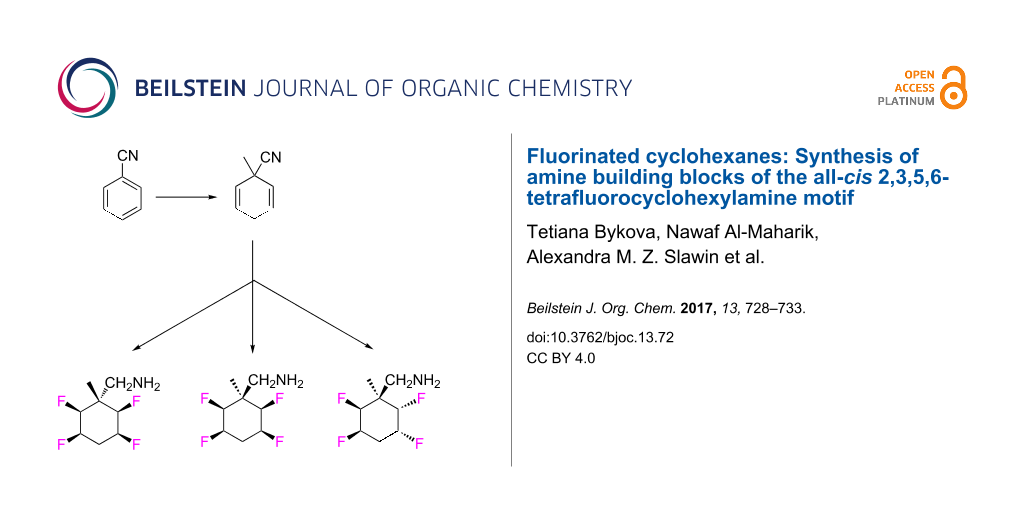
Abstract
This paper reports the synthesis of three amine stereoisomers 5a–c of the tetrafluorocyclohexyl ring system, as building blocks for discovery chemistry programmes. The synthesis starts from a Birch reduction of benzonitrile, followed by an in situ methyl iodide quench. The resultant 2,5-cyclohexadiene was progressed via double epoxidations and then hydrofluorination ring opening reactions. The resultant fluorohydrin moieties were then converted to different stereoisomers of the tetrafluorocyclohexyl ring system, and then reductive hydrogenation of the nitrile delivered three amine stereoisomers. It proved necessary to place a methyl group on the cyclohexane ring in order to stabilise the compound against subsequent HF elimination. The two all-cis tetrafluorocyclohexyl isomers 5a and 5b constitute facially polarized cyclohexane rings, with fluorines on the electronegative face and hydrogens on the electropositive face.

Graphical Abstract
Introduction
The all-cis-2,3,5,6-tetrafluorocyclohexane 1 ring has been introduced recently as a polarized cyclohexane ring and it has been a focus of our research group to elaborate new building blocks that enable the introduction of this motif into organic discovery programmes (Figure 1) [1-3]. The cyclohexane ring has four C–F bonds on one face, two of which are aligned 1,3-diaxial. Due to the particularly polar nature of the C–F bond, the alignment of those two bonds results in a large molecular dipole moment [4-6]. For the parent cyclohexane ring 1 the magnitude of the molecular dipole is 5.2 D [2]. The motif has the unique property of inducing facial polarity to the ring system [2,7]. The nature of the interaction of this ring system with protein targets remains to be examined, and its incorporation into organic materials is in its infancy. Access to this motif by the wider research community requires that a range of building blocks be prepared. It has proven relatively straightforward to prepare the phenyl derivative 2 and then subsequent elaboration to a range of functionalized analogues by standard electrophilic aromatic substitution reactions (Figure 1) [1,8-10]. A greater challenge involved the preparation of the tetrafluorocyclohexane ring derivatives without the attached aromatic ring. We have recently demonstrated that both diastereoisomers of alcohols 3 and their corresponding aldehydes 4 (Figure 1), could be prepared after a Birch reduction on benzoic acid, quenching with methyl iodide, and then subsequent conversion of the cyclohexadiene product to a tetrafluorocycloheane motif [11]. The methyl group was a design feature to block hydrogen fluoride elimination from the position alpha to the aldehyde. The diastereoisomers of all cis-tetrafluorocyclohexane aldehydes 4 were used successfully in Ugi multicomponent reactions [11,12]. In this paper we report the preparation of amines of this series starting from a Birch reduction on benzonitrile, and with a similar methyl iodide quench, to generate amines 5, which are stable to hydrogen fluoride elimination.
![[1860-5397-13-72-1]](/bjoc/content/figures/1860-5397-13-72-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The derivatives of all-cis-2,3,5,6-tetrafluorocyclohexane.
Figure 1: The derivatives of all-cis-2,3,5,6-tetrafluorocyclohexane.
Results and Discussion
The Birch reduction of benzonitrile 6 followed by in situ methylation with iodomethane generated cyclohexadiene 7 as previously described [13,14]. Cyclohexadiene 7 was then subjected to a double epoxidation protocol using mCPBA [1,11,15,16]. This generated three diastereoisomers of 8, two of which have the meso diepoxides syn and anti in relation to the nitrile functional group (8a and 8b) and a racemic 8c diepoxide with trans configuration. These isomers were generated in a ratio of 10:15:13 (8a:8b:8c) (Scheme 1) [15]. Diepoxide 8a was isolated by chromatography (18% yield), however, diepoxides 8b and 8c (35%) could not be separated, and therefore were taken as a mixture of isomers to the next step in the reaction sequence.
![[1860-5397-13-72-i1]](/bjoc/content/inline/1860-5397-13-72-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reagents and conditions: a) Li (2.5 equiv), NH3, t-BuOH (1 equiv), MeI (2 equiv), 3 h, −78 °C, 18 h, rt, 31%; b) mCPBA (4.9 equiv), DCM, 48 h, 35 °C, 18% (8a), 35% (8b and 8c), ratio 8a:8b:8c: 10:15:13.
Scheme 1: Reagents and conditions: a) Li (2.5 equiv), NH3, t-BuOH (1 equiv), MeI (2 equiv), 3 h, −78 °C, 18 h...
Treatment of 8a with Et3N·3HF at 140 °C resulted in its full conversion to the hydrofluorinated ring-opened diol 9a as a single regioisomer, a product which was treated directly with triflic anhydride in pyridine, to generate ditriflate 10a in a 30% yield over two steps (Scheme 2). We have found that related treatments with the more acidic Olah’s reagent (HF·pyridine) were less satisfactory generating product mixtures [2,17].
![[1860-5397-13-72-i2]](/bjoc/content/inline/1860-5397-13-72-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reagents and conditions: a) Et3N·3HF (8 equiv), 18 h, 140 °C; b) Tf2O (4 equiv), pyridine, 1 h, 0 °C, 3 h, rt, 30%; c) Et3N·3HF (10 equiv), 4 days, 120 °C, 30%; d) NaBH4 (10 equiv)/NiCl2·6H2O (5 equiv), MeOH, 1 h, 0 °C, 18 h, rt, 50%; e) 10% Pd/C (10 mol %), H2, Et3N/CHOOH: molar ratio 1:37, THF, 18 h, rt, 78%.
Scheme 2: Reagents and conditions: a) Et3N·3HF (8 equiv), 18 h, 140 °C; b) Tf2O (4 equiv), pyridine, 1 h, 0 °...
Treatment of 10a with Et3N·3HF at 120 °C generated the tetrafluorocyclohexane 11a, the structure of which was confirmed by X-ray crystallography (Scheme 2). Although the conversion of 10a to 11a was high as judged by 19F NMR, the isolated yield was modest as the compound was volatile and sublimed easily under reduced pressure. Hydrogenation of 11a over 10% Pd/C in ethyl acetate gave the desired amine 5a but in very low yield (0–15%) [18-20]. The structure of 5a was confirmed by X-ray crystallography (Scheme 2). Adding a few drops of formic acid and triethylamine (molar ratio 37:1) to the hydrogenation, furnished formamide 12 in 78% yield, a compound also confirmed by X-ray crystallography [20]. Ultimately treatment of nitrile 11a with nickel boride generated in situ from nickel chloride and sodium borohydride, resulted in its full reduction to amine 5a in 50% yield (Scheme 2) [21,22]. The analogous protocol was then applied to diastereoisomers 8b and 8c as illustrated in Scheme 3. Fluorination of the isomer mixture 8b/8c with Et3N·3HF at 140 °C gave 9b and the racemic 9c as an inseparable mixture. Triflation of this product mixture generated 10b and 10c, isomeric products which could now be separated by chromatography. Finally fluorination of 10b and 10c in separate reactions with Et3N·3HF at 120 °C furnished nitrile 11b (51% yield) and 11c (31% yield) respectively.
![[1860-5397-13-72-i3]](/bjoc/content/inline/1860-5397-13-72-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reagents and conditions: a) Et3N·3HF (8 equiv), 18 h, 140 °C; b) Tf2O (4 equiv), pyridine, 1 h, 0 °C, 3 h, rt, 40% (10b), 15% (10c); c) Et3N·3HF (10 equiv), 4 days, 120 °C, 51% (11b), 31% (11c); d) NaBH4 (10 equiv)/NiCl2·6H2O (5 equiv), MeOH, 1 h, 0 °C, 18 h, rt, 65% (5b), 23% (5c).
Scheme 3: Reagents and conditions: a) Et3N·3HF (8 equiv), 18 h, 140 °C; b) Tf2O (4 equiv), pyridine, 1 h, 0 °...
The structure and stereochemistry of both products were unambiguously confirmed by X-ray structure analysis (Scheme 3). Reduction of 11b with nickel boride delivered amine 5b in 65% yield as a crystalline solid and an analogous reduction of 11c generated the racemic amine 5c as a colourless liquid. The structure of 11b was confirmed by X-ray structure analysis (Scheme 3).
Amines 5a and 5b were reacted with terephthaloyl chloride as a means of preparing higher order, bis-amide systems to explore intermolecular packing of the cyclohexane rings in the solid state. To extend the study a comparison was also made with the ester derivative from the previously prepared alcohol 3a [11].
Treatment of 2.1 equivalents of 2,3,5,6-tetrafluorocyclohexane derivatives 5a, 5b and 3a with 1 equivalent of terephthaloyl chloride, in the presence of triethylamine (4 equiv) and DMAP resulted in bis-systems 13, 14 and 15 in 79%, 48%, and 88% yields respectively (Scheme 4) [23,24]. All of the above compounds were found to be solid materials with low solubility. However suitable crystals were obtained from acetone/acetonitrile for X-ray structure analysis.
![[1860-5397-13-72-i4]](/bjoc/content/inline/1860-5397-13-72-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reagents and conditions: a) terephthaloyl chloride (1 equiv), Et3N (4 equiv), DMAP (20 mol %), DCM, 18 h, rt, 79% (13), 48% (14), 88% (15).
Scheme 4: Reagents and conditions: a) terephthaloyl chloride (1 equiv), Et3N (4 equiv), DMAP (20 mol %), DCM,...
The X-ray structures of 13, 14 and 15 are illustrated in Figure 2. It was assumed that the facial polarity of the tetrafluorocyclohexane rings would be apparent in the molecular ordering in the solid state, with intercalations between negative fluorous faces and positive protic faces of the rings. This was the case as can be seen from the packing structures in Figure 2. For compounds 13 and 14 intermolecular hydrogen bonding between the amide groups also contributes to this stacking structure but for compound 15, there are no amides to stabilize such a structure, and the facially polarized cyclohexane rings presumably form the strongest intermolecular interactions in this case (Figure 2).
![[1860-5397-13-72-2]](/bjoc/content/figures/1860-5397-13-72-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray structures and crystal packing of compounds 13, 14 and 15.
Figure 2: X-ray structures and crystal packing of compounds 13, 14 and 15.
