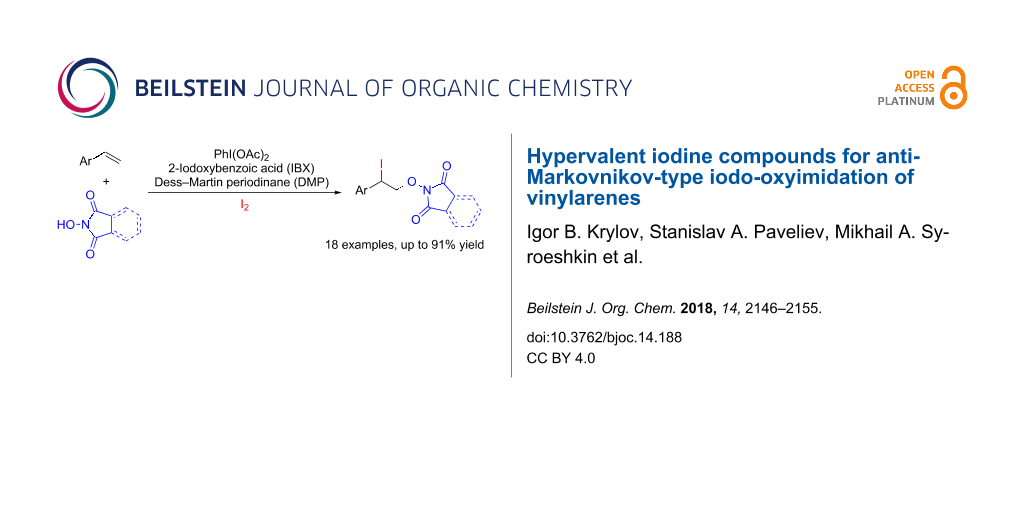
Abstract
The iodo-oxyimidation of styrenes with the N-hydroxyimide/I2/hypervalent iodine oxidant system was proposed. Among the examined hypervalent iodine oxidants (PIDA, PIFA, IBX, DMP) PhI(OAc)2 proved to be the most effective; yields of iodo-oxyimides are 34–91%. A plausible reaction pathway includes the addition of an imide-N-oxyl radical to the double C=C bond and trapping of the resultant benzylic radical by iodine. It was shown that the iodine atom in the prepared iodo-oxyimides can be substituted by various nucleophiles.

Graphical Abstract
Introduction
The presented work opens a new chapter in the chemistry of N-hydroxyimides in combination with hypervalent iodine compounds with formation of imide-N-oxyl radicals. These radicals were used as reagents for the addition to a terminal position of the double bond of styrenes with subsequent iodination of the resulting benzylic radical.
It is important to note, that nitroxyl radicals are widely used in organic and biological chemistry, and in material design [1-3]. These radicals are applied in the development of monomolecular magnets [4,5], spintronics [2,6], magneto-LC effect studies [7,8], organic voltaic cells [9], electrodes for electrochemical synthesis [2], and as mediators of living polymerization [10,11]. In organic synthesis more stable types of N-oxyl radicals can be used as carbon-centered radical scavengers [12], oxidation catalysts, mainly for conversion of alcohols to carbonyl compounds [11,13-17]. Less stable imide-N-oxyl radicals are used as effective mediators for CH-functionalization with formation of C–C, C–O, C–S, and C–N bonds [11,16,18-42].
Phthalimide-N-oxyl (PINO) is one of the most known imide-N-oxyl radicals that is generated from an inexpensive N-hydroxyphthalimide (NHPI). This radical was used in various aerobic oxidations of bulk chemicals [18,19,43,44].
In the present work imide-N-oxyl radicals were used for the addition to the C=C bonds of styrenes with subsequent functionalization of the resulting benzylic radicals.
Recently, the precursors of N-oxyl radicals, such as N-hydroxyphthalimide (NHPI), N-hydroxysuccinimide (NHSI), N-hydroxybenzotriazole (HOBt) and hydroxamic acids, have been used in the reactions of radical oxygenation of styrenes [45]. Growth of interest is observed concerning the reactions of styrenes with imide-N-oxyl radicals, in which the latter add to the terminal position of the double C=C bond with the formation of stabilized benzyl radicals, which undergo the subsequent functionalization. In the presence of oxygen or tert-butyl hydroperoxide, oxidation proceeds to form the C–O [46-51] or the C=O [52-55] moiety. More complex reagents and reaction systems allows to form C–C [56,57] and C–N [58,59] bonds.
Among the above-mentioned methods, there are no examples of C–Hal bond formation despite the wide usage of organohalides in chemical syntheses. In the row of organohalides, iodides are the most reactive and versatile reagents for the following transformations [60].
One of the purposes of our work was to introduce iodine in the process of difunctionalization of styrenes with imide-N-oxyl radicals. Iodine atom in the product can act as a versatile leaving group for further transformations. The involvement of the iodine in the radical reactions of styrenes is complicated by the fact that unsaturated compounds readily undergo electrophilic iodination with the addition of an external nucleophile [61,62]. The oxidants used for the preparation of imide-N-oxyl radicals, in particular the hypervalent iodine compounds and peroxides [63-73], also generate electrophilic iodinating intermediates (Scheme 1).
![[1860-5397-14-188-i1]](/bjoc/content/inline/1860-5397-14-188-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Difunctionalization of double C=C bond with the formation of C–O and C–I bonds.
Scheme 1: Difunctionalization of double C=C bond with the formation of C–O and C–I bonds.
For several decades, a number of papers on the electrophilic iodination of C=C bonds by iodine-containing oxidative systems with the addition of various nucleophiles have been published, all of these processes have common mechanism and the same regioselectivity. The free-radical approach developed in the present work affords the opposite (anti-Markovnikov) regioselectivity of the addition to the double bond.
Results and Discussion
In the present work, the reaction of styrenes 1a–k and N-hydroxyimides 2a,b with the formation of iodo-oxyimidated products 3aa–ka, 3ab–db, 3fb, 3hb and 3kb was carried out (Scheme 2).
![[1860-5397-14-188-i2]](/bjoc/content/inline/1860-5397-14-188-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Iodo-oxyimidation of styrenes 1a–k with preparation of products 3aa–ka, 3ab–db, 3fb, 3hb, and 3kb.
Scheme 2: Iodo-oxyimidation of styrenes 1a–k with preparation of products 3aa–ka, 3ab–db, 3fb, 3hb, and 3kb.
The key feature of the developed process is the non-standard regioselectivity of the formation of C–I and C–O bonds, which implies the radical pathway of the reaction.
The iodo-oxyimidation of styrenes was studied in the model reaction of styrene (1a) with N-hydroxyphthalimide (2a). During the optimization, the oxidant and the iodine source, as well as the nature of the solvent and the reaction time were varied (Table 1).
Table 1: Optimization of the synthesis of iodo-oxyimidation product 3a from styrene 1a and N-hydroxyimide 2a a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-188-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | oxidant (molar ratio: mol/mol of 1a) | solvent | time | yield of 3aab (%) |
|---|---|---|---|---|
| 1 | PhI(OAc)2 (0.6) | DCM | 5 min | 63 |
| 2 | PhI(OAc)2 (0.6) | DCM | 10 min | 90 |
| 3 | PhI(OAc)2 (0.6) | DCM | 24 h | 84 |
| 4 | PhI(OAc)2 (1.5) | DCM | 10 min | 73 |
| 5 | PhI(OAc)2 (0.6) | MeCN | 10 min | 73 |
| 6 | PhI(OAc)2 (0.6) | AcOH | 10 min | 65 |
| 7 | PhI(OAc)2 (0.6) | PhMe | 10 min | 84 |
| 8c | PhI(OAc)2 (2) | DCM | 10 min | 7 |
| 9d | PhI(OAc)2 (2) | DCM | 10 min | 52 |
| 10 | PhI(OCOCF3)2 (0.6) | DCM | 10 min | 31 |
| 11 | IBX (1.0) | DCM | 24 h | 54 |
| 12 | IBX (0.3) | DCM | 24 h | 32 |
| 13 | DMP (0.6) | DCM | 30 min | 52 |
| 14 | DMP (0.3) | DCM | 30 min | 52 |
| 15 | Oxone (2) | DCM/H2O (2:1) | 4 h | 44 |
| 16 | 2-iodobenzoic acid (0.1), Oxone (2) | DCM/H2O (2:1) | 4 h | 44 |
| 17 | TBHP (70% aq) (2) | DCM | 12 h | ND |
| 18 | TBHP (70% aq) (2) | MeCN | 12 h | ND |
| 19 | TBHP (70% aq) (2) | AcOH | 12 h | ND |
| 20 | TBAI (0.1), TBHP (70% aq) (2) | MeCN | 12 h | ND |
| 21 | (NH4)2S2O8 (1.5) | DCM/H2O (2:1) | 12 h | 5 |
| 22 | DDQ (2) | MeCN | 30 min | 5 |
aReaction conditions: 1a (1 mmol), 2a (1 mmol), I2 (0.5 mmol), oxidant (0.3–2 mmol), solvent (6.0 mL), 20–25 °C, 5 min–24 h, under air. For entries where a mixture of solvents was used, the v/v ratio is given in parentheses. bIsolated yield. ND = not detected. cNaI·2H2O (1 mmol) was employed instead of I2. dTBAI (1 mmol) was employed instead of I2.
In general, the iodo-oxyimidation reaction is characterized by the following: The product 3aa is formed regardless what kind of hypervalent iodine compound is used (Table 1, entries 1-14) and Oxone (Table 1, entries 15 and 16) as the oxidant. The best yield of 3aa (90%) was obtained using PhI(OAc)2 (Table 1, entry 2). Other iodine-based oxidants, such as PhI(OCOCF3)2 (Table 1, entry 10, yield 31%), IBX (Table 1, entries 11 and 12, yield 32–54%), DMP (Table 1, entries 13–14, yield 52%), showed less efficacy in this process. Peroxide oxidants, such as TBHP, TBAI/TBHP system [74], (NH4)2S2O8, and DDQ were ineffective in the studied process (Table 1, entries 17–22). A satisfactory yield of 3aa (44%) was achieved using Oxone as the oxidant (Table 1, entry 15). The addition of a catalytic amount of 2-iodobenzoic acid, which forms hypervalent iodine compounds in the presence of Oxone [75], did not lead to an increased yield of 3aa (Table 1, entry 16).
Dichloromethane proved to be the best solvent for the reaction, as carrying out the reaction in other solvents led to a decrease in the yield of 3aa (Table 1, entries 5–7). Increasing the amount of PhI(OAc)2 from 0.6 mmol to 1.5 mmol (Table 1, entry 4) leads to a decrease in the yield of 3aa presumably due to the enhancing the role of side oxidation processes. The optimal reaction time was 10 min, a reaction of 5 min resulted in a significant decrease in the yield of the desired product (Table 1, entry 1). Prolongation of the reaction time to 24 h led to a slight decrease in the yield of 3aa (Table 1, entry 3) due to its instability under the reaction conditions.
The possibility of using iodide salts (NaI and TBAI) was shown in Entries 8 and 9, however, the yield of 3aa in that cases is lower than in the case of molecular iodine.
In the optimized reaction conditions (Table 1, entry 2) iodo-oxyimidation of various vinylarenes were performed in order to study the scope of the developed method (Figure 1).
![[1860-5397-14-188-1]](/bjoc/content/figures/1860-5397-14-188-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Scope of the iodo-oxyimidation of vinylarenes with I2/PhI(OAc)2 system. Reaction conditions: vinylarene 1a–k (0.5 mmol), N-hydroxyimide 2a,b (0.5 mmol), I2 (0.25 mmol), PhI(OAc)2 (0.3 mmol), DCM (3.0 mL), 20–25 °C, 10 min, under air. rr = regioisomers ratio. aIBX (0.5 mmol) was used instead of PhI(OAc)2, reaction time: 24 h. bDMP (0.15 mmol) was used instead of PhI(OAc)2, reaction time 30 min.
Figure 1: Scope of the iodo-oxyimidation of vinylarenes with I2/PhI(OAc)2 system. Reaction conditions: vinyla...
The iodo-oxyimidation successfully proceeded using styrenes having both electron-withdrawing (Cl, F, Br) substituents in the aromatic ring (products 3ca, 3da, 3fa, 3ha, 3cb–hb, yield 34–91%), and an electron-donating methyl group (products 3ba, 3ea, 3ga, 3bb, yield 39–85%). Good yields (60–79%) were achieved with a cyclic analogue of styrene – indene (1k, compounds 3ka, 3kb). β-Substituted styrenes (β-methyl styrene (1i) and (E)-stilbene (1j) also underwent the studied transformation giving iodo-oxyimides 3ia (yield 51%) and 3ja (yield 83%). The reaction of NHPI (2a) with p-methoxystyrene under standard conditions led to a complex mixture of products, possibly due to an increased tendency of the substrate to electrophilic addition of iodine. The use of allylbenzene in the reaction did not result in the formation of the desired iodo-oxyimide, presumably due to a side process of oxidation of the allylic methylene fragment. The use of N-hydroxyphthalimide (2a) gives iodo-oxymidation products with yields generally higher (25% on average) than that of N-hydroxysuccinimide (2b).
Structures of the iodo-oxyimides 3aa–ka, 3ab–db, 3fb, 3hb and 3kb were confirmed by 1D and 2D NMR spectroscopy, IR spectroscopy, high-resolution mass spectrometry and elemental analysis. An additional confirmation of the structure of 3ca was made using X-ray crystallographic analysis (Figure 2). Details of the data collection and refinement are provided in Supporting Information File 1 and can be obtained free of charge via the web at https://www.ccdc.cam.ac.uk/structures/ (CCDC-1845323).
![[1860-5397-14-188-2]](/bjoc/content/figures/1860-5397-14-188-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of 3ca. Atoms are presented as anisotropic displacement parameters (ADP) ellipsoids (50% probability). For clarity, only one set of positions of the disordered ethylene bridge and Ph groups is shown.
Figure 2: Molecular structure of 3ca. Atoms are presented as anisotropic displacement parameters (ADP) ellips...
Proposed mechanism of the iodo-oxyimidation
Based on the literature data describing the formation of the phthalimide-N-oxyl radical (PINO) from NHPI under the action of PhI(OAc)2 [34,55,59,76,77], and based on information about the reaction of the PINO radical with styrenes [45] and interaction of benzyl radicals with iodine [78,79], a mechanism for the reaction of iodo-oxyimidation of styrenes under the action of the NHPI/I2/PhI(OAc)2 system was proposed (Scheme 3).
![[1860-5397-14-188-i3]](/bjoc/content/inline/1860-5397-14-188-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The proposed mechanism of iodo-oxyimidation of styrene (1a) using the NHPI/I2/PhI(OAc)2 system with the formation of product 3aa.
Scheme 3: The proposed mechanism of iodo-oxyimidation of styrene (1a) using the NHPI/I2/PhI(OAc)2 system with...
On the first step, NHPI (2a) is oxidized by PhI(OAc)2 to form PINO radical. The addition of PINO to the double C=C bond of styrene (1a) leads to the formation of intermediate A. At the final step the iodine traps benzyl radical A to form the final product 3aa.
Electrochemical studies
Cyclic voltammetry (CV) experiments on a working glassy-carbon electrode were carried out for deeper understanding of the plausible reaction mechanism. As CH2Cl2 is not suitable as a solvent due to the poor solubility of NHPI, thus MeCN was used. Tetrabutylammonium tetrafluoroborate, which cannot be oxidized in such experimental conditions [80], was chosen as a supporting electrolyte. Cyclic voltammograms of styrene (1a), NHPI (2a), I2 and PhI(OAc)2 in MeCN solution were registered (Figure 3).
![[1860-5397-14-188-3]](/bjoc/content/figures/1860-5397-14-188-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: CV curves of styrene (1a, purple), NHPI (2a, red), I2 (blue) and PhI(OAc)2 (green) in 0.1 M n-Bu4NBF4/MeCN at a scan rate of 100 mV/s on a working glassy-carbon electrode.
Figure 3: CV curves of styrene (1a, purple), NHPI (2a, red), I2 (blue) and PhI(OAc)2 (green) in 0.1 M n-Bu4NBF...
The NHPI oxidation peak is observed at +2.18 V, whereas iodine is oxidized at slightly higher potential (+2.27 V), and styrene oxidation peak is not so pronounced. Therefore, we can conclude that under experimental conditions NHPI is oxidized preferentially over iodine providing PINO radicals that leads to the observed regioselectivity. The contribution of the oxidation of styrene to the overall process is unlikely.
Practical application of the iodo-oxyimidation
The applicability of the developed method for the gram-scale preparation was demonstrated by the synthesis of 3aa (3.1 g, 79%) without column chromatography (Scheme 4).
![[1860-5397-14-188-i4]](/bjoc/content/inline/1860-5397-14-188-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Gram-scale synthesis of compound 3aa.
Scheme 4: Gram-scale synthesis of compound 3aa.
The synthetic utility of the obtained products 3aa and 3ab was demonstrated by the substitution of the iodine atom with O- (methanol), S- (benzenesulfinate) and N- (azide) nucleophiles (Scheme 5).
![[1860-5397-14-188-i5]](/bjoc/content/inline/1860-5397-14-188-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthetic utility of the iodo-oxyimides 3aa and 3ab.
Scheme 5: Synthetic utility of the iodo-oxyimides 3aa and 3ab.
It is noteworthy that the reaction of compound 3aa with sodium benzenesulfinate results in the nucleophilic substitution of both the iodine atom and the oxyphthalimide moiety to form a vicinal disulfone 4b.
Conclusion
Iodo-oxyimidation of vinylarenes using N-hydroxyphthalimide and N-hydroxysuccinimide was developed. PhI(OAc)2 was the best oxidant for the synthesis of the target products (yields up to 91%). In contrast to previous studies in which oxidants promote the electrophilic addition of iodine to the C=C bond, radical addition predominates in the discovered process. Radical pathway starts from the attack of imide-N-oxyl radicals on the double C=C bond, which allows for anti-Markovnikov type regioselectivity of C–O and C–I bond formation. Electrochemical mechanistic studies based on cyclic voltammetry (CV) data confirm proposed reaction mechanism. Possible ways of using the obtained iodo-oxyimidated products via substitution of iodine atom were demonstrated.
Experimental
General procedure for the synthesis of compounds 3aa–ka, 3ab–db, 3fb, 3hb and 3kb (Figure 1)
Iodine (64 mg, 0.25 mmol) was added to a stirred mixture of vinylarene 1a–k (52–97 mg, 0.50 mmol) and N-hydroxyimide 2a,b (58–82 mg, 0.50 mmol) in CH2Cl2 (3 mL) at 20–25 °С. Then, PhI(OAc)2 (97 mg, 0.30 mmol) was added. In the additional experiments compounds 3ca and 3ga were prepared using IBX (140 mg, 0.50 mmol) or DMP (64 mg, 0.15 mmol) instead of PhI(OAc)2. After stirring for 10 min under air atmosphere at 20–25 °С, CH2Cl2 (30 mL) was added. The resulting mixture was washed with an aqueous solution of Na2S2O3·5H2O (200 mg in 20 mL of water), saturated aqueous NaHCO3 solution (20 mL), and with water (20 mL), dried over anhydrous MgSO4 and filtered. CH2Cl2 was evaporated at 20–25 °С under water-jet vacuum (20–30 mm Hg). Products 3aa–ka, 3ab–db, 3fb, 3hb and 3kb were isolated by column chromatography on silica gel using with EtOAc/DCM eluent (with the volume part of EtOAc gradually increased from 0% to 2.5%).
Iodo-oxyimides 3aa–ka, 3ab–db, 3fb, 3hb and 3kb should be stored in a freezer and handled with minimal heat due to their instability at elevated temperatures.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H, 13C and 19F NMR spectra, copies of HRMS and FT-IR spectra and the ORTEP diagram and X-ray data for compound 3ca. | ||
| Format: PDF | Size: 8.5 MB | Download |
| Supporting Information File 2: X-ray structure analysis data for 3ca (CCDC-1845323). | ||
| Format: CIF | Size: 322.5 KB | Download |
References
-
Hicks, R. G. Org. Biomol. Chem. 2007, 5, 1321. doi:10.1039/b617142g
Return to citation in text: [1] -
Mas-Torrent, M.; Crivillers, N.; Rovira, C.; Veciana, J. Chem. Rev. 2012, 112, 2506. doi:10.1021/cr200233g
Return to citation in text: [1] [2] [3] -
Berliner, L. J. History of the Use of Nitroxides (Aminoxyl Radicals) in Biochemistry: Past, Present and Future of Spin Label and Probe Method. In Nitroxides - Theory, Experiment and Applications; Kokorin, A. I., Ed.; InTech: Croatia, 2012; pp 3–24.
Return to citation in text: [1] -
Tretyakov, E. V.; Ovcharenko, V. I. Russ. Chem. Rev. 2009, 78, 971. doi:10.1070/RC2009v078n11ABEH004093
Return to citation in text: [1] -
Train, C.; Norel, L.; Baumgarten, M. Coord. Chem. Rev. 2009, 253, 2342. doi:10.1016/j.ccr.2008.10.004
Return to citation in text: [1] -
Sugawara, T.; Komatsu, H.; Suzuki, K. Chem. Soc. Rev. 2011, 40, 3105. doi:10.1039/c0cs00157k
Return to citation in text: [1] -
Suzuki, K.; Uchida, Y.; Tamura, R.; Shimono, S.; Yamauchi, J. J. Mater. Chem. 2012, 22, 6799. doi:10.1039/c2jm16278d
Return to citation in text: [1] -
Uchida, Y.; Suzuki, K.; Tamura, R. J. Phys. Chem. B 2012, 116, 9791. doi:10.1021/jp301930k
Return to citation in text: [1] -
Suga, T.; Konishi, H.; Nishide, H. Chem. Commun. 2007, 1730. doi:10.1039/b618710b
Return to citation in text: [1] -
Hawker, C. J.; Bosman, A. W.; Harth, E. Chem. Rev. 2001, 101, 3661. doi:10.1021/cr990119u
Return to citation in text: [1] -
Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 5034. doi:10.1002/anie.201002547
Return to citation in text: [1] [2] [3] -
Bagryanskaya, E. G.; Marque, S. R. A. Chem. Rev. 2014, 114, 5011. doi:10.1021/cr4000946
Return to citation in text: [1] -
Ciriminna, R.; Pagliaro, M. Org. Process Res. Dev. 2010, 14, 245. doi:10.1021/op900059x
Return to citation in text: [1] -
Hamada, S.; Furuta, T.; Wada, Y.; Kawabata, T. Angew. Chem., Int. Ed. 2013, 52, 8093. doi:10.1002/anie.201302261
Return to citation in text: [1] -
Ryland, B. L.; Stahl, S. S. Angew. Chem., Int. Ed. 2014, 53, 8824. doi:10.1002/anie.201403110
Return to citation in text: [1] -
Wertz, S.; Studer, A. Green Chem. 2013, 15, 3116. doi:10.1039/c3gc41459k
Return to citation in text: [1] [2] -
Muramatsu, W.; Nakano, K. Org. Lett. 2015, 17, 1549. doi:10.1021/acs.orglett.5b00434
Return to citation in text: [1] -
Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800. doi:10.1021/cr040170k
Return to citation in text: [1] [2] -
Galli, C.; Gentili, P.; Lanzalunga, O. Angew. Chem., Int. Ed. 2008, 47, 4790. doi:10.1002/anie.200704292
Return to citation in text: [1] [2] -
Wentzel, B. B.; Donners, M. P. J.; Alsters, P. L.; Feiters, M. C.; Nolte, R. J. M. Tetrahedron 2000, 56, 7797. doi:10.1016/s0040-4020(00)00679-7
Return to citation in text: [1] -
Amaoka, Y.; Kamijo, S.; Hoshikawa, T.; Inoue, M. J. Org. Chem. 2012, 77, 9959. doi:10.1021/jo301840e
Return to citation in text: [1] -
Sakaguchi, S.; Hirabayashi, T.; Ishii, Y. Chem. Commun. 2002, 516. doi:10.1039/b110638d
Return to citation in text: [1] -
Sakaguchi, S.; Eikawa, M.; Ishii, Y. Tetrahedron Lett. 1997, 38, 7075. doi:10.1016/s0040-4039(97)01652-3
Return to citation in text: [1] -
Ishii, Y.; Iwahama, T.; Sakaguchi, S.; Nakayama, K.; Nishiyama, Y. J. Org. Chem. 1996, 61, 4520. doi:10.1021/jo951970l
Return to citation in text: [1] -
Minisci, F.; Punta, C.; Recupero, F. J. Mol. Catal. A: Chem. 2006, 251, 129. doi:10.1016/j.molcata.2006.02.011
Return to citation in text: [1] -
Lee, J. M.; Park, E. J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2008, 130, 7824. doi:10.1021/ja8031218
Return to citation in text: [1] -
Terent'ev, A. O.; Krylov, I. B.; Sharipov, M. Y.; Kazanskaya, Z. M.; Nikishin, G. I. Tetrahedron 2012, 68, 10263. doi:10.1016/j.tet.2012.10.018
Return to citation in text: [1] -
Terent'ev, A. O.; Krylov, I. B.; Timofeev, V. P.; Starikova, Z. A.; Merkulova, V. M.; Ilovaisky, A. I.; Nikishin, G. I. Adv. Synth. Catal. 2013, 355, 2375. doi:10.1002/adsc.201300341
Return to citation in text: [1] -
Krylov, I. B.; Vil', V. A.; Terent'ev, A. O. Beilstein J. Org. Chem. 2015, 11, 92. doi:10.3762/bjoc.11.13
Return to citation in text: [1] -
Ananikov, V. P.; Eremin, D. B.; Yakukhnov, S. A.; Dilman, A. D.; Levin, V. V.; Egorov, M. P.; Karlov, S. S.; Kustov, L. M.; Tarasov, A. L.; Greish, A. A.; Shesterkina, A. A.; Sakharov, A. M.; Nysenko, Z. N.; Sheremetev, A. B.; Stakheev, A. Y.; Mashkovsky, I. S.; Sukhorukov, A. Y.; Ioffe, S. L.; Terent’ev, A. O.; Vil’, V. A.; Tomilov, Y. V.; Novikov, R. A.; Zlotin, S. G.; Kucherenko, A. S.; Ustyuzhanina, N. E.; Krylov, V. B.; Tsvetkov, Y. E.; Gening, M. L.; Nifantiev, N. E. Mendeleev Commun. 2017, 27, 425. doi:10.1016/j.mencom.2017.09.001
Return to citation in text: [1] -
Krylov, I. B.; Paveliev, S. A.; Shelimov, B. N.; Lokshin, B. V.; Garbuzova, I. A.; Tafeenko, V. A.; Chernyshev, V. V.; Budnikov, A. S.; Nikishin, G. I.; Terent'ev, A. O. Org. Chem. Front. 2017, 4, 1947. doi:10.1039/c7qo00447h
Return to citation in text: [1] -
Guo, Z.; Jin, C.; Zhou, J.; Su, W. RSC Adv. 2016, 6, 79016. doi:10.1039/c6ra14697j
Return to citation in text: [1] -
Lv, Y.; Sun, K.; Wang, T.; Li, G.; Pu, W.; Chai, N.; Shen, H.; Wu, Y. RSC Adv. 2015, 5, 72142. doi:10.1039/c5ra12691f
Return to citation in text: [1] -
Qian, P.-C.; Liu, Y.; Song, R.-J.; Hu, M.; Yang, X.-H.; Xiang, J.-N.; Li, J.-H. Eur. J. Org. Chem. 2015, 1680. doi:10.1002/ejoc.201403616
Return to citation in text: [1] [2] -
Dian, L.; Wang, S.; Zhang-Negrerie, D.; Du, Y. Adv. Synth. Catal. 2015, 357, 3836. doi:10.1002/adsc.201500623
Return to citation in text: [1] -
Dinda, M.; Bose, C.; Ghosh, T.; Maity, S. RSC Adv. 2015, 5, 44928. doi:10.1039/c5ra05719a
Return to citation in text: [1] -
Guo, Z.; Jiang, X.; Jin, C.; Zhou, J.; Sun, B.; Su, W. Synlett 2017, 28, 1321. doi:10.1055/s-0036-1588760
Return to citation in text: [1] -
Lv, Y.; Sun, K.; Pu, W.; Mao, S.; Li, G.; Niu, J.; Chen, Q.; Wang, T. RSC Adv. 2016, 6, 93486. doi:10.1039/c6ra22653a
Return to citation in text: [1] -
Tan, B.; Toda, N.; Barbas, C. F., III. Angew. Chem., Int. Ed. 2012, 51, 12538. doi:10.1002/anie.201205921
Return to citation in text: [1] -
Xu, X.; Li, P.; Huang, Y.; Tong, C.; Yan, Y.; Xie, Y. Tetrahedron Lett. 2017, 58, 1742. doi:10.1016/j.tetlet.2017.03.064
Return to citation in text: [1] -
Xu, X.; Sun, J.; Lin, Y.; Cheng, J.; Li, P.; Yan, Y.; Shuai, Q.; Xie, Y. Org. Biomol. Chem. 2017, 15, 9875. doi:10.1039/C7OB02249B
Return to citation in text: [1] -
Siddaraju, Y.; Prabhu, K. R. Org. Biomol. Chem. 2015, 13, 11651. doi:10.1039/C5OB01929J
Return to citation in text: [1] -
Kompanets, M. O.; Kushch, O. V.; Litvinov, Y. E.; Pliekhov, O. L.; Novikova, K. V.; Novokhatko, A. O.; Shendrik, A. N.; Vasilyev, A. V.; Opeida, I. O. Catal. Commun. 2014, 57, 60. doi:10.1016/j.catcom.2014.08.005
Return to citation in text: [1] -
Kasperczyk, K.; Orlińska, B.; Zawadiak, J. Cent. Eur. J. Chem. 2014, 12, 1176. doi:10.2478/s11532-014-0565-8
Return to citation in text: [1] -
Bag, R.; De, P. B.; Pradhan, S.; Punniyamurthy, T. Eur. J. Org. Chem. 2017, 5424. doi:10.1002/ejoc.201700512
Return to citation in text: [1] [2] -
Xia, X.-F.; Zhu, S.-L.; Hu, Q.-T.; Chen, C. Tetrahedron 2016, 72, 8000. doi:10.1016/j.tet.2016.10.029
Return to citation in text: [1] -
Bag, R.; Sar, D.; Punniyamurthy, T. Org. Biomol. Chem. 2016, 14, 3246. doi:10.1039/c6ob00210b
Return to citation in text: [1] -
Yamamoto, D.; Soga, M.; Ansai, H.; Makino, K. Org. Chem. Front. 2016, 3, 1420. doi:10.1039/c6qo00318d
Return to citation in text: [1] -
Luo, J.; Zhang, J. J. Org. Chem. 2016, 81, 9131. doi:10.1021/acs.joc.6b01704
Return to citation in text: [1] -
Samanta, S.; Ravi, C.; Joshi, A.; Pappula, V.; Adimurthy, S. Tetrahedron Lett. 2017, 58, 721. doi:10.1016/j.tetlet.2016.12.073
Return to citation in text: [1] -
Xia, X.-F.; Zhu, S.-L.; Gu, Z.; Wang, H.; Li, W.; Liu, X.; Liang, Y.-M. J. Org. Chem. 2015, 80, 5572. doi:10.1021/acs.joc.5b00460
Return to citation in text: [1] -
Bag, R.; Sar, D.; Punniyamurthy, T. Org. Lett. 2015, 17, 2010. doi:10.1021/acs.orglett.5b00770
Return to citation in text: [1] -
Andia, A. A.; Miner, M. R.; Woerpel, K. A. Org. Lett. 2015, 17, 2704. doi:10.1021/acs.orglett.5b01120
Return to citation in text: [1] -
Zhang, J.-Z.; Tang, Y. Adv. Synth. Catal. 2016, 358, 752. doi:10.1002/adsc.201500732
Return to citation in text: [1] -
Samanta, S.; Donthiri, R. R.; Ravi, C.; Adimurthy, S. J. Org. Chem. 2016, 81, 3457. doi:10.1021/acs.joc.6b00266
Return to citation in text: [1] [2] -
Huang, L.; Zheng, S.-C.; Tan, B.; Liu, X.-Y. Org. Lett. 2015, 17, 1589. doi:10.1021/acs.orglett.5b00479
Return to citation in text: [1] -
Li, Y.-X.; Wang, Q.-Q.; Yang, L. Org. Biomol. Chem. 2017, 15, 1338. doi:10.1039/c7ob00030h
Return to citation in text: [1] -
Li, Y.; Zhou, X.; Zheng, G.; Zhang, Q. Beilstein J. Org. Chem. 2015, 11, 2721. doi:10.3762/bjoc.11.293
Return to citation in text: [1] -
Xia, X.-F.; Gu, Z.; Liu, W.; Wang, H.; Xia, Y.; Gao, H.; Liu, X.; Liang, Y.-M. J. Org. Chem. 2015, 80, 290. doi:10.1021/jo502327r
Return to citation in text: [1] [2] -
Wirth, T.; Singh, F. V. Synthesis by substitution of other halogens. Science of Synthesis Knowledge Updates: 2015/2; Thieme, 2015; pp 407–414.
Return to citation in text: [1] -
Myint, Y. Y.; Pasha, M. A. Synth. Commun. 2004, 34, 4477. doi:10.1081/scc-200043180
Return to citation in text: [1] -
Sanseverino, A. M.; de Mattos, M. C. S. Synthesis 1998, 1584. doi:10.1055/s-1998-2187
Return to citation in text: [1] -
Courtneidge, J. L.; Lusztyk, J.; Pagé, D. Tetrahedron Lett. 1994, 35, 1003. doi:10.1016/s0040-4039(00)79950-3
Return to citation in text: [1] -
Achar, T. K.; Maiti, S.; Mal, P. Org. Biomol. Chem. 2016, 14, 4654. doi:10.1039/c6ob00532b
Return to citation in text: [1] -
Gottam, H.; Vinod, T. K. J. Org. Chem. 2011, 76, 974. doi:10.1021/jo102051z
Return to citation in text: [1] -
Moorthy, J. N.; Senapati, K.; Kumar, S. J. Org. Chem. 2009, 74, 6287. doi:10.1021/jo9007892
Return to citation in text: [1] -
Yusubov, M. S.; Drygunova, L. A.; Zhdankin, V. V. Synthesis 2004, 2289. doi:10.1055/s-2004-831175
Return to citation in text: [1] -
Yusubov, M. S.; Yusubova, R. J.; Filimonov, V. D.; Chi, K.-W. Synth. Commun. 2004, 34, 443. doi:10.1081/scc-120027283
Return to citation in text: [1] -
Yusubov, M. S.; Yusubova, R. Y.; Filimonov, V. D.; Chi, K.-W. Russ. J. Org. Chem. 2002, 38, 902. doi:10.1023/a:1020320011241
Return to citation in text: [1] -
De Corso, A. R.; Panunzi, B.; Tingoli, M. Tetrahedron Lett. 2001, 42, 7245. doi:10.1016/s0040-4039(01)01509-x
Return to citation in text: [1] -
Hokamp, T.; Storm, A. T.; Yusubov, M.; Wirth, T. Synlett 2018, 29, 415. doi:10.1055/s-0036-1589119
Return to citation in text: [1] -
Terent’ev, A. O.; Krylov, I. B.; Borisov, D. A.; Nikishin, G. I. Synthesis 2007, 2979. doi:10.1055/s-2007-990776
Return to citation in text: [1] -
Terent’ev, A. O.; Borisov, A. M.; Platonov, M. M.; Starikova, Z. A.; Chernyshev, V. V.; Nikishin, G. I. Synthesis 2009, 4159. doi:10.1055/s-0029-1217062
Return to citation in text: [1] -
Wu, X.-F.; Gong, J.-L.; Qi, X. Org. Biomol. Chem. 2014, 12, 5807. doi:10.1039/c4ob00276h
Return to citation in text: [1] -
Thottumkara, A. P.; Bowsher, M. S.; Vinod, T. K. Org. Lett. 2005, 7, 2933. doi:10.1021/ol050875o
Return to citation in text: [1] -
Xia, X.-F.; Zhu, S.-L.; Zhang, D. Tetrahedron 2015, 71, 8517. doi:10.1016/j.tet.2015.09.040
Return to citation in text: [1] -
Krylov, I. B.; Kompanets, M. O.; Novikova, K. V.; Opeida, I. O.; Kushch, O. V.; Shelimov, B. N.; Nikishin, G. I.; Levitsky, D. O.; Terent'ev, A. O. J. Phys. Chem. A 2016, 120, 68. doi:10.1021/acs.jpca.5b10722
Return to citation in text: [1] -
Rafiee, M.; Wang, F.; Hruszkewycz, D. P.; Stahl, S. S. J. Am. Chem. Soc. 2018, 140, 22. doi:10.1021/jacs.7b09744
Return to citation in text: [1] -
Nair, V.; Augustine, A.; George, T. G.; Nair, L. G. Tetrahedron Lett. 2001, 42, 6763. doi:10.1016/s0040-4039(01)01377-6
Return to citation in text: [1] -
Jörissen, J.; Speiser, B. Preparative Electrolysis on the Laboratory Scale. In Organic Electrochemistry, Fifth Edition: Revised and Expanded; Hammerich, O.; Speiser, B., Eds.; CRC Press, 2015; pp 265–329.
Return to citation in text: [1]
| 1. | Hicks, R. G. Org. Biomol. Chem. 2007, 5, 1321. doi:10.1039/b617142g |
| 2. | Mas-Torrent, M.; Crivillers, N.; Rovira, C.; Veciana, J. Chem. Rev. 2012, 112, 2506. doi:10.1021/cr200233g |
| 3. | Berliner, L. J. History of the Use of Nitroxides (Aminoxyl Radicals) in Biochemistry: Past, Present and Future of Spin Label and Probe Method. In Nitroxides - Theory, Experiment and Applications; Kokorin, A. I., Ed.; InTech: Croatia, 2012; pp 3–24. |
| 9. | Suga, T.; Konishi, H.; Nishide, H. Chem. Commun. 2007, 1730. doi:10.1039/b618710b |
| 56. | Huang, L.; Zheng, S.-C.; Tan, B.; Liu, X.-Y. Org. Lett. 2015, 17, 1589. doi:10.1021/acs.orglett.5b00479 |
| 57. | Li, Y.-X.; Wang, Q.-Q.; Yang, L. Org. Biomol. Chem. 2017, 15, 1338. doi:10.1039/c7ob00030h |
| 7. | Suzuki, K.; Uchida, Y.; Tamura, R.; Shimono, S.; Yamauchi, J. J. Mater. Chem. 2012, 22, 6799. doi:10.1039/c2jm16278d |
| 8. | Uchida, Y.; Suzuki, K.; Tamura, R. J. Phys. Chem. B 2012, 116, 9791. doi:10.1021/jp301930k |
| 58. | Li, Y.; Zhou, X.; Zheng, G.; Zhang, Q. Beilstein J. Org. Chem. 2015, 11, 2721. doi:10.3762/bjoc.11.293 |
| 59. | Xia, X.-F.; Gu, Z.; Liu, W.; Wang, H.; Xia, Y.; Gao, H.; Liu, X.; Liang, Y.-M. J. Org. Chem. 2015, 80, 290. doi:10.1021/jo502327r |
| 2. | Mas-Torrent, M.; Crivillers, N.; Rovira, C.; Veciana, J. Chem. Rev. 2012, 112, 2506. doi:10.1021/cr200233g |
| 6. | Sugawara, T.; Komatsu, H.; Suzuki, K. Chem. Soc. Rev. 2011, 40, 3105. doi:10.1039/c0cs00157k |
| 46. | Xia, X.-F.; Zhu, S.-L.; Hu, Q.-T.; Chen, C. Tetrahedron 2016, 72, 8000. doi:10.1016/j.tet.2016.10.029 |
| 47. | Bag, R.; Sar, D.; Punniyamurthy, T. Org. Biomol. Chem. 2016, 14, 3246. doi:10.1039/c6ob00210b |
| 48. | Yamamoto, D.; Soga, M.; Ansai, H.; Makino, K. Org. Chem. Front. 2016, 3, 1420. doi:10.1039/c6qo00318d |
| 49. | Luo, J.; Zhang, J. J. Org. Chem. 2016, 81, 9131. doi:10.1021/acs.joc.6b01704 |
| 50. | Samanta, S.; Ravi, C.; Joshi, A.; Pappula, V.; Adimurthy, S. Tetrahedron Lett. 2017, 58, 721. doi:10.1016/j.tetlet.2016.12.073 |
| 51. | Xia, X.-F.; Zhu, S.-L.; Gu, Z.; Wang, H.; Li, W.; Liu, X.; Liang, Y.-M. J. Org. Chem. 2015, 80, 5572. doi:10.1021/acs.joc.5b00460 |
| 4. | Tretyakov, E. V.; Ovcharenko, V. I. Russ. Chem. Rev. 2009, 78, 971. doi:10.1070/RC2009v078n11ABEH004093 |
| 5. | Train, C.; Norel, L.; Baumgarten, M. Coord. Chem. Rev. 2009, 253, 2342. doi:10.1016/j.ccr.2008.10.004 |
| 52. | Bag, R.; Sar, D.; Punniyamurthy, T. Org. Lett. 2015, 17, 2010. doi:10.1021/acs.orglett.5b00770 |
| 53. | Andia, A. A.; Miner, M. R.; Woerpel, K. A. Org. Lett. 2015, 17, 2704. doi:10.1021/acs.orglett.5b01120 |
| 54. | Zhang, J.-Z.; Tang, Y. Adv. Synth. Catal. 2016, 358, 752. doi:10.1002/adsc.201500732 |
| 55. | Samanta, S.; Donthiri, R. R.; Ravi, C.; Adimurthy, S. J. Org. Chem. 2016, 81, 3457. doi:10.1021/acs.joc.6b00266 |
| 11. | Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 5034. doi:10.1002/anie.201002547 |
| 13. | Ciriminna, R.; Pagliaro, M. Org. Process Res. Dev. 2010, 14, 245. doi:10.1021/op900059x |
| 14. | Hamada, S.; Furuta, T.; Wada, Y.; Kawabata, T. Angew. Chem., Int. Ed. 2013, 52, 8093. doi:10.1002/anie.201302261 |
| 15. | Ryland, B. L.; Stahl, S. S. Angew. Chem., Int. Ed. 2014, 53, 8824. doi:10.1002/anie.201403110 |
| 16. | Wertz, S.; Studer, A. Green Chem. 2013, 15, 3116. doi:10.1039/c3gc41459k |
| 17. | Muramatsu, W.; Nakano, K. Org. Lett. 2015, 17, 1549. doi:10.1021/acs.orglett.5b00434 |
| 18. | Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800. doi:10.1021/cr040170k |
| 19. | Galli, C.; Gentili, P.; Lanzalunga, O. Angew. Chem., Int. Ed. 2008, 47, 4790. doi:10.1002/anie.200704292 |
| 43. | Kompanets, M. O.; Kushch, O. V.; Litvinov, Y. E.; Pliekhov, O. L.; Novikova, K. V.; Novokhatko, A. O.; Shendrik, A. N.; Vasilyev, A. V.; Opeida, I. O. Catal. Commun. 2014, 57, 60. doi:10.1016/j.catcom.2014.08.005 |
| 44. | Kasperczyk, K.; Orlińska, B.; Zawadiak, J. Cent. Eur. J. Chem. 2014, 12, 1176. doi:10.2478/s11532-014-0565-8 |
| 12. | Bagryanskaya, E. G.; Marque, S. R. A. Chem. Rev. 2014, 114, 5011. doi:10.1021/cr4000946 |
| 45. | Bag, R.; De, P. B.; Pradhan, S.; Punniyamurthy, T. Eur. J. Org. Chem. 2017, 5424. doi:10.1002/ejoc.201700512 |
| 10. | Hawker, C. J.; Bosman, A. W.; Harth, E. Chem. Rev. 2001, 101, 3661. doi:10.1021/cr990119u |
| 11. | Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 5034. doi:10.1002/anie.201002547 |
| 2. | Mas-Torrent, M.; Crivillers, N.; Rovira, C.; Veciana, J. Chem. Rev. 2012, 112, 2506. doi:10.1021/cr200233g |
| 11. | Tebben, L.; Studer, A. Angew. Chem., Int. Ed. 2011, 50, 5034. doi:10.1002/anie.201002547 |
| 16. | Wertz, S.; Studer, A. Green Chem. 2013, 15, 3116. doi:10.1039/c3gc41459k |
| 18. | Recupero, F.; Punta, C. Chem. Rev. 2007, 107, 3800. doi:10.1021/cr040170k |
| 19. | Galli, C.; Gentili, P.; Lanzalunga, O. Angew. Chem., Int. Ed. 2008, 47, 4790. doi:10.1002/anie.200704292 |
| 20. | Wentzel, B. B.; Donners, M. P. J.; Alsters, P. L.; Feiters, M. C.; Nolte, R. J. M. Tetrahedron 2000, 56, 7797. doi:10.1016/s0040-4020(00)00679-7 |
| 21. | Amaoka, Y.; Kamijo, S.; Hoshikawa, T.; Inoue, M. J. Org. Chem. 2012, 77, 9959. doi:10.1021/jo301840e |
| 22. | Sakaguchi, S.; Hirabayashi, T.; Ishii, Y. Chem. Commun. 2002, 516. doi:10.1039/b110638d |
| 23. | Sakaguchi, S.; Eikawa, M.; Ishii, Y. Tetrahedron Lett. 1997, 38, 7075. doi:10.1016/s0040-4039(97)01652-3 |
| 24. | Ishii, Y.; Iwahama, T.; Sakaguchi, S.; Nakayama, K.; Nishiyama, Y. J. Org. Chem. 1996, 61, 4520. doi:10.1021/jo951970l |
| 25. | Minisci, F.; Punta, C.; Recupero, F. J. Mol. Catal. A: Chem. 2006, 251, 129. doi:10.1016/j.molcata.2006.02.011 |
| 26. | Lee, J. M.; Park, E. J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2008, 130, 7824. doi:10.1021/ja8031218 |
| 27. | Terent'ev, A. O.; Krylov, I. B.; Sharipov, M. Y.; Kazanskaya, Z. M.; Nikishin, G. I. Tetrahedron 2012, 68, 10263. doi:10.1016/j.tet.2012.10.018 |
| 28. | Terent'ev, A. O.; Krylov, I. B.; Timofeev, V. P.; Starikova, Z. A.; Merkulova, V. M.; Ilovaisky, A. I.; Nikishin, G. I. Adv. Synth. Catal. 2013, 355, 2375. doi:10.1002/adsc.201300341 |
| 29. | Krylov, I. B.; Vil', V. A.; Terent'ev, A. O. Beilstein J. Org. Chem. 2015, 11, 92. doi:10.3762/bjoc.11.13 |
| 30. | Ananikov, V. P.; Eremin, D. B.; Yakukhnov, S. A.; Dilman, A. D.; Levin, V. V.; Egorov, M. P.; Karlov, S. S.; Kustov, L. M.; Tarasov, A. L.; Greish, A. A.; Shesterkina, A. A.; Sakharov, A. M.; Nysenko, Z. N.; Sheremetev, A. B.; Stakheev, A. Y.; Mashkovsky, I. S.; Sukhorukov, A. Y.; Ioffe, S. L.; Terent’ev, A. O.; Vil’, V. A.; Tomilov, Y. V.; Novikov, R. A.; Zlotin, S. G.; Kucherenko, A. S.; Ustyuzhanina, N. E.; Krylov, V. B.; Tsvetkov, Y. E.; Gening, M. L.; Nifantiev, N. E. Mendeleev Commun. 2017, 27, 425. doi:10.1016/j.mencom.2017.09.001 |
| 31. | Krylov, I. B.; Paveliev, S. A.; Shelimov, B. N.; Lokshin, B. V.; Garbuzova, I. A.; Tafeenko, V. A.; Chernyshev, V. V.; Budnikov, A. S.; Nikishin, G. I.; Terent'ev, A. O. Org. Chem. Front. 2017, 4, 1947. doi:10.1039/c7qo00447h |
| 32. | Guo, Z.; Jin, C.; Zhou, J.; Su, W. RSC Adv. 2016, 6, 79016. doi:10.1039/c6ra14697j |
| 33. | Lv, Y.; Sun, K.; Wang, T.; Li, G.; Pu, W.; Chai, N.; Shen, H.; Wu, Y. RSC Adv. 2015, 5, 72142. doi:10.1039/c5ra12691f |
| 34. | Qian, P.-C.; Liu, Y.; Song, R.-J.; Hu, M.; Yang, X.-H.; Xiang, J.-N.; Li, J.-H. Eur. J. Org. Chem. 2015, 1680. doi:10.1002/ejoc.201403616 |
| 35. | Dian, L.; Wang, S.; Zhang-Negrerie, D.; Du, Y. Adv. Synth. Catal. 2015, 357, 3836. doi:10.1002/adsc.201500623 |
| 36. | Dinda, M.; Bose, C.; Ghosh, T.; Maity, S. RSC Adv. 2015, 5, 44928. doi:10.1039/c5ra05719a |
| 37. | Guo, Z.; Jiang, X.; Jin, C.; Zhou, J.; Sun, B.; Su, W. Synlett 2017, 28, 1321. doi:10.1055/s-0036-1588760 |
| 38. | Lv, Y.; Sun, K.; Pu, W.; Mao, S.; Li, G.; Niu, J.; Chen, Q.; Wang, T. RSC Adv. 2016, 6, 93486. doi:10.1039/c6ra22653a |
| 39. | Tan, B.; Toda, N.; Barbas, C. F., III. Angew. Chem., Int. Ed. 2012, 51, 12538. doi:10.1002/anie.201205921 |
| 40. | Xu, X.; Li, P.; Huang, Y.; Tong, C.; Yan, Y.; Xie, Y. Tetrahedron Lett. 2017, 58, 1742. doi:10.1016/j.tetlet.2017.03.064 |
| 41. | Xu, X.; Sun, J.; Lin, Y.; Cheng, J.; Li, P.; Yan, Y.; Shuai, Q.; Xie, Y. Org. Biomol. Chem. 2017, 15, 9875. doi:10.1039/C7OB02249B |
| 42. | Siddaraju, Y.; Prabhu, K. R. Org. Biomol. Chem. 2015, 13, 11651. doi:10.1039/C5OB01929J |
| 63. | Courtneidge, J. L.; Lusztyk, J.; Pagé, D. Tetrahedron Lett. 1994, 35, 1003. doi:10.1016/s0040-4039(00)79950-3 |
| 64. | Achar, T. K.; Maiti, S.; Mal, P. Org. Biomol. Chem. 2016, 14, 4654. doi:10.1039/c6ob00532b |
| 65. | Gottam, H.; Vinod, T. K. J. Org. Chem. 2011, 76, 974. doi:10.1021/jo102051z |
| 66. | Moorthy, