A.; Nödling, A. R.; Hilt, G. Angew. Chem., Int. Ed. 2015, 54, 801. doi:10.1002/anie.201409902 |
© 2017 Suleymanov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)