Abstract
The alkynylbenziodoxole derivatives are recently developed alkynylation reagents in organic synthesis, which demonstrate excellent radical alkynylation reactivity in photoredox catalysis reactions. Herein we report the synthesis of alkynylbenziodoxole derivatives with difluoro, monofluoro, monomethoxy, and dimethoxy substitution on the benziodoxole moiety, and investigated their radical alkynylation reactivity for the first time. A series of mechanistic experiments were conducted to study the radical acceptor and oxidative quencher reactivity of alkynylbenziodoxoles, in which unsubstituted alkynylbenziodoxoles played balancing roles in both processes, while electron-rich benziodoxole derivatives demonstrate synthetic advantages in some cases.

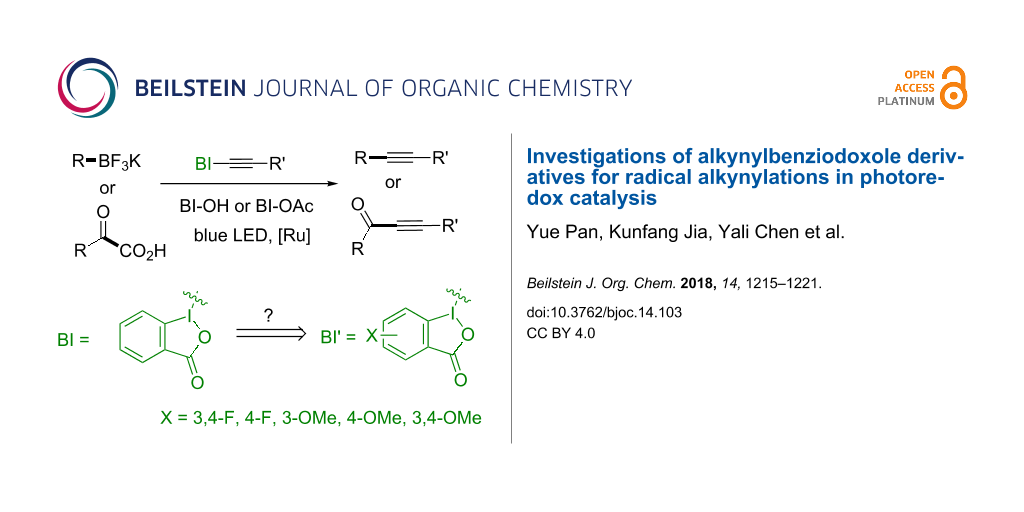
Graphical Abstract
Introduction
The introduction of the alkynyl group to organic molecules is an important synthetic transformation in organic synthesis [1-4]. Recently, cyclic iodine(III) reagents (CIR)-substituted alkynes, alkynylbenziodoxoles, were developed with readily preparation and shelf-stableness [5-10]. The alkynylbenziodoxoles were first synthesized by the Ochiai group, and later studied by Waser and other groups for the use in electrophilic alkynylation reactions [11-18]. In 2012, the Li group first used alkynylbenziodoxoles for decarboxylative radical alkynylation under silver salt and persulfate conditions [19]. In 2014, the Chen group discovered that alkynylbenziodoxoles (BI-alkyne) readily participated in photoredox catalysis as the radical alkynylation reagent [20], after which various applications in photoredox catalysis were reported [21-27].
Currently, the use of BI-alkyne for radical alkynylation is limited to unsubstituted alkynylbenziodoxoles. While effective, its reactivity with some radical precursors was compromised [19-27]. The Waser group pioneered the study of substituted alkynylbenziodoxoles for the electrophilic alkynylation reactivity, however, no significant improvements were observed by the derivatizations [28-32]. Herein, we report the synthesis of alkynylbenziodoxole derivatives and investigate their reactivity toward alkyl radical and acyl radical additions in photoredox catalysis. The mechanistic investigations were carried out to study the derivatization of BI-alkynes in radical acceptor and oxidative quencher reactivity, and the electron-rich benziodoxole derivatives demonstrated synthetic advantages in some cases (Scheme 1).
![[1860-5397-14-103-i1]](/bjoc/content/inline/1860-5397-14-103-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Investigation of alkynylbenziodoxole derivatives for radical alkynylations.
