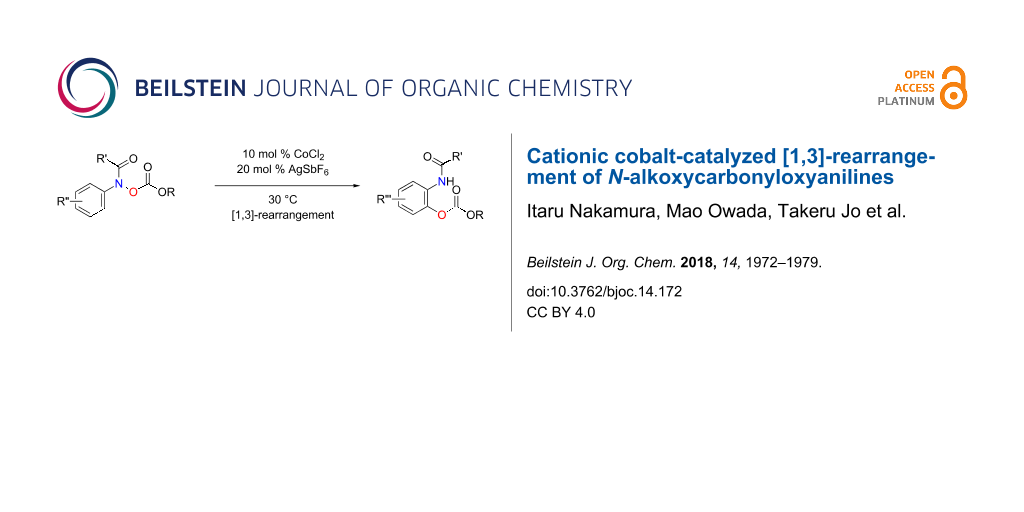
Abstract
A cationic cobalt catalyst efficiently promoted the reaction of N-alkoxycarbonyloxyanilines at 30 °C, affording the corresponding ortho-aminophenols in good to high yields. As reported previously, our mechanistic studies including oxygen-18 labelling experiments indicate that the rearrangement of the alkoxycarbonyloxy group proceeds in [1,3]-manner. In this article, we discuss the overall picture of the cobalt-catalysed [1,3]-rearrangement reaction including details of the reaction conditions and substrate scope.

Graphical Abstract
Introduction
The 2-aminophenol moiety is ubiquitously found as a core structure of biologically active compounds, such as tigecycline [1], iguratimod [2], and phosalone (Scheme 1) [3]. The scaffolds have also been frequently utilized as synthetic intermediates not only in pharmaceutical chemistry but also in materials science. Thus, it is of great importance to efficiently synthesize functionalized 2-aminophenols under mild reaction conditions in a regioselective manner. Among numerous methods, the [3,3]-rearrangement of O-acyl-N-arylhydroxylamines 1 driven by cleavage of the N–O bond is an ideal approach to selectively synthesize O-protected 2-aminophenols 2 while maintaining the oxidation state during the transformation (Scheme 2a) [4-11]. However, there is a significant drawback, these [3,3]-rearrangements of carboxylic acyloxy and alkoxylcarbonyloxy groups generally require long heating times at elevated reaction temperatures (>140 °C) or microwave irradiation (Scheme 2a). In contrast, N-sulfonyloxyanilines are known to readily undergo the [3,3]-rearrangement during the preparation of the starting material below −20 °C due to the strongly electron-withdrawing nature of the sulfonyl group (Scheme 2b) [12]. Accordingly, we envisioned that appropriate Lewis acidic metal catalysts would promote the rearrangement reaction of stable N-acyloxyanilines to afford readily deprotectable 2-acyloxyanilines under much milder reaction conditions with high functional group tolerance. Based on this concept, we disclosed that cationic cobalt catalysts efficiently promote the reaction of O-alkoxylcarbonyl-N-arylhydroxylamines 1 at 30 °C, affording the corresponding 2-aminophenol derivatives 2 in good to high yields [13]. Our mechanistic studies revealed that the rearrangement of the alkoxycarbonyloxy group proceeded in an unprecedented [1,3]-manner (Scheme 2c). In this article, we describe the overall picture of the intriguing [1,3]-rearrangement reaction, particularly the detail of the reaction, which were not sufficiently discussed in our preliminary communication.
![[1860-5397-14-172-i1]](/bjoc/content/inline/1860-5397-14-172-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-172-i2]](/bjoc/content/inline/1860-5397-14-172-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Rearrangement of N-acyloxyanilines.
Scheme 2: Rearrangement of N-acyloxyanilines.
Results and Discussion
At the beginning of this investigation, N,O-di(methoxycarbonyl)hydroxylaniline (1a) was treated with catalytic amounts of several copper salts in 1,2-dichloroethane (DCE) at 60 °C (Table 1, entries 1–7), according to our previous copper-catalysed cascade reaction involving rearrangement via N–O bond cleavage [14]. While divalent copper acetate and copper chloride did not show any catalytic activities (Table 1, entries 1 and 2), more Lewis acidic copper complexes, such as [Cu(MeCN)4](PF6)2 and [Cu(OTf)]2·toluene, afforded the corresponding 2-aminophenol derivative 2a (Table 1, entries 3 and 4). Moreover, a cationic copper catalyst generated from CuCl2 and two equivalents of AgSbF6 was effective to afford 2a in good yield (Table 1, entry 5), even at 30 °C (Table 1, entry 8). The use of a ligand, such as 1,10-phenanthroline (phen) and 1,3-bis(diphenylphosphino)propane (dppp), totally diminished the activity of cationic cobalt catalyst (Table 1, entries 6 and 7). Among metal chlorides examined, CoCl2 exhibited the best catalytic activity at 30 °C, affording the corresponding 2a in 68% yield (Table 1, entry 9), as reported previously [13]. A divalent cationic zinc catalyst also promoted the present reaction, albeit with lower chemical yield than Co(II) (Table 1, entries 10 and 11), while the use of Fe(II) and Pd(II) resulted in low chemical yield due to the formation of the para-isomer 3a (Table 1, entries 12 and 13). Indeed the para-isomer 3a was obtained as a major product when the reaction of 1a was conducted using trivalent metal salts, such as FeCl3 and RuCl3, and tetravalent salts, such as ZrCl4, as a catalyst (Table 1, entries 15–18). Although we quite recently disclosed that cationic NHC-copper catalysts efficiently promoted the [1,3]-alkoxy rearrangement of N-alkoxyaniline [15], the cationic NHC-Cu catalyst generated from IPrCuBr and AgSbF6 was totally inefficient for the present reaction; 1a was decomposed under the reaction conditions (Table 1, entry 19). Whereas AgSbF6 promoted the reaction at 60 °C (Table 1, entry 20), the catalytic activity was diminished at 30 °C (Table 1, entry 21). Neutral CoCl2 did not promote the present reaction (Table 1, entry 22). Brønsted acids, such as trifluoromethanesulfonic acid and diphenylphosphoric acid, were much less active (Table 1, entries 23 and 24). The kind of the counteranion significantly affected the reaction efficiency; hexafluoroantimonate and bis(trifluoromethanesulfonyl)imidate were efficient (Table 1, entries 9 and 25), while the use of hexafluorophosphate and trifluoromethanesulfonate did not promote the reaction at all (Table 1, entries 26 and 27). The use of an equal amount of AgSbF6 to CoCl2 resulted in slightly decreasing the chemical yield (Table 1, entry 28).
Table 1: Catalytic activity.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-172-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | catalyst (mol %) | temp. (°C) | 2a (%)a | 3a (%)a | 1a (%)a |
| 1 | CuCl2 (10) | 60 | <1 | <1 | >99 |
| 2 | Cu(OAc)2 (10) | 60 | <1 | <1 | >99 |
| 3 | [Cu(MeCN)4](PF6) (10) | 60 | 5 | <1 | 75 |
| 4 | [Cu(OTf)]2·C6H5CH3 (10) | 60 | 50 | 4 | 23 |
| 5 | CuCl2 (10), AgSbF6 (20) | 60 | 52 | <1 | 14 |
| 6 | CuCl2 (10), AgSbF6 (20), phen (20) | 60 | <1 | <1 | >99 |
| 7 | CuCl2 (10), AgSbF6 (20), dppp (20) | 60 | <1 | <1 | 80 |
| 8b | CuCl2 (10), AgSbF6 (20) | 30 | 57 | 10 | <1 |
| 9b | CoCl2 (10), AgSbF6 (20) | 30 | 63 (68) | <3 | <1 |
| 10b | ZnCl2 (10), AgSbF6 (20) | 30 | 54 | 8 | <1 |
| 11 | ZnCl2 (10), AgSbF6 (10) | 30 | 54 | 8 | 1 |
| 12b | PdCl2 (10), AgSbF6 (20) | 30 | 24 | 7 | <1 |
| 13b | FeCl2 (10), AgSbF6 (20) | 30 | 32 | 21 | 1 |
| 14b | FeCl3 (10), AgSbF6 (30) | 30 | 22 | 26 | <1 |
| 15b | RuCl3 (10), AgSbF6 (30) | 30 | 18 | 35 | <1 |
| 16 | RuCl3 (10), AgSbF6 (20) | 30 | 21 | 35 | <1 |
| 17 | IrCl3 (10), AgSbF6 (30) | 30 | 15 | 37 | <1 |
| 18 | ZrCl4 (10), AgSbF6 (40) | 30 | 16 | 28 | <1 |
| 19 | IPrCuBr (10), AgSbF6 (10) | 30 | <1 | <1 | 12 |
| 20 | AgSbF6 (10) | 60 | 53 | 17 | 7 |
| 21b | AgSbF6 (10) | 30 | <3 | <3 | 80 |
| 22b | CoCl2 (10) | 30 | <1 | <1 | 90 |
| 23 | TfOH (10) | 30 | 5 | 6 | 74 |
| 24 | (PhO)2P(O)OH | 30 | <1 | <1 | 90 |
| 25b | CoCl2 (10), AgNTf2 (20) | 30 | 50 | 1 | 3 |
| 26b | CoCl2 (10), AgPF6 (20) | 30 | <1 | <1 | 98 |
| 27b | CoCl2 (10), AgOTf (20) | 30 | <1 | <1 | 97 |
| 28 | CoCl2 (10), AgSbF6 (10) | 30 | 53 | 3 | <1 |
| 29 | CoCl2 (5), AgSbF6 (10) | 30 | 59 | 2 | <1 |
aYields were determined by 1H NMR using CH2Br2 as an internal standard. Isolated yield in parenthesis. bReported in the Supporting Information of our previous paper [13], except for yields of recovered 1a.
Next solvent and concentration effects were examined as summarized in Table 2. 1,2-Dichloroethane (DCE) gave the best result (Table 2, entry 1), as described previously [13]. Other halogen solvents, such as CHCl3, CH2Cl2, and PhCl, ethereal solvent, such as Et2O and tert-butyl methyl ether (MTBE), and toluene were less efficient (Table 2, entries 2–7), while the use of polar solvents, such as tetrahydrofuran (THF), acetonitrile, and N,N-dimethylformamide (DMF), resulted in quantitative recovery of the starting material 1a (Table 2, entries 8–10). A protic solvent, such as methanol, was ineffective (Table 2, entry 11). Slight dilution of the reaction solution (0.25 M) improved the chemical yield (Table 2, entry 12).
Table 2: Solvent and concentration effects.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-172-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | concentration (M) | 2a (%)a | 3a (%)a | 1a (%)a |
| 1b | DCE | 0.5 | 63 | 3 | <1 |
| 2b | CHCl3 | 0.5 | 49 | 7 | 19 |
| 3b | CH2Cl2 | 0.5 | 40 | 2 | <1 |
| 4b | PhCl | 0.5 | 39 | 2 | 25 |
| 5b | toluene | 0.5 | 43 | 1 | 11 |
| 6b | Et2O | 0.5 | 38 | <1 | 1 |
| 7 | MTBE | 0.5 | 49 | 1 | 4 |
| 8 | THF | 0.5 | <1 | <1 | >99 |
| 9 | CH3CN | 0.5 | <1 | <1 | 98 |
| 10 | DMF | 0.5 | <1 | <1 | >99 |
| 11 | MeOH | 0.5 | 4 | <1 | 81 |
| 12 | DCE | 1.0 | 51 | 2 | <1 |
| 13b | DCE | 0.25 | 72 | 3 | <1 |
| 14b,c | DCE | 0.05 | 53 | 9 | 11 |
aYields were determined by 1H NMR using CH2Br2 as an internal standard. bReported in the Supporting Information of our previous paper (ref. [13]), except for yields of recovered 1a. cFor 5 days.
As mentioned previously [13], carbamate-type groups, such as methoxycarbonyl, Alloc and Cbz were tolerated as a protective group on the nitrogen atom, affording the desired products 2 in good yields (Table 3, entries 1–3). The reaction of 1e having a 2,2,2-trichloroethoxycarbonyl (Troc) group, however, resulted in decomposing 1e (Table 3, entry 4). The use of aroyl groups gave the desired product in good yields (Table 3, entries 5–7), while the acetyl group required a prolonged reaction time (Table 3, entry 8). Substrate 1j having a tosyl group on the nitrogen resulted in decomposition of 1j (Table 3, entry 9). The alkoxycarbonyl groups, such as Cbz, methoxycarbonyl, and 2-chloroethoxy groups, were employed as good migrating groups (Table 3, entries 7–11), while 1m having a Boc group on the oxygen atom did not give the desired product, due to decomposition of 1m (Table 3, entry 12). It is noteworthy that the substrate having a highly electron-withdrawing Troc group on the oxygen atom was readily isomerized to the ortho-aminophenol derivative under its preparing conditions in the absence of the cationic cobalt catalyst. In sharp contrast to alkoxycarbonyloxy groups, acyloxy groups, such as the benzoyloxy group, were not migrated to the ortho-position, resulting in decomposing the starting material (Table 3, entry 13), as mentioned previously [13].
Table 3: Substituent effect at the hydroxylamine moiety.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-172-i8.svg?max-width=637&scale=1.0)
|
||||||
| entry | 1 | R1 | R2 | time (h) | 2 | yield (%)b |
| 1c | 1b | OMe | Cbz | 5 | 2b | 64 |
| 2c | 1c | OMe | Alloc | 4 | 2c | 45 |
| 3c | 1d | OMe | Boc | 6 | 2d | 64 |
| 4 | 1e | OMe | Troc | 18 | – | <1 |
| 5c | 1f | OMe | p-MeOC6H4C(O) | 2 | 2f | 60d |
| 6c | 1g | OMe | Bz | 3 | 2g | 75 |
| 7c | 1h | OMe | p-F3CC6H4C(O) | 24 | 2h | 61 |
| 8c | 1i | OMe | Ac | 120 | 2i | 44 |
| 9 | 1j | OMe | Ts | 11 | – | <1 |
| 10c | 1k | OBn | Bz | 2 | 2k | 82 |
| 11 | 1l | O-CH2CH2Cl | Bz | 2 | 2l | 56 |
| 12 | 1m | O-tBu | Bz | 10 | – | <1 |
| 13c | 1n | Ph | Bz | 120 | – | <1 |
aThe reactions of 1 (0.4 mmol) were conducted in the presence of 10 mol % CoCl2 and 20 mol % of AgSbF6 in DCE (1.6 mL) at 30 °C. bIsolated yield. cPreviously reported in [13]. d1H NMR yield using dibromomethane as an internal standard.
The reaction was applied to 1o–ab having various substituents at the para-position, as summarized in Table 4. As reported previously [13], reactive functional groups, such as bromo, iodo, and alkynyl groups were tolerated, affording the desired products in good to high yields (Table 4, entries 5–7). The substrate 1v having a methoxycarbonyl group afforded 2v in good yield, while cyano and acetyl groups interrupted the present reaction presumably due to deactivation of the catalyst, recovering the starting materials quantitatively (Table 4, entries 9 and 10). Notably, our catalytic conditions successfully promoted the rearrangement of 1y’, having a highly electron-deficient p-trifluromethylphenyl group, which have not been employed in the thermal [3,3]-rearrangement reaction, when using a p-nitrobenzyloxycarbonyl group in place of Cbz as the migrating group (Table 4, entry 11). In addition, compatibility of the protective group on the oxygen atom was tested (Table 4, entries 12–14), since it is expected that the cationic cobalt would make the protective group labile as well as the protective group would deactivate the cationic cobalt catalyst. As results, tert-butyldimethylsilyl (TBS) and methoxymethyl (MOM) groups were tolerated under the cationic cobalt-catalyzed reaction conditions to afford the desired product in good yields (Table 4, entries 13 and 14). The reaction using a benzoyl group was sluggish, affording the desired product 2z in moderate yield with formation of inseparable byproducts (Table 4, entry 12). Thus, the use of silyl- and acetal-type protective groups is suitable for the present reaction.
Table 4: Co-catalyzed reaction of N-alkoxycarbonyloxyanilines 1o–ab.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-172-i9.svg?max-width=637&scale=1.0)
|
||||||
| entry | 1 | R | R1 | time (h) | 2 | yield (%)b |
| 1c | 1o | Me | Bn | 3 | 2o | 74 |
| 2c | 1p | F | Bn | 11 | 2p | 66d |
| 3c | 1q | Cl | Bn | 1 | 2q | 88 |
| 4c | 1r | Cl | Me | 3 | 2r | 79 |
| 5c | 1s | Br | Bn | 1 | 2s | 86 |
| 6c | 1t | I | Bn | 1 | 2t | 77 |
| 7c | 1u | TMSC≡C | Bn | 2 | 2u | 62 |
| 8c | 1v | CO2Me | Bn | 15 | 2v | 84d |
| 9 | 1w | Ac | Bn | >120 | – | <1e |
| 10 | 1x | CN | Bn | >120 | – | <1e |
| 11c | 1y’ | CF3 | p-O2NC6H4CH2 | 48 | 2y’ | 76d,f |
| 12 | 1z | BzO(CH2)2 | Bn | 72 | 2z | 50d,g |
| 13 | 1aa | TBSO(CH2)2 | Bn | 14 | 2aa | 64 |
| 14 | 1ab | MOM(CH2)2 | Bn | 14 | 2ab | 59 |
aThe reactions of 1 (0.4 mmol) were conducted in the presence of 10 mol % CoCl2 and 20 mol % of AgSbF6 in DCE (1.6 mL) at 30 °C. bIsolated yield. cPreviously reported in [13]. d1H NMR yield using dibromomethane as an internal standard. See Supporting Information File 1 for details. eThe starting material was quantitatively recovered. fYield brsm (28% of 1y’ was recovered). gIsolation of 2z was unsuccessful due to contamination by inseparable byproducts (see Supporting Information File 1).
As reported previously [13], the fact that the present rearrangement reaction proceeds in a [1,3]-manner was confirmed by a crossover experiment and oxygen-18 labeling experiments. That is, the reaction of a 1:1 mixture of equally-reactive substrates 1h and 1r under the standard reaction conditions afforded only the products 2h and 2r derived from the starting materials (Scheme 3a). Thus, we confirmed that the present reaction proceeds in an intramolecular manner. Next, 18-oxygen-labelling experiments were conducted using substrate 1h-18O, of which the oxygen-18 content at the hydroxylamine oxygen atom was 62% [16,17]. The reaction of 1h-18O in the presence of the cationic cobalt catalyst at 30 °C followed by hydrogenative cleavage of the Cbz group afforded the phenol 4h-18O, of which the oxygen-18 content was 64% (Scheme 3b). The result clearly indicates that the rearrangement of the CbzO group in the presence of cationic cobalt catalysts proceeds in a concerted [1,3]-manner [18-24]. In addition, the reaction of 1h-18O (23% 18O) in the absence of the cationic cobalt catalyst at 140 °C followed by hydrogenative deprotection afforded 4h, of which the oxygen-18 content was less than 2% (Scheme 3c). Therefore, we concluded that the cationic cobalt catalyst not only made the reaction much milder than the thermally-induced reaction but also changed the rearrangement mode to an unprecedented [1,3]-manner. In addition, intermolecular and intramolecular competitive experiments using deuterium-labelled substrates resulted in no kinetic effect (Scheme 4). These results suggest that the C–O bond would form prior to cleavage of the C–H bond in the [1,3]-rearrangement reaction.
![[1860-5397-14-172-i3]](/bjoc/content/inline/1860-5397-14-172-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Mechanistic studies, reported in [13].
Scheme 3: Mechanistic studies, reported in [13].
![[1860-5397-14-172-i4]](/bjoc/content/inline/1860-5397-14-172-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Competitive experiments, reported in [13].
Scheme 4: Competitive experiments, reported in [13].
Due to the fact that the reaction of 1a in the presence of tri- and tetravalent cationic metal catalysts afforded the para-isomer 3a as a major product (Table 1, entries 14–18), the reaction of ortho-aminophenol derivative 2a in the presence of catalytic amounts of RuCl3 and AgSbF6 was conducted. However, the para-isomer 3a was not afforded; 73% of 2a was recovered (Scheme 5a). The result indicates that the para-isomer 3a was not formed through the ortho-isomer 2a. It is assumed that 3a was furnished through direct C–O bond formation at the para-position through ionic cleavage of the N–O bond by cationic Ru(III) as a much stronger Lewis acid, while it is also possible that the second migration of the alkoxycarbonyloxy group from ortho to para occurs prior to proton transfer (Scheme 5b) [25]. Further mechanistic studies are underway in our laboratory.
![[1860-5397-14-172-i5]](/bjoc/content/inline/1860-5397-14-172-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanism for rearrangement to the para-position.
Scheme 5: Mechanism for rearrangement to the para-position.
Conclusion
The cationic cobalt catalysts enabled the rearrangement reaction of N-alkoxycarbonyloxyanilines to proceed under much milder reaction conditions, expanding the substrate scope to more electron-deficient anilines. More importantly, the cobalt catalyst changes the mode of the rearrangement to an unprecedented [1,3]-manner.
Experimental
To a mixture of 1k (138.9 mg, 0.4 mmol), CoCl2 (5.2 mg, 0.04 mmol), and AgSbF6 (27.5 mg, 0.08 mmol) under an argon atmosphere in a pressure vial was added 1,2-dichloroethane (1.6 mL). Then, the mixture was stirred at 30 °C for 2 hours. After complete consumption of the starting material 1k, the mixture was passed through a small pad of silica gel with ethyl acetate. After removing the solvents in vacuo, the residue was purified by flash silica gel column chromatography using hexane/ethyl acetate (3:1) as eluent to obtain 2k (113.9 mg, 82%).
Supporting Information
| Supporting Information File 1: General procedure and analytic data for obtained products. | ||
| Format: PDF | Size: 4.0 MB | Download |
References
-
Sum, P.-E.; Peterson, P. Bioorg. Med. Chem. Lett. 1999, 9, 1459. doi:10.1016/S0960-894X(99)00216-4
Return to citation in text: [1] -
Aikawa, Y.; Yamamoto, M.; Yamamoto, T.; Morimoto, K.; Tanaka, K. Inflammation Res. 2002, 51, 188. doi:10.1007/PL00000291
Return to citation in text: [1] -
Ahmad, R.; Kookana, R. S.; Alsoton, A. M.; Skjemstad, J. O. Environ. Sci. Technol. 2001, 35, 878. doi:10.1021/es001446i
Return to citation in text: [1] -
Leffler, J. E.; Bradford Bond, W. J. Am. Chem. Soc. 1956, 78, 335. doi:10.1021/ja01583a023
Return to citation in text: [1] -
Denney, D. B.; Denney, D. Z. J. Am. Chem. Soc. 1960, 82, 1389. doi:10.1021/ja01491a026
Return to citation in text: [1] -
Oae, S.; Sakurai, T.; Kimura, H.; Kozuka, S. Chem. Lett. 1974, 3, 671. doi:10.1246/cl.1974.671
Return to citation in text: [1] -
Oae, S.; Sakurai, T. Tetrahedron 1976, 32, 2289. doi:10.1016/0040-4020(76)88003-9
Return to citation in text: [1] -
Ohta, T.; Shudo, K.; Okamoto, T. Tetrahedron Lett. 1978, 19, 1983. doi:10.1016/S0040-4039(01)94727-6
Return to citation in text: [1] -
Porzelle, A.; Woodrow, M. D.; Tomkinson, N. C. O. Eur. J. Org. Chem. 2008, 5135. doi:10.1002/ejoc.200800672
Return to citation in text: [1] -
Porzelle, A.; Woodrow, M. D.; Tomkinson, N. C. O. Org. Lett. 2010, 12, 812. doi:10.1021/ol902885j
Return to citation in text: [1] -
Tabolin, A. A.; Ioffe, S. L. Chem. Rev. 2014, 114, 5426. doi:10.1021/cr400196x
Return to citation in text: [1] -
Gutschke, D.; Heesing, A.; Heuschkel, U. Tetrahedron Lett. 1979, 20, 1363–1364. doi:10.1016/S0040-4039(01)86151-7
Return to citation in text: [1] -
Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] -
Nakamura, I.; Kudo, Y.; Terada, M. Angew. Chem., Int. Ed. 2013, 52, 7536. doi:10.1002/anie.201302751
Return to citation in text: [1] -
Nakamura, I.; Jo, T.; Ishida, Y.; Tashiro, H.; Terada, M. Org. Lett. 2017, 19, 3059. doi:10.1021/acs.orglett.7b01110
Return to citation in text: [1] -
The percentage of oxygen-18 content was determined by intensity of the mass spectrum.
Return to citation in text: [1] -
Rajendran, G.; Santini, R. E.; Van Etten, R. L. J. Am. Chem. Soc. 1987, 109, 4357. doi:10.1021/ja00248a037
Return to citation in text: [1] -
Oae, S.; Kitao, T.; Kitaoka, Y. Tetrahedron 1963, 19, 827. doi:10.1016/S0040-4020(01)99334-2
Return to citation in text: [1] -
Nasveschuk, C. G.; Rovis, T. Org. Biomol. Chem. 2008, 6, 240. doi:10.1039/B714881J
Return to citation in text: [1] -
Hou, S.; Li, X.; Xu, J. J. Org. Chem. 2012, 77, 10856. doi:10.1021/jo302210t
Return to citation in text: [1] -
Wada, N.; Kaneko, K.; Ukaji, Y.; Inomata, K. Chem. Lett. 2011, 40, 440. doi:10.1246/cl.2011.440
Return to citation in text: [1] -
Xu, J. Curr. Org. Synth. 2017, 14, 511. doi:10.2174/1570179413666161021103952
Return to citation in text: [1] -
Hou, S.; Li, X.; Xu, J. Org. Biomol. Chem. 2014, 12, 4952. doi:10.1039/C4OB00080C
Return to citation in text: [1] -
Yang, Z.; Hou, S.; He, W.; Cheng, B.; Jiao, P.; Xu, J. Tetrahedron 2016, 72, 2186. doi:10.1016/j.tet.2016.03.019
Return to citation in text: [1] -
The reaction of a mixture of 1a and 1h in the presence of RuCl3 (10 mol %) and AgSbF6 (30 mol %) gave only the products 2a and 2h derived from the starting materials; crossover products were not detected (<3%) by HRMS. This result indicates that the rearrangement to the para-position proceeds in an intramolecular manner.
Return to citation in text: [1]
| 16. | The percentage of oxygen-18 content was determined by intensity of the mass spectrum. |
| 17. | Rajendran, G.; Santini, R. E.; Van Etten, R. L. J. Am. Chem. Soc. 1987, 109, 4357. doi:10.1021/ja00248a037 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 1. | Sum, P.-E.; Peterson, P. Bioorg. Med. Chem. Lett. 1999, 9, 1459. doi:10.1016/S0960-894X(99)00216-4 |
| 12. | Gutschke, D.; Heesing, A.; Heuschkel, U. Tetrahedron Lett. 1979, 20, 1363–1364. doi:10.1016/S0040-4039(01)86151-7 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 4. | Leffler, J. E.; Bradford Bond, W. J. Am. Chem. Soc. 1956, 78, 335. doi:10.1021/ja01583a023 |
| 5. | Denney, D. B.; Denney, D. Z. J. Am. Chem. Soc. 1960, 82, 1389. doi:10.1021/ja01491a026 |
| 6. | Oae, S.; Sakurai, T.; Kimura, H.; Kozuka, S. Chem. Lett. 1974, 3, 671. doi:10.1246/cl.1974.671 |
| 7. | Oae, S.; Sakurai, T. Tetrahedron 1976, 32, 2289. doi:10.1016/0040-4020(76)88003-9 |
| 8. | Ohta, T.; Shudo, K.; Okamoto, T. Tetrahedron Lett. 1978, 19, 1983. doi:10.1016/S0040-4039(01)94727-6 |
| 9. | Porzelle, A.; Woodrow, M. D.; Tomkinson, N. C. O. Eur. J. Org. Chem. 2008, 5135. doi:10.1002/ejoc.200800672 |
| 10. | Porzelle, A.; Woodrow, M. D.; Tomkinson, N. C. O. Org. Lett. 2010, 12, 812. doi:10.1021/ol902885j |
| 11. | Tabolin, A. A.; Ioffe, S. L. Chem. Rev. 2014, 114, 5426. doi:10.1021/cr400196x |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 3. | Ahmad, R.; Kookana, R. S.; Alsoton, A. M.; Skjemstad, J. O. Environ. Sci. Technol. 2001, 35, 878. doi:10.1021/es001446i |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 2. | Aikawa, Y.; Yamamoto, M.; Yamamoto, T.; Morimoto, K.; Tanaka, K. Inflammation Res. 2002, 51, 188. doi:10.1007/PL00000291 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 15. | Nakamura, I.; Jo, T.; Ishida, Y.; Tashiro, H.; Terada, M. Org. Lett. 2017, 19, 3059. doi:10.1021/acs.orglett.7b01110 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 25. | The reaction of a mixture of 1a and 1h in the presence of RuCl3 (10 mol %) and AgSbF6 (30 mol %) gave only the products 2a and 2h derived from the starting materials; crossover products were not detected (<3%) by HRMS. This result indicates that the rearrangement to the para-position proceeds in an intramolecular manner. |
| 14. | Nakamura, I.; Kudo, Y.; Terada, M. Angew. Chem., Int. Ed. 2013, 52, 7536. doi:10.1002/anie.201302751 |
| 18. | Oae, S.; Kitao, T.; Kitaoka, Y. Tetrahedron 1963, 19, 827. doi:10.1016/S0040-4020(01)99334-2 |
| 19. | Nasveschuk, C. G.; Rovis, T. Org. Biomol. Chem. 2008, 6, 240. doi:10.1039/B714881J |
| 20. | Hou, S.; Li, X.; Xu, J. J. Org. Chem. 2012, 77, 10856. doi:10.1021/jo302210t |
| 21. | Wada, N.; Kaneko, K.; Ukaji, Y.; Inomata, K. Chem. Lett. 2011, 40, 440. doi:10.1246/cl.2011.440 |
| 22. | Xu, J. Curr. Org. Synth. 2017, 14, 511. doi:10.2174/1570179413666161021103952 |
| 23. | Hou, S.; Li, X.; Xu, J. Org. Biomol. Chem. 2014, 12, 4952. doi:10.1039/C4OB00080C |
| 24. | Yang, Z.; Hou, S.; He, W.; Cheng, B.; Jiao, P.; Xu, J. Tetrahedron 2016, 72, 2186. doi:10.1016/j.tet.2016.03.019 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
| 13. | Nakamura, I.; Owada, M.; Jo, T.; Terada, M. Org. Lett. 2017, 19, 2194. doi:10.1021/acs.orglett.7b00700 |
© 2018 Nakamura et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)