Conclusion
In summary we report a method for the preparation of 2,3,5,6-tetrafluorocyclohexane amines 5a, 5b and 5c, through a synthesis sequence starting from the Birch reduction of benzonitrile. Bis-compounds 13–15 were readily prepared from amines 5a, 5b and an alcohol 3a in reactions with terephthaloyl chloride. The structures of which indicate arrangements in the solid state consistent with electrostatic ordering of the cyclohexane rings. These polarized cyclohexyl derivatives should prove valuable as potential building blocks in drug discovery and agrochemistry research programs [3,25].
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.0 MB | Download |
References
-
Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354
Return to citation in text: [1] [2] [3] -
Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f
Return to citation in text: [1] [2] [3] [4] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] [2] -
O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/B711844A
Return to citation in text: [1] -
Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910
Return to citation in text: [1] -
Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O'Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232
Return to citation in text: [1] -
Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2011, 47, 8265–8267. doi:10.1039/c1cc13016a
Return to citation in text: [1] -
Durie, A. J.; Fujiwara, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. J. Org. Chem. 2014, 79, 8228–8233. doi:10.1021/jo501432x
Return to citation in text: [1] -
Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2015, 11, 2671–2676. doi:10.3762/bjoc.11.287
Return to citation in text: [1] -
Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2015, 13, 5621–5624. doi:10.1039/C5OB00650C
Return to citation in text: [1] -
Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2016, 14, 1117–1123. doi:10.1039/C5OB02334C
Return to citation in text: [1] [2] [3] [4] -
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] -
Ohkata, K.; Tamura, Y.; Shetuni, B. B.; Takagi, R.; Miyanaga, W.; Kojima, S.; Paquette, L. A. J. Am. Chem. Soc. 2004, 126, 16783–16792. doi:10.1021/ja047027t
Return to citation in text: [1] -
Birch, A. J. Pure Appl. Chem. 1996, 68, 553–556. doi:10.1351/pac199668030553
Return to citation in text: [1] -
Schwartz, N. N.; Blumbergs, J. H. J. Org. Chem. 1964, 29, 1976–1979. doi:10.1021/jo01030a078
Return to citation in text: [1] [2] -
Prileschajew, N. Ber. Dtsch. Chem. Ges. 1909, 42, 4811–4815. doi:10.1002/cber.190904204100
Return to citation in text: [1] -
Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. J. Fluorine Chem. 2015, 179, 188–192. doi:10.1016/j.jfluchem.2015.08.003
Return to citation in text: [1] -
Vilches-Herrera, M.; Werkmeister, S.; Junge, K.; Börner, A.; Beller, M. Catal. Sci. Technol. 2014, 4, 629–632. doi:10.1039/c3cy00854a
Return to citation in text: [1] -
Johnson, D. C., II; Widlanski, T. S. Org. Lett. 2004, 6, 4643–4646. doi:10.1021/ol048426w
Return to citation in text: [1] -
Cortese, N. A.; Heck, R. F. J. Org. Chem. 1977, 42, 3491–3494. doi:10.1021/jo00442a008
Return to citation in text: [1] [2] -
Magoo, D. Synlett 2010, 2525–2526. doi:10.1055/s-0030-1258550
Return to citation in text: [1] -
Alcaide, B.; Almendros, P.; Cabrero, G.; Ruiz, M. P. J. Org. Chem. 2007, 72, 7980–7991. doi:10.1021/jo701452a
Return to citation in text: [1] -
Khan, G. S.; Dickson, B. D.; Barker, D. Tetrahedron 2012, 68, 1790–1801. doi:10.1016/j.tet.2011.12.030
Return to citation in text: [1] -
Scriven, E. F. V. Chem. Soc. Rev. 1983, 12, 129–161. doi:10.1039/cs9831200129
Return to citation in text: [1] -
Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007
Return to citation in text: [1]
| 3. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 25. | Kirk, K. L. J. Fluorine Chem. 2006, 127, 1013–1029. doi:10.1016/j.jfluchem.2006.06.007 |
| 1. | Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354 |
| 2. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f |
| 3. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 1. | Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354 |
| 8. | Durie, A. J.; Fujiwara, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. J. Org. Chem. 2014, 79, 8228–8233. doi:10.1021/jo501432x |
| 9. | Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Beilstein J. Org. Chem. 2015, 11, 2671–2676. doi:10.3762/bjoc.11.287 |
| 10. | Ayoup, M. S.; Cordes, D. B.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2015, 13, 5621–5624. doi:10.1039/C5OB00650C |
| 11. | Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2016, 14, 1117–1123. doi:10.1039/C5OB02334C |
| 2. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f |
| 7. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2011, 47, 8265–8267. doi:10.1039/c1cc13016a |
| 23. | Khan, G. S.; Dickson, B. D.; Barker, D. Tetrahedron 2012, 68, 1790–1801. doi:10.1016/j.tet.2011.12.030 |
| 24. | Scriven, E. F. V. Chem. Soc. Rev. 1983, 12, 129–161. doi:10.1039/cs9831200129 |
| 2. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f |
| 20. | Cortese, N. A.; Heck, R. F. J. Org. Chem. 1977, 42, 3491–3494. doi:10.1021/jo00442a008 |
| 4. | O’Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/B711844A |
| 5. | Biffinger, J. C.; Kim, H. W.; DiMagno, S. G. ChemBioChem 2004, 5, 622–627. doi:10.1002/cbic.200300910 |
| 6. | Keddie, N. S.; Slawin, A. M. Z.; Lebl, T.; Philp, D.; O'Hagan, D. Nat. Chem. 2015, 7, 483–488. doi:10.1038/nchem.2232 |
| 21. | Magoo, D. Synlett 2010, 2525–2526. doi:10.1055/s-0030-1258550 |
| 22. | Alcaide, B.; Almendros, P.; Cabrero, G.; Ruiz, M. P. J. Org. Chem. 2007, 72, 7980–7991. doi:10.1021/jo701452a |
| 1. | Durie, A. J.; Fujiwara, T.; Cormanich, R.; Bühl, M.; Slawin, A. M. Z.; O’Hagan, D. Chem. – Eur. J. 2014, 20, 6259–6263. doi:10.1002/chem.201400354 |
| 11. | Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2016, 14, 1117–1123. doi:10.1039/C5OB02334C |
| 15. | Schwartz, N. N.; Blumbergs, J. H. J. Org. Chem. 1964, 29, 1976–1979. doi:10.1021/jo01030a078 |
| 16. | Prileschajew, N. Ber. Dtsch. Chem. Ges. 1909, 42, 4811–4815. doi:10.1002/cber.190904204100 |
| 2. | Durie, A. J.; Slawin, A. M. Z.; Lebl, T.; Kirsch, P.; O’Hagan, D. Chem. Commun. 2012, 48, 9643–9645. doi:10.1039/c2cc34679f |
| 17. | Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. J. Fluorine Chem. 2015, 179, 188–192. doi:10.1016/j.jfluchem.2015.08.003 |
| 13. | Ohkata, K.; Tamura, Y.; Shetuni, B. B.; Takagi, R.; Miyanaga, W.; Kojima, S.; Paquette, L. A. J. Am. Chem. Soc. 2004, 126, 16783–16792. doi:10.1021/ja047027t |
| 14. | Birch, A. J. Pure Appl. Chem. 1996, 68, 553–556. doi:10.1351/pac199668030553 |
| 18. | Vilches-Herrera, M.; Werkmeister, S.; Junge, K.; Börner, A.; Beller, M. Catal. Sci. Technol. 2014, 4, 629–632. doi:10.1039/c3cy00854a |
| 19. | Johnson, D. C., II; Widlanski, T. S. Org. Lett. 2004, 6, 4643–4646. doi:10.1021/ol048426w |
| 20. | Cortese, N. A.; Heck, R. F. J. Org. Chem. 1977, 42, 3491–3494. doi:10.1021/jo00442a008 |
| 11. | Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2016, 14, 1117–1123. doi:10.1039/C5OB02334C |
| 12. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 11. | Bykova, T.; Al-Maharik, N.; Slawin, A. M. Z.; O’Hagan, D. Org. Biomol. Chem. 2016, 14, 1117–1123. doi:10.1039/C5OB02334C |
| 15. | Schwartz, N. N.; Blumbergs, J. H. J. Org. Chem. 1964, 29, 1976–1979. doi:10.1021/jo01030a078 |
© 2017 Bykova et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)