Scheme 1: Investigation of alkynylbenziodoxole derivatives for radical alkynylations.
Results and Discussion
We started the synthesis of BI-alkyne derivatives with substituted o-iodobenzoic acids 1 bearing 3,4-difluoro, 4-fluoro, 3-methoxy, 4-methoxy, or 3,4-dimethoxy substitutions (Scheme 2). Using a slightly modified Ochiai procedure [11], the substituted hydroxybenziodoxoles 2a–f were prepared with periodate oxidation in 75–90% yield, in which the electronic effect did not have much influence on the reaction [33]. Subsequently, the treatment with trimethylsilyl p-tolylacetylene in the presence of trimethylsilyl trifluoromethanesulfonate afforded p-tolylacetylenic benziodoxoles 3a–f in 25–65% yield, in which the electron-donating substitutions were beneficial for the reaction. The two-step synthesis of BI’-alkyne derivatives 3a,b,d–f were in the range of 23–50% yield in gram scale, which was comparable to the synthesis of unsubstituted BI-alkyne 3c.
![[1860-5397-14-103-i2]](/bjoc/content/inline/1860-5397-14-103-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and characterization of BI-alkyne derivatives 3a–f.
Scheme 2: Synthesis and characterization of BI-alkyne derivatives 3a–f.
The 13C NMR spectra of BI-alkynes 3a–f were studied with the focus on the α-carbon, which position directly underwent α-radical addition [19]. The electron density of the α-carbon was decreased in 3a with electron-deficient 3,4-difluoro groups on the benziodoxole, and was increased for 3f with electron-donating 3,4-dimethoxy groups. Cyclic voltammetry measurements were also carried out for BI-alkynes 3a–f, in which the reduction potential (Ep re) indicated the electron-accepting capacity of the BI-alkynes. As expected, the reduction potential of 3a was increased with electron-deficient 3,4-difluoro substituents on the benziodoxole, and was decreased for 3f with electron-donating 3,4-dimethoxy groups. It is interesting to note that the effect of single substitution on benziodoxoles in 3b, 3d and 3e was insignificant and sporadical in both 13C NMR spectroscopy and cyclic voltammetry experiments.
We next tested the reactivity of tolylacetylenic benziodoxole derivatives 3a–f for deboronative alkynylation under photoredox catalysis conditions (Scheme 3) [20,34]. Using the tertiary alkyl trifluoroborate 4a as the alkyl radical precursor under literature conditions, the unsubstituted BI-alkyne 3c only results in 50% yield of alkynylation adduct 5a, which is consistent with the literature report that tertiary alkyl trifluoroborates did not give satisfying results [20]. Using BI’-alkyne 3a with 3,4-difluoro substitutions, the alkynylation adduct 5a was obtained in decreased 47% yield. In contrast, 74% yield of 5a was obtained with 3,4-dimethoxy-substituted BI’-alkyne 3f. Being consistent with the 13C NMR spectroscopy and cyclic voltammetry experiments, the effect of single substitution on the benziodoxole was insignificant and no improvement was observed. We also tested the secondary alkyl trifluoroborate 4b and primary alkyl trifluoroborate 4c, in which the deboronative alkynylation with unsubstituted BI-alkynes already gave good results. The electronic effects on the benziodoxoles were less significant and fluctuated within the 5% yield range: The alkynylation adducts 5b and 5c were obtained in decreased 80% and 65% yields using BI’-alkyne 3a, while 86% and 73% yields of 5b and 5c were obtained with BI’-alkyne 3f. We then tested benzyl trifluoroborate 4d and oxygen-substituted alkyl trifluoroborate 4e, which were not reported for deboronative alkynylations before. The 3,4-dimethoxy-substituted BI’-alkyne 3f gave the optimal 70% and 82% yields of alkynes 5d and 5e, which observed ≈10% yield improvement comparing to the unsubstituted BI-alkyne 3c.
![[1860-5397-14-103-i3]](/bjoc/content/inline/1860-5397-14-103-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of alkynylbenziodoxole derivatives for deboronative alkynylation in photoredox catalysis. Reaction conditions: a) alkyl potassium trifluoroborate 4 (0.15 mmol, 1.5 equiv), alkynylbenziodoxole 3 (0.10 mmol, 1.0 equiv), Ru(bpy)3(PF6)2 (1.7 mg, 0.002 mmol, 0.02 equiv), hydroxybenziodoxole (BI-OH, 0.05 mmol, 0.5 equiv), and Na2CO3 (0.2 mmol, 2.0 equiv) in 1.0 mL CH2Cl2 and 1.0 mL H2O for 20 h under a nitrogen atmosphere, unless otherwise noted; b) 4 (0.3 mmol, 3.0 equiv), Na2CO3 (0.4 mmol, 4.0 equiv). Yields are isolated yields. N/P = not performed.
Scheme 3: Reaction of alkynylbenziodoxole derivatives for deboronative alkynylation in photoredox catalysis. ...
We then tested if the propensity of BI-alkyne derivatives toward alkyl radical additions was general and could extend to other alkyl radical precursors (Scheme 4). Tertiary alcohols 6 were reported to be activated by cyclic iodine(III) reagents under photoredox conditions to generate alkoxyl radicals, and subsequently underwent β-fragmentation and alkynylation to yield 7 after eliminating the arylketone [25]. With tertiary alcohol 6a as the alkyl radical precursor, the unsubstituted BI-alkyne 3c gave 74% yield of 7a, which was consistent with the literature report [25]. Under otherwise identical reaction conditions, 67% yield of 7a was obtained with 3,4-difluoro BI’-alkyne 3a, while optimal 85% yield of 7a was obtained with 3,4-dimethoxy BI’-alkyne 3f. We then tested the secondary alkyl radical precursor 6b and observed 74% yield of alkyne 5b using unsubstituted BI-alkyne 3c. In contrast, 3,4-dimethoxy BI'-alkyne 3f gave improved 80% yields of 5b.
![[1860-5397-14-103-i4]](/bjoc/content/inline/1860-5397-14-103-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of alkynylbenziodoxole derivatives for radical alkynylations in photoredox catalysis. Reaction conditions: tertiary alcohol 6 (0.25 mmol, 2.5 equiv), alkynylbenziodoxole 3 (0.10 mmol, 1.0 equiv), Ru(bpy)3(PF6)2 (0.002 mmol, 0.02 equiv), and BI-OAc (0.25 mmol, 2.5 equiv) in 2.0 mL DCE for 24 h under a nitrogen atmosphere, unless otherwise noted. Yields are isolated yields.
Scheme 4: Reaction of alkynylbenziodoxole derivatives for radical alkynylations in photoredox catalysis. Reac...
We finally moved to test the BI-alkyne derivatives toward acyl radical additions (Scheme 5). With ketoacid 8 as the acyl radical precursor, the decarboxylative alkynylation with BI-alkyne derivatives afforded ynone 9 under the photoredox conditions [21]. Both the unsubstituted and 3,4-dimethoxy substituted BI’-alkynes 3c and 3f gave ynone 9 in similar 77–79% yields, while the 3,4-difluoro substituted BI’-alkyne 3a gave a slightly lower 63% yield of 9 [21]. β-Ketone alcohols 10 were reported to be activated by cyclic iodine(III) reagents under photoredox conditions to generate alkoxyl radicals, and subsequently underwent β-fragmentation and alkynylation to yield ynone 9 [26]. The unsubstituted BI-alkyne 3c gave 84% yield of 9 consistent with the literature report, while 62% yield of 9 was obtained with 3,4-difluoro BI’-alkyne 3a and 84% yield of 9 was obtained with 3,4-dimethoxy BI’-alkyne 3f.
![[1860-5397-14-103-i5]](/bjoc/content/inline/1860-5397-14-103-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of alkynylbenziodoxole derivatives for acyl radical alkynylation in photoredox catalysis. Reaction conditions: a) ketoacid 8 (0.15 mmol, 1.5 equiv), alkynylbenziodoxole 3 (0.10 mmol, 1.0 equiv), Ru(bpy)3(PF6)2 (0.002 mmol, 0.02 equiv), and BIOAc (0.10 mmol, 1.0 equiv) in 2.0 mL DCM for 5 h under a nitrogen atmosphere; b) β-ketone alcohol 10 (0.20 mmol, 2.0 equiv), alkynylbenziodoxole 3 (0.10 mmol, 1.0 equiv), Ru(bpy)3(PF6)2 (0.002 mmol, 0.02 equiv), and BI-OAc (0.20 mmol, 2.0 equiv) in 2.0 mL DCM for 24 h under a nitrogen atmosphere. Yields are isolated yields.
Scheme 5: Reaction of alkynylbenziodoxole derivatives for acyl radical alkynylation in photoredox catalysis. ...
With the preliminary hypothesis that the electron-withdrawing and electron-donating substituents on the benziodoxole have opposite effects for the radical alkynylation, we first conducted the fluorescence quenching experiments of tolylacetylenic benziodoxole derivatives 3a–f and found none of them oxidatively quenched the photoexcited Ru(bpy)32+* complex (see Supporting Information File 1, Scheme S1). We next investigated if the benziodoxole radical released from the radical alkynylation of BI-alkynes affected the reaction (Scheme 6). Using the combination of substituted hydroxybenziodoxoles (BI’-OH) and substituted BI’-alkynes, we found the 3,4-difluoro electron-withdrawing substituents either on BI’-OH or BI’-alkyne decreased the reaction yields, while the use of both further decreased the formation of 5a to 39% yield. In contrast, the use of electron-donating 3,4-dimethoxy group either BI'-OH or BI'-alkyne increased the yields of 5a to 74% and 72% yields, while the use of both increased the formation of 5a to optimal 80% yield.
![[1860-5397-14-103-i6]](/bjoc/content/inline/1860-5397-14-103-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Mechanistic investigations of alkynylbenziodoxole for radical acceptor and oxidative quenching reactivity. Yields are isolated yields. N/P = not performed.
Scheme 6: Mechanistic investigations of alkynylbenziodoxole for radical acceptor and oxidative quenching reac...
Based on mechanistic investigations above, we propose that the electronic effect on benziodoxoles affected both the radical acceptor and oxidative quencher reactivity of BI-alkyne derivatives (Scheme 7). In the alkyl or acyl radical addition step to BI’-alkyne (step 1) and the oxidative quenching step by benziodoxole radical (step 2), the electron-donating substituents on BI’-alkynes are both beneficial, while the electron-withdrawing substituents have opposite effects.
![[1860-5397-14-103-i7]](/bjoc/content/inline/1860-5397-14-103-i7.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The role of alkynylbenziodoxole derivatives for radical alkynylation in photoredox catalysis.
Scheme 7: The role of alkynylbenziodoxole derivatives for radical alkynylation in photoredox catalysis.
Conclusion
In conclusion, we have developed and investigated novel alkynylbenziodoxole derivatives as radical alkynylation reagents in photoredox catalysis reactions. Alkynylbenziodoxole derivatives with electron-rich benziodoxoles demonstrate synthetic advantages in some situations. The mechanistic investigations suggested both the radical acceptor (step 1) and oxidative quencher reactivity (step 2) were affected by BI-alkyne derivatization. We envision these alkynylbenziodoxole derivatives will provide alternative radical alkynylation reagents in photoredox catalysis and other synthetic applications.
Supporting Information
| Supporting Information File 1: Experimental details, and copies of 1H NMR and 13C NMR spectra for all new compounds. | ||
| Format: PDF | Size: 4.2 MB | Download |
References
-
Jones, R. G.; Gilman, H. Chem. Rev. 1954, 54, 835–890. doi:10.1021/cr60171a004
Return to citation in text: [1] -
Orita, A.; Otera, J. Chem. Rev. 2006, 106, 5387–5412. doi:10.1021/cr050560m
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084–5121. doi:10.1039/c1cs15071e
Return to citation in text: [1] -
Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Tetrahedron 1998, 54, 10927–10966. doi:10.1016/S0040-4020(98)00410-4
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Chem. Soc. Rev. 2012, 41, 4165–4179. doi:10.1039/c2cs35034c
Return to citation in text: [1] -
Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073
Return to citation in text: [1] -
Ochiai, M.; Masaki, Y.; Shiro, M. J. Org. Chem. 1991, 56, 5511–5513. doi:10.1021/jo00019a007
Return to citation in text: [1] [2] -
Zhdankin, V. V.; Kuehl, C. J.; Krasutsky, A. P.; Bolz, J. T.; Simonsen, A. J. J. Org. Chem. 1996, 61, 6547–6551. doi:10.1021/jo960927a
Return to citation in text: [1] -
Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419
Return to citation in text: [1] -
Collins, K. D.; Lied, F.; Glorius, F. Chem. Commun. 2014, 50, 4459–4461. doi:10.1039/c4cc01141d
Return to citation in text: [1] -
Feng, C.; Loh, T.-P. Angew. Chem., Int. Ed. 2014, 53, 2722–2726. doi:10.1002/anie.201309198
Return to citation in text: [1] -
Wang, Z. F.; Li, L.; Huang, Y. J. Am. Chem. Soc. 2014, 136, 12233–12236. doi:10.1021/ja506352b
Return to citation in text: [1] -
Xie, F.; Qi, Z.; Yu, S.; Li, X. J. Am. Chem. Soc. 2014, 136, 4780–4787. doi:10.1021/ja501910e
Return to citation in text: [1] -
Abegg, D.; Frei, R.; Cerato, L.; Prasad Hari, D.; Wang, C.; Waser, J.; Adibekian, A. Angew. Chem., Int. Ed. 2015, 54, 10852–10857. doi:10.1002/anie.201505641
Return to citation in text: [1] -
Liu, X.; Wang, Z.; Cheng, X.; Li, C. J. Am. Chem. Soc. 2012, 134, 14330–14333. doi:10.1021/ja306638s
Return to citation in text: [1] [2] [3] -
Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280–2283. doi:10.1021/ja413208y
Return to citation in text: [1] [2] [3] [4] -
Huang, H.; Zhang, G.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. doi:10.1002/anie.201502369
Return to citation in text: [1] [2] [3] [4] -
Tan, H.; Li, H.; Ji, W.; Wang, L. Angew. Chem., Int. Ed. 2015, 54, 8374–8377. doi:10.1002/anie.201503479
Return to citation in text: [1] [2] -
Le Vaillant, F.; Courant, T.; Waser, J. Angew. Chem., Int. Ed. 2015, 54, 11200–11204. doi:10.1002/anie.201505111
Return to citation in text: [1] [2] -
Zhou, Q.-Q.; Guo, W.; Ding, W.; Wu, X.; Chen, X.; Lu, L.-Q.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 11196–11199. doi:10.1002/anie.201504559
Return to citation in text: [1] [2] -
Jia, K.; Zhang, F.; Huang, H.; Chen, Y. J. Am. Chem. Soc. 2016, 138, 1514–1517. doi:10.1021/jacs.5b13066
Return to citation in text: [1] [2] [3] [4] -
Jia, K.; Pan, Y.; Chen, Y. Angew. Chem., Int. Ed. 2017, 56, 2478–2481. doi:10.1002/anie.201611897
Return to citation in text: [1] [2] [3] -
Jia, K.; Li, J.; Chen, Y. Chem. – Eur. J. 2018, 24, 3174–3177. doi:10.1002/chem.201800202
Return to citation in text: [1] [2] -
Togo, H.; Taguchi, R.; Yamaguchi, K.; Yokoyama, M. J. Chem. Soc., Perkin Trans. 1 1995, 2135–2139. doi:10.1039/p19950002135
Return to citation in text: [1] -
Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem. – Eur. J. 2012, 18, 5655–5666. doi:10.1002/chem.201200200
Return to citation in text: [1] -
Fernández González, D.; Brand, J. P.; Mondière, R.; Waser, J. Adv. Synth. Catal. 2013, 355, 1631–1639. doi:10.1002/adsc.201300266
Return to citation in text: [1] -
Lu, B.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2014, 136, 11598–11601. doi:10.1021/ja5059795
Return to citation in text: [1] -
Hari, D. P.; Waser, J. J. Am. Chem. Soc. 2017, 139, 8420–8423. doi:10.1021/jacs.7b04756
Return to citation in text: [1] -
While not all the substituted o-iodobenzoic acids are commercially available, the substituted o-aminobenzoic acids are readily available in reasonable price and can synthesize the corresponding substituted o-iodobenzoic acids by Sandmeyer reaction in one step in 85–90% yields.
Return to citation in text: [1] -
Li, G.-X.; Morales-Rivera, C. A.; Wang, Y.; Gao, F.; He, G.; Liu, P.; Chen, G. Chem. Sci. 2016, 7, 6407–6412. doi:10.1039/C6SC02653B
Return to citation in text: [1]
| 21. | Huang, H.; Zhang, G.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. doi:10.1002/anie.201502369 |
| 26. | Jia, K.; Pan, Y.; Chen, Y. Angew. Chem., Int. Ed. 2017, 56, 2478–2481. doi:10.1002/anie.201611897 |
| 1. | Jones, R. G.; Gilman, H. Chem. Rev. 1954, 54, 835–890. doi:10.1021/cr60171a004 |
| 2. | Orita, A.; Otera, J. Chem. Rev. 2006, 106, 5387–5412. doi:10.1021/cr050560m |
| 3. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 4. | Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084–5121. doi:10.1039/c1cs15071e |
| 20. | Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280–2283. doi:10.1021/ja413208y |
| 25. | Jia, K.; Zhang, F.; Huang, H.; Chen, Y. J. Am. Chem. Soc. 2016, 138, 1514–1517. doi:10.1021/jacs.5b13066 |
| 19. | Liu, X.; Wang, Z.; Cheng, X.; Li, C. J. Am. Chem. Soc. 2012, 134, 14330–14333. doi:10.1021/ja306638s |
| 21. | Huang, H.; Zhang, G.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. doi:10.1002/anie.201502369 |
| 11. | Ochiai, M.; Masaki, Y.; Shiro, M. J. Org. Chem. 1991, 56, 5511–5513. doi:10.1021/jo00019a007 |
| 12. | Zhdankin, V. V.; Kuehl, C. J.; Krasutsky, A. P.; Bolz, J. T.; Simonsen, A. J. J. Org. Chem. 1996, 61, 6547–6551. doi:10.1021/jo960927a |
| 13. | Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419 |
| 14. | Collins, K. D.; Lied, F.; Glorius, F. Chem. Commun. 2014, 50, 4459–4461. doi:10.1039/c4cc01141d |
| 15. | Feng, C.; Loh, T.-P. Angew. Chem., Int. Ed. 2014, 53, 2722–2726. doi:10.1002/anie.201309198 |
| 16. | Wang, Z. F.; Li, L.; Huang, Y. J. Am. Chem. Soc. 2014, 136, 12233–12236. doi:10.1021/ja506352b |
| 17. | Xie, F.; Qi, Z.; Yu, S.; Li, X. J. Am. Chem. Soc. 2014, 136, 4780–4787. doi:10.1021/ja501910e |
| 18. | Abegg, D.; Frei, R.; Cerato, L.; Prasad Hari, D.; Wang, C.; Waser, J.; Adibekian, A. Angew. Chem., Int. Ed. 2015, 54, 10852–10857. doi:10.1002/anie.201505641 |
| 20. | Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280–2283. doi:10.1021/ja413208y |
| 5. | Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+ |
| 6. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c |
| 7. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 8. | Zhdankin, V. V.; Stang, P. J. Tetrahedron 1998, 54, 10927–10966. doi:10.1016/S0040-4020(98)00410-4 |
| 9. | Brand, J. P.; Waser, J. Chem. Soc. Rev. 2012, 41, 4165–4179. doi:10.1039/c2cs35034c |
| 10. | Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073 |
| 25. | Jia, K.; Zhang, F.; Huang, H.; Chen, Y. J. Am. Chem. Soc. 2016, 138, 1514–1517. doi:10.1021/jacs.5b13066 |
| 11. | Ochiai, M.; Masaki, Y.; Shiro, M. J. Org. Chem. 1991, 56, 5511–5513. doi:10.1021/jo00019a007 |
| 19. | Liu, X.; Wang, Z.; Cheng, X.; Li, C. J. Am. Chem. Soc. 2012, 134, 14330–14333. doi:10.1021/ja306638s |
| 28. | Togo, H.; Taguchi, R.; Yamaguchi, K.; Yokoyama, M. J. Chem. Soc., Perkin Trans. 1 1995, 2135–2139. doi:10.1039/p19950002135 |
| 29. | Brand, J. P.; Chevalley, C.; Scopelliti, R.; Waser, J. Chem. – Eur. J. 2012, 18, 5655–5666. doi:10.1002/chem.201200200 |
| 30. | Fernández González, D.; Brand, J. P.; Mondière, R.; Waser, J. Adv. Synth. Catal. 2013, 355, 1631–1639. doi:10.1002/adsc.201300266 |
| 31. | Lu, B.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2014, 136, 11598–11601. doi:10.1021/ja5059795 |
| 32. | Hari, D. P.; Waser, J. J. Am. Chem. Soc. 2017, 139, 8420–8423. doi:10.1021/jacs.7b04756 |
| 20. | Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280–2283. doi:10.1021/ja413208y |
| 34. | Li, G.-X.; Morales-Rivera, C. A.; Wang, Y.; Gao, F.; He, G.; Liu, P.; Chen, G. Chem. Sci. 2016, 7, 6407–6412. doi:10.1039/C6SC02653B |
| 19. | Liu, X.; Wang, Z.; Cheng, X.; Li, C. J. Am. Chem. Soc. 2012, 134, 14330–14333. doi:10.1021/ja306638s |
| 20. | Huang, H.; Zhang, G.; Gong, L.; Zhang, S.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 2280–2283. doi:10.1021/ja413208y |
| 21. | Huang, H.; Zhang, G.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. doi:10.1002/anie.201502369 |
| 22. | Tan, H.; Li, H.; Ji, W.; Wang, L. Angew. Chem., Int. Ed. 2015, 54, 8374–8377. doi:10.1002/anie.201503479 |
| 23. | Le Vaillant, F.; Courant, T.; Waser, J. Angew. Chem., Int. Ed. 2015, 54, 11200–11204. doi:10.1002/anie.201505111 |
| 24. | Zhou, Q.-Q.; Guo, W.; Ding, W.; Wu, X.; Chen, X.; Lu, L.-Q.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 11196–11199. doi:10.1002/anie.201504559 |
| 25. | Jia, K.; Zhang, F.; Huang, H.; Chen, Y. J. Am. Chem. Soc. 2016, 138, 1514–1517. doi:10.1021/jacs.5b13066 |
| 26. | Jia, K.; Pan, Y.; Chen, Y. Angew. Chem., Int. Ed. 2017, 56, 2478–2481. doi:10.1002/anie.201611897 |
| 27. | Jia, K.; Li, J.; Chen, Y. Chem. – Eur. J. 2018, 24, 3174–3177. doi:10.1002/chem.201800202 |
| 21. | Huang, H.; Zhang, G.; Chen, Y. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. doi:10.1002/anie.201502369 |
| 22. | Tan, H.; Li, H.; Ji, W.; Wang, L. Angew. Chem., Int. Ed. 2015, 54, 8374–8377. doi:10.1002/anie.201503479 |
| 23. | Le Vaillant, F.; Courant, T.; Waser, J. Angew. Chem., Int. Ed. 2015, 54, 11200–11204. doi:10.1002/anie.201505111 |
| 24. | Zhou, Q.-Q.; Guo, W.; Ding, W.; Wu, X.; Chen, X.; Lu, L.-Q.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 11196–11199. doi:10.1002/anie.201504559 |
| 25. | Jia, K.; Zhang, F.; Huang, H.; Chen, Y. J. Am. Chem. Soc. 2016, 138, 1514–1517. doi:10.1021/jacs.5b13066 |
| 26. | Jia, K.; Pan, Y.; Chen, Y. Angew. Chem., Int. Ed. 2017, 56, 2478–2481. doi:10.1002/anie.201611897 |
| 27. | Jia, K.; Li, J.; Chen, Y. Chem. – Eur. J. 2018, 24, 3174–3177. doi:10.1002/chem.201800202 |
| 33. | While not all the substituted o-iodobenzoic acids are commercially available, the substituted o-aminobenzoic acids are readily available in reasonable price and can synthesize the corresponding substituted o-iodobenzoic acids by Sandmeyer reaction in one step in 85–90% yields. |
© 2018 Pan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)