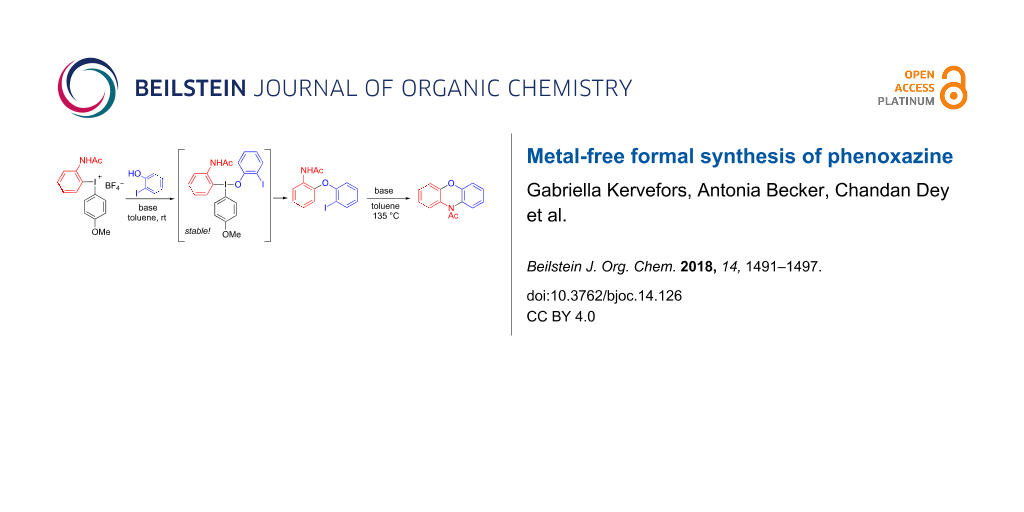
Abstract
A transition metal-free formal synthesis of phenoxazine is presented. The key step of the sequence is a high-yielding O-arylation of a phenol with an unsymmetrical diaryliodonium salt to provide an ortho-disubstituted diaryl ether. This species was cyclized to acetylphenoxazine in moderate yield. The overall yield in the three-step sequence is 72% based on recovered diaryl ether. An interesting, unusually stable iodine(III) intermediate in the O-arylation was observed by NMR and could be converted to the product upon longer reaction time.

Graphical Abstract
Introduction
Phenoxazine (1) is a tricyclic compound consisting of an oxazine ring fused between two benzene rings. A range of compounds with interesting biological or photophysical properties contain the phenoxazine core, where the amine moiety is either functionalized or oxidized to the corresponding imine [1-4]. Phenoxazine derivatives can display antitumor activity [5-8], are present in a variety of dyes [9], and can be applied in chemosensors and dye-sensitized solar cells (Figure 1) [10-12]. N-Arylphenoxazines were recently employed as photoredox catalysts in metal-free polymerizations [13].
![[1860-5397-14-126-1]](/bjoc/content/figures/1860-5397-14-126-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Compounds containing a phenoxazine moiety.
Figure 1: Compounds containing a phenoxazine moiety.
The first synthesis of phenoxazine dates back more than 100 years [14], and a range of synthetic routes to this target has since been developed [1]. Transition metal-free routes include the synthesis from 2-aminophenols and 3,4-dihaloarenes decorated with electron-withdrawing substituents, which proceed through a Smiles rearrangement (Scheme 1a) [15-17]. More recently, Bolm and co-workers reported a metal-free cyclization of iodo-substituted diaryl ethers with a broad scope (Scheme 1b) [18]. Transition metal-catalyzed cross couplings have also been employed to form the required C–O and C–N bonds, e.g., by Cu-catalyzed cyclization of 2-(2-bromophenoxy)anilines [17,19,20]. A Pd-catalyzed double N-arylation using di(2-bromoaryl) ethers and primary amines was recently developed (Scheme 1c) [21]. Furthermore, N-functionalization of the phenoxazine core can be performed under metal-free conditions [22].
![[1860-5397-14-126-i1]](/bjoc/content/inline/1860-5397-14-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reported syntheses of phenoxazine derivatives.
Scheme 1: Reported syntheses of phenoxazine derivatives.
Our research group has reported highly efficient O-arylations of phenols using diaryliodonium salts [23-25], which are reactive electrophilic arylation reagents [26-28]. As diaryliodonium salts can be easily synthesized in a one-pot manner [28], we envisioned that phenoxazines could be obtained in a straightforward and efficient manner from commercially available starting materials.
According to the retrosynthesis depicted in Scheme 2, target 1 would be formed from acetyl derivative 2, which can be derived from the functionalized diaryl ether 3 using Bolm’s N-arylation (see Scheme 1c) [18]. The synthesis of diaryl ether 3 is the key step of the sequence, proceeding via arylation of phenols with diaryliodonium salts using our reported methodology [23-25]. In reactions with unsymmetrical iodonium salts, this type of O-arylation is known to have an ortho-effect, i.e., chemoselective transfer of the ortho-substituted aryl moiety [29,30]. The use of unsymmetrical iodonium salts facilitates the synthesis of the reagents and avoids waste of an expensive iodoarene, and we hence envisioned chemoselective transfer of the desired aryl moiety from an unsymmetrical salt with a suitable “dummy” aryl group.
![[1860-5397-14-126-i2]](/bjoc/content/inline/1860-5397-14-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Two different approaches to reach 3 are illustrated, either employing 2-iodophenol (4) and 2-acetamido-substituted salt 5 (route A) or N-functionalized phenol 6 with 2-iodophenyl salt 7 (route B). In route A, the chemoselective transfer of the 2-amido aryl group over the other aryl group would require a quite electron-rich dummy group or the use of a symmetric iodonium salt (R = 2-NHAc). In route B, the challenge would instead be the synthesis of iodonium salt 7, as the iodo substituent might be prone to oxidation. Furthermore, selective O-arylation in the presence of an amide moiety could be challenging, as N-arylation of amides with diaryliodonium salts proceeds at room temperature [31,32].
Results and Discussion
We initially focused on route A and decided to explore an anisyl moiety as dummy group in iodonium salt 5. The electronic difference between the aryl groups in the formed salt should be sufficient to allow chemoselective transfer of the desired aryl moiety, and the ortho-effect exerted by the 2-amido substituent was expected to improve the chemoselectivity further. Salt 5a was synthesized in good yield using our reported arylboronic acid methodology [33] (Scheme 3). Attempts to form the corresponding tosylate salt, either from iodoarene 8 with anisole and mCPBA/TsOH [34] or via anion exchange of 5a were in vain.
![[1860-5397-14-126-i3]](/bjoc/content/inline/1860-5397-14-126-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The O-arylation of 2-iodophenol (4) with salt 5a was investigated using our previously reported methodology [24,25]. As expected, the reaction proceeded well, both in THF and toluene, of which the latter was most appealing as it might allow a tandem arylation and cyclization to reach N-acetylphenoxazine (2) in one pot. The arylations were analyzed by crude 1H NMR yields using an internal standard and revealed that a minor amount of a byproduct was formed along with the expected diaryl ether 3 (Table 1, entry 1). This was initially believed to be 2-iodophenyl 4-anisyl ether, i.e., the diaryl ether that would form upon transfer of the anisyl moiety to the nucleophile. The reaction was thus further investigated to find conditions that would allow high yield of 3 with complete chemoselectivity.
Table 1: Optimization of the O-arylation of route A.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-126-i8.svg?max-width=637&scale=1.0)
|
|||||||
| entry |
phenol 4
(equiv) |
salt 5a
(equiv) |
base
(equiv) |
T (°C) | time (h) | yield 3 (%)b | yield byproduct (%)c |
| 1 | 1 | 1.1 | t-BuOK (1.1) | rt | 18 | 80 | 3 |
| 2d | 1 | 1.1 | t-BuOK (1.1) | rt | 18 | 67 | 2 |
| 3 | 1 | 1.1 | NaH (1.1) | rt | 18 | 14 | 2 |
| 4 | 1 | 1.1 | K2CO3 (1.1) | rt | 18 | 65 | 2 |
| 5 | 1 | 1.5 | t-BuOK (1.1) | rt | 18 | 77 | 4 |
| 6 | 1 | 1.5 | t-BuOK (1.5) | rt | 18 | 74 | 14 |
| 7 | 1.5 | 1 | t-BuOK (1.5) | rt | 18 | 89 (91) | 5 |
| 8 | 1.5 | 1 | t-BuOK (1.5) | rt | 1 | 9 (58) | 52 |
| 9 | 1 | 1 | t-BuOK (1.5) | 40 | 1 | 49 | 38 |
| 10 | 1.5 | 1 | t-BuOK (1.5) | 80 | 1 | 79 | 4 |
| 11e | 1.5 | 1 | t-BuOK (1.5) | rt | 1 | 7 | trace |
aPhenol 4 (0.2 mmol) and base were stirred in anhydrous toluene (1 mL) for 30 min at rt before addition of 5a. The reaction was stirred at the tabulated temperature and time and quenched with brine (see Supporting Information File 1 for details). b1H NMR yield using dibromomethane as internal standard (isolated yield in parenthesis). c1H NMR yield based on the assumption of one OMe group in byproduct, see Supporting Information File 1. d0.5 mL DME as co-solvent. eQuenched with sat. ammonium chloride solution instead of brine.
The amount of byproduct remained approximately constant when solvents and bases were screened (Table 1, entries 1–4, see also Supporting Information File 1), whereas certain changes in stoichiometry influenced the yield (Table 1, entries 5–7). Diaryl ether 3 was efficiently formed using the conditions in entry 7, and could be easily isolated by flash chromatography in 91% yield. A shortening of the reaction time to 1 h profoundly increased the amount of byproduct to 52% (Table 1, entry 8). Surprisingly, the desired product 3 could be isolated in 58% yield from this experiment after prolonged storage of the neat crude product at rt, along with 52% of 4-iodoanisol (see Supporting Information File 1).
This observation is in line with a ligand-coupling (LC) mechanism [30,35,36] where the observed byproduct could be the T-shaped intermediate 9 or the corresponding 4-coordinated species with two phenoxide units [37]. This type of intermediate is expected in LC reactions but can rarely be detected. The groups of Quideau and Muñiz have reported such compounds using an ortho-nitro substituted phenol and tetrafluorophthalimide as nucleophiles, respectively [38,39]. Those compounds were isolable at room temperature and could be converted to the arylated products upon heating. The reactivity of 9 is unusual, as it proved stable to neutral work-up with brine, formed product 3 upon longer reaction time or rt storage in CDCl3 or neat, but decomposed to starting materials upon work-up with sat. ammonium chloride solution (Table 1, entry 11).
Due to the successful outcome of route A, pathway B was only briefly explored. Several dummy groups were considered for the iodonium salt 7. A phenyl dummy might give sufficient chemoselectivity due to the ortho-effect, and the synthesis of salt 7a was straightforward using 2-iodophenylboronic acid (Scheme 4a). The use of an anisyl dummy proved more difficult, and tetrafluoroborate 7b was isolated with impurities using the arylboronic acid methodology [33] (Scheme 4b). The mCPBA/TsOH methodology [34] with 1,2-diiodobenzene (10) and anisole or trimethoxybenzene (TMP-H) as dummy surprisingly delivered the corresponding bisiodonium ditosylates [40-42] instead of the desired 7 (Scheme 4c, see also Supporting Information File 1). The stoichiometry of the reaction did not allow complete formation of 11, but the yield based on TMP-H was 89%.
![[1860-5397-14-126-i4]](/bjoc/content/inline/1860-5397-14-126-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The arylation of phenol 6 with unsymmetrical diaryliodonium salt 7a provided the desired diaryl ether 3 in modest yield and limited chemoselectivity (Scheme 5). Competitive N-arylation of the amide moiety [31,32] was not observed. Preliminary attempts to employ the bisiodonium salt 11 in the arylation of 6 were fruitless (see Supporting Information File 1), and we hence concluded that route A was most suitable for further investigations.
![[1860-5397-14-126-i5]](/bjoc/content/inline/1860-5397-14-126-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
With high-yielding conditions for the O-arylation step at hand (Table 1, entry 7), the literature-reported [18] cyclization of 3 to N-acetylphenoxazine (2) was performed (Scheme 6a). In our hands, however, product 2 could only be isolated in 49% yield as the cyclization did not reach completion. The ratio of product 2 to starting material 3 remained 1:1 also upon modified conditions (see Supporting Information File 1). Still, the reaction was clean and the yield based on recovered starting material was 96%. The limited conversion into product 2 complicated our aim to perform the arylation and cyclization in one pot. The tandem reaction set up depicted in Scheme 6b delivered the O-arylated 3 in 87% yield, rather than product 2, further illustrating the sensitivity of the cyclization step. Attempts to perform the cyclization with t-BuOK and DMEDA only resulted in byproduct formation.
![[1860-5397-14-126-i6]](/bjoc/content/inline/1860-5397-14-126-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: a) Cyclization of diaryl ether 3. b) Attempted one pot-synthesis of 2. aBased on recovered 3.
Scheme 6: a) Cyclization of diaryl ether 3. b) Attempted one pot-synthesis of 2. aBased on recovered 3.
The deprotection of 2 is reported to proceed in high yield [18], and the synthesis of 2 hence constitutes a metal-free formal synthesis of phenoxazine (1). The complete route is depicted in Scheme 7 and delivered N-acetylphenoxazine (2) in 72% overall yield over 3 steps. Combined with the literature yield for the deprotection, this would provide the target 1 in 65% overall yield based on recovered 3. The required starting materials in this route can easily be further substituted; hence we expect the methodology to be applicable also for phenoxazine derivatives.
![[1860-5397-14-126-i7]](/bjoc/content/inline/1860-5397-14-126-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Formal synthesis of phenoxazine (1). aBased on recovered 3.
Scheme 7: Formal synthesis of phenoxazine (1). aBased on recovered 3.
Conclusion
A transition metal-free formal synthesis of phenoxazine is presented, relying on the O-arylation of phenol 4 with unsymmetrical diaryliodonium salt 5a to provide substituted diaryl ether 3 as the key step. This species underwent an intramolecular N-arylation to provide the cyclized product 2. The overall yield in this three-step sequence is 72% based on recovered diaryl ether. Interestingly, an unusually stable iodine(III) intermediate was formed in the O-arylation. This species survived neutral work-up and could be converted to product upon longer reaction time.
Supporting Information
| Supporting Information File 1: Experimental details for the synthesis of starting materials and products, analytical data for products 2, 3, 5a, 7a, 9 and 11. | ||
| Format: PDF | Size: 1.4 MB | Download |
| Supporting Information File 2: NMR spectra for products 2, 3, 5a, 7a and 11. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Katsamakas, S.; Zografos, A. L.; Sarli, V. Curr. Med. Chem. 2016, 23, 2972–2999. doi:10.2174/0929867323666160510123023
Return to citation in text: [1] [2] -
Hernandez-Olmos, V.; Abdelrahman, A.; El-Tayeb, A.; Freudendahl, D.; Weinhausen, S.; Müller, C. E. J. Med. Chem. 2012, 55, 9576–9588. doi:10.1021/jm300845v
Return to citation in text: [1] -
Thimmaiah, K. N.; Easton, J. B.; Germain, G. S.; Morton, C. L.; Kamath, S.; Buolamwini, J. K.; Houghton, P. J. J. Biol. Chem. 2005, 280, 31924–31935. doi:10.1074/jbc.M507057200
Return to citation in text: [1] -
Moosmann, B.; Skutella, T.; Beyer, K.; Behl, C. Biol. Chem. 2001, 382, 1601–1612. doi:10.1515/BC.2001.195
Return to citation in text: [1] -
Zhao, Y.-q.; Yin, Y.-q.; Liu, J.; Wang, G.-h.; Huang, J.; Zhu, L.-j.; Wang, J.-h. Acta Pharmacol. Sin. 2016, 37, 1237–1250. doi:10.1038/aps.2016.60
Return to citation in text: [1] -
Prinz, H.; Chamasmani, B.; Vogel, K.; Böhm, K. J.; Aicher, B.; Gerlach, M.; Günther, E. G.; Amon, P.; Ivanov, I.; Müller, K. J. Med. Chem. 2011, 54, 4247–4263. doi:10.1021/jm200436t
Return to citation in text: [1] -
Avendaño, C.; Menéndez, J. C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, 2008; pp 93–138. doi:10.1016/B978-0-444-52824-7.00004-4
Return to citation in text: [1] -
Gao, S.; Takano, T.; Sada, K.; He, J.; Noda, C.; Hori-Tamura, N.; Tomoda, A.; Yamamura, H. Br. J. Pharmacol. 2002, 137, 749–755. doi:10.1038/sj.bjp.0704939
Return to citation in text: [1] -
Martinez, V.; Henary, M. Chem. – Eur. J. 2016, 22, 13764–13782. doi:10.1002/chem.201601570
Return to citation in text: [1] -
Yang, X.-B.; Yang, B.-X.; Ge, J.-F.; Xu, Y.-J.; Xu, Q.-F.; Liang, J.; Lu, J.-M. Org. Lett. 2011, 13, 2710–2713. doi:10.1021/ol2008022
Return to citation in text: [1] -
Karlsson, K. M.; Jiang, X.; Eriksson, S. K.; Gabrielsson, E.; Rensmo, H.; Hagfeldt, A.; Sun, L. Chem. – Eur. J. 2011, 17, 6415–6424. doi:10.1002/chem.201003730
Return to citation in text: [1] -
Tian, H.; Yang, X.; Cong, J.; Chen, R.; Liu, J.; Hao, Y.; Hagfeldt, A.; Sun, L. Chem. Commun. 2009, 6288–6290. doi:10.1039/b912746a
Return to citation in text: [1] -
Pearson, R. M.; Lim, C.-H.; McCarthy, B. G.; Musgrave, C. B.; Miyake, G. M. J. Am. Chem. Soc. 2016, 138, 11399–11407. doi:10.1021/jacs.6b08068
Return to citation in text: [1] -
Bernthsen, A. Ber. Dtsch. Chem. Ges. 1887, 20, 942–944. doi:10.1002/cber.188702001214
Return to citation in text: [1] -
Eastmond, G. C.; Gilchrist, T. L.; Paprotny, J.; Steiner, A. New J. Chem. 2001, 25, 385–390. doi:10.1039/b008503k
Return to citation in text: [1] -
Schmidt, D. M.; Bonvicino, G. E. J. Org. Chem. 1984, 49, 1664–1666. doi:10.1021/jo00183a039
Return to citation in text: [1] -
Bonvicino, G. E.; Yogodzinski, L. H.; Hardy, R. A., Jr. J. Org. Chem. 1961, 26, 2797–2803. doi:10.1021/jo01066a042
Return to citation in text: [1] [2] -
Thomé, I.; Bolm, C. Org. Lett. 2012, 14, 1892–1895. doi:10.1021/ol3005134
Return to citation in text: [1] [2] [3] [4] -
Liu, N.; Wang, B.; Chen, W.; Liu, C.; Wang, X.; Hu, Y. RSC Adv. 2014, 4, 51133–51139. doi:10.1039/C4RA09593F
Return to citation in text: [1] -
Eregowda, G. B.; Kalpana, H. N.; Hegde, R.; Thimmaiah, K. N. Indian J. Chem. 2000, 39B, 243–259.
Return to citation in text: [1] -
Zhang, L.; Huang, X.; Zhen, S.; Zhao, J.; Li, H.; Yuan, B.; Yang, G. Org. Biomol. Chem. 2017, 15, 6306–6309. doi:10.1039/C7OB01540B
Return to citation in text: [1] -
Louillat-Habermeyer, M.-L.; Jin, R.; Patureau, F. W. Angew. Chem., Int. Ed. 2015, 54, 4102–4104. doi:10.1002/anie.201500089
Return to citation in text: [1] -
Lindstedt, E.; Ghosh, R.; Olofsson, B. Org. Lett. 2013, 15, 6070–6073. doi:10.1021/ol402960f
Return to citation in text: [1] [2] -
Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645
Return to citation in text: [1] [2] [3] -
Jalalian, N.; Ishikawa, E. E.; Silva, L. F., Jr.; Olofsson, B. Org. Lett. 2011, 13, 1552–1555. doi:10.1021/ol200265t
Return to citation in text: [1] [2] [3] -
Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369
Return to citation in text: [1] -
Olofsson, B. Top. Curr. Chem. 2016, 373, 135–166. doi:10.1007/128_2015_661
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689
Return to citation in text: [1] [2] -
Stuart, D. R. Chem. – Eur. J. 2017, 23, 15852–15863. doi:10.1002/chem.201702732
Return to citation in text: [1] -
Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Chem. – Eur. J. 2013, 19, 10334–10342. doi:10.1002/chem.201300860
Return to citation in text: [1] [2] -
Tinnis, F.; Stridfeldt, E.; Lundberg, H.; Adolfsson, H.; Olofsson, B. Org. Lett. 2015, 17, 2688–2691. doi:10.1021/acs.orglett.5b01079
Return to citation in text: [1] [2] -
Wang, M.; Huang, Z. Org. Biomol. Chem. 2016, 14, 10185–10188. doi:10.1039/C6OB01649A
Return to citation in text: [1] [2] -
Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974
Return to citation in text: [1] [2] -
Zhu, M.; Jalalian, N.; Olofsson, B. Synlett 2008, 592–596. doi:10.1055/s-2008-1032050
Return to citation in text: [1] [2] -
Ochiai, M. Top. Curr. Chem. 2003, 224, 5–68. doi:10.1007/3-540-46114-0_2
Return to citation in text: [1] -
Pinto de Magalhães, H.; Lüthi, H. P.; Togni, A. Org. Lett. 2012, 14, 3830–3833. doi:10.1021/ol3014039
Return to citation in text: [1] -
Stridfeldt, E.; Lindstedt, E.; Reitti, M.; Blid, J.; Norrby, P.-O.; Olofsson, B. Chem. – Eur. J. 2017, 23, 13249–13258. doi:10.1002/chem.201703057
Return to citation in text: [1] -
Ozanne-Beaudenon, A.; Quideau, S. Angew. Chem., Int. Ed. 2005, 44, 7065–7069. doi:10.1002/anie.200501638
Return to citation in text: [1] -
Lucchetti, N.; Scalone, M.; Fantasia, S.; Muñiz, K. Angew. Chem., Int. Ed. 2016, 55, 13335–13339. doi:10.1002/anie.201606599
Return to citation in text: [1] -
Dohi, T.; Kato, D.; Hyodo, R.; Yamashita, D.; Shiro, M.; Kita, Y. Angew. Chem., Int. Ed. 2011, 50, 3784–3787. doi:10.1002/anie.201007640
Return to citation in text: [1] -
Stang, P. J.; Zhdankin, V. V. J. Am. Chem. Soc. 1993, 115, 9808–9809. doi:10.1021/ja00074a061
Return to citation in text: [1] -
Kitamura, T.; Furuki, R.; Nagata, K.; Taniguchi, H.; Stang, P. J. J. Org. Chem. 1992, 57, 6810–6814. doi:10.1021/jo00051a025
Return to citation in text: [1]
| 40. | Dohi, T.; Kato, D.; Hyodo, R.; Yamashita, D.; Shiro, M.; Kita, Y. Angew. Chem., Int. Ed. 2011, 50, 3784–3787. doi:10.1002/anie.201007640 |
| 41. | Stang, P. J.; Zhdankin, V. V. J. Am. Chem. Soc. 1993, 115, 9808–9809. doi:10.1021/ja00074a061 |
| 42. | Kitamura, T.; Furuki, R.; Nagata, K.; Taniguchi, H.; Stang, P. J. J. Org. Chem. 1992, 57, 6810–6814. doi:10.1021/jo00051a025 |
| 31. | Tinnis, F.; Stridfeldt, E.; Lundberg, H.; Adolfsson, H.; Olofsson, B. Org. Lett. 2015, 17, 2688–2691. doi:10.1021/acs.orglett.5b01079 |
| 32. | Wang, M.; Huang, Z. Org. Biomol. Chem. 2016, 14, 10185–10188. doi:10.1039/C6OB01649A |
| 1. | Katsamakas, S.; Zografos, A. L.; Sarli, V. Curr. Med. Chem. 2016, 23, 2972–2999. doi:10.2174/0929867323666160510123023 |
| 2. | Hernandez-Olmos, V.; Abdelrahman, A.; El-Tayeb, A.; Freudendahl, D.; Weinhausen, S.; Müller, C. E. J. Med. Chem. 2012, 55, 9576–9588. doi:10.1021/jm300845v |
| 3. | Thimmaiah, K. N.; Easton, J. B.; Germain, G. S.; Morton, C. L.; Kamath, S.; Buolamwini, J. K.; Houghton, P. J. J. Biol. Chem. 2005, 280, 31924–31935. doi:10.1074/jbc.M507057200 |
| 4. | Moosmann, B.; Skutella, T.; Beyer, K.; Behl, C. Biol. Chem. 2001, 382, 1601–1612. doi:10.1515/BC.2001.195 |
| 13. | Pearson, R. M.; Lim, C.-H.; McCarthy, B. G.; Musgrave, C. B.; Miyake, G. M. J. Am. Chem. Soc. 2016, 138, 11399–11407. doi:10.1021/jacs.6b08068 |
| 28. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 10. | Yang, X.-B.; Yang, B.-X.; Ge, J.-F.; Xu, Y.-J.; Xu, Q.-F.; Liang, J.; Lu, J.-M. Org. Lett. 2011, 13, 2710–2713. doi:10.1021/ol2008022 |
| 11. | Karlsson, K. M.; Jiang, X.; Eriksson, S. K.; Gabrielsson, E.; Rensmo, H.; Hagfeldt, A.; Sun, L. Chem. – Eur. J. 2011, 17, 6415–6424. doi:10.1002/chem.201003730 |
| 12. | Tian, H.; Yang, X.; Cong, J.; Chen, R.; Liu, J.; Hao, Y.; Hagfeldt, A.; Sun, L. Chem. Commun. 2009, 6288–6290. doi:10.1039/b912746a |
| 9. | Martinez, V.; Henary, M. Chem. – Eur. J. 2016, 22, 13764–13782. doi:10.1002/chem.201601570 |
| 23. | Lindstedt, E.; Ghosh, R.; Olofsson, B. Org. Lett. 2013, 15, 6070–6073. doi:10.1021/ol402960f |
| 24. | Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645 |
| 25. | Jalalian, N.; Ishikawa, E. E.; Silva, L. F., Jr.; Olofsson, B. Org. Lett. 2011, 13, 1552–1555. doi:10.1021/ol200265t |
| 5. | Zhao, Y.-q.; Yin, Y.-q.; Liu, J.; Wang, G.-h.; Huang, J.; Zhu, L.-j.; Wang, J.-h. Acta Pharmacol. Sin. 2016, 37, 1237–1250. doi:10.1038/aps.2016.60 |
| 6. | Prinz, H.; Chamasmani, B.; Vogel, K.; Böhm, K. J.; Aicher, B.; Gerlach, M.; Günther, E. G.; Amon, P.; Ivanov, I.; Müller, K. J. Med. Chem. 2011, 54, 4247–4263. doi:10.1021/jm200436t |
| 7. | Avendaño, C.; Menéndez, J. C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, 2008; pp 93–138. doi:10.1016/B978-0-444-52824-7.00004-4 |
| 8. | Gao, S.; Takano, T.; Sada, K.; He, J.; Noda, C.; Hori-Tamura, N.; Tomoda, A.; Yamamura, H. Br. J. Pharmacol. 2002, 137, 749–755. doi:10.1038/sj.bjp.0704939 |
| 26. | Aradi, K.; Tóth, B. L.; Tolnai, G. L.; Novák, Z. Synlett 2016, 27, 1456–1485. doi:10.1055/s-0035-1561369 |
| 27. | Olofsson, B. Top. Curr. Chem. 2016, 373, 135–166. doi:10.1007/128_2015_661 |
| 28. | Merritt, E. A.; Olofsson, B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. doi:10.1002/anie.200904689 |
| 21. | Zhang, L.; Huang, X.; Zhen, S.; Zhao, J.; Li, H.; Yuan, B.; Yang, G. Org. Biomol. Chem. 2017, 15, 6306–6309. doi:10.1039/C7OB01540B |
| 15. | Eastmond, G. C.; Gilchrist, T. L.; Paprotny, J.; Steiner, A. New J. Chem. 2001, 25, 385–390. doi:10.1039/b008503k |
| 16. | Schmidt, D. M.; Bonvicino, G. E. J. Org. Chem. 1984, 49, 1664–1666. doi:10.1021/jo00183a039 |
| 17. | Bonvicino, G. E.; Yogodzinski, L. H.; Hardy, R. A., Jr. J. Org. Chem. 1961, 26, 2797–2803. doi:10.1021/jo01066a042 |
| 22. | Louillat-Habermeyer, M.-L.; Jin, R.; Patureau, F. W. Angew. Chem., Int. Ed. 2015, 54, 4102–4104. doi:10.1002/anie.201500089 |
| 1. | Katsamakas, S.; Zografos, A. L.; Sarli, V. Curr. Med. Chem. 2016, 23, 2972–2999. doi:10.2174/0929867323666160510123023 |
| 14. | Bernthsen, A. Ber. Dtsch. Chem. Ges. 1887, 20, 942–944. doi:10.1002/cber.188702001214 |
| 17. | Bonvicino, G. E.; Yogodzinski, L. H.; Hardy, R. A., Jr. J. Org. Chem. 1961, 26, 2797–2803. doi:10.1021/jo01066a042 |
| 19. | Liu, N.; Wang, B.; Chen, W.; Liu, C.; Wang, X.; Hu, Y. RSC Adv. 2014, 4, 51133–51139. doi:10.1039/C4RA09593F |
| 20. | Eregowda, G. B.; Kalpana, H. N.; Hegde, R.; Thimmaiah, K. N. Indian J. Chem. 2000, 39B, 243–259. |
| 31. | Tinnis, F.; Stridfeldt, E.; Lundberg, H.; Adolfsson, H.; Olofsson, B. Org. Lett. 2015, 17, 2688–2691. doi:10.1021/acs.orglett.5b01079 |
| 32. | Wang, M.; Huang, Z. Org. Biomol. Chem. 2016, 14, 10185–10188. doi:10.1039/C6OB01649A |
| 23. | Lindstedt, E.; Ghosh, R.; Olofsson, B. Org. Lett. 2013, 15, 6070–6073. doi:10.1021/ol402960f |
| 24. | Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645 |
| 25. | Jalalian, N.; Ishikawa, E. E.; Silva, L. F., Jr.; Olofsson, B. Org. Lett. 2011, 13, 1552–1555. doi:10.1021/ol200265t |
| 29. | Stuart, D. R. Chem. – Eur. J. 2017, 23, 15852–15863. doi:10.1002/chem.201702732 |
| 30. | Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Chem. – Eur. J. 2013, 19, 10334–10342. doi:10.1002/chem.201300860 |
| 33. | Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974 |
| 34. | Zhu, M.; Jalalian, N.; Olofsson, B. Synlett 2008, 592–596. doi:10.1055/s-2008-1032050 |
| 37. | Stridfeldt, E.; Lindstedt, E.; Reitti, M.; Blid, J.; Norrby, P.-O.; Olofsson, B. Chem. – Eur. J. 2017, 23, 13249–13258. doi:10.1002/chem.201703057 |
| 38. | Ozanne-Beaudenon, A.; Quideau, S. Angew. Chem., Int. Ed. 2005, 44, 7065–7069. doi:10.1002/anie.200501638 |
| 39. | Lucchetti, N.; Scalone, M.; Fantasia, S.; Muñiz, K. Angew. Chem., Int. Ed. 2016, 55, 13335–13339. doi:10.1002/anie.201606599 |
| 24. | Jalalian, N.; Petersen, T. B.; Olofsson, B. Chem. – Eur. J. 2012, 18, 14140–14149. doi:10.1002/chem.201201645 |
| 25. | Jalalian, N.; Ishikawa, E. E.; Silva, L. F., Jr.; Olofsson, B. Org. Lett. 2011, 13, 1552–1555. doi:10.1021/ol200265t |
| 30. | Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Chem. – Eur. J. 2013, 19, 10334–10342. doi:10.1002/chem.201300860 |
| 35. | Ochiai, M. Top. Curr. Chem. 2003, 224, 5–68. doi:10.1007/3-540-46114-0_2 |
| 36. | Pinto de Magalhães, H.; Lüthi, H. P.; Togni, A. Org. Lett. 2012, 14, 3830–3833. doi:10.1021/ol3014039 |
| 33. | Bielawski, M.; Aili, D.; Olofsson, B. J. Org. Chem. 2008, 73, 4602–4607. doi:10.1021/jo8004974 |
| 34. | Zhu, M.; Jalalian, N.; Olofsson, B. Synlett 2008, 592–596. doi:10.1055/s-2008-1032050 |
© 2018 Kervefors et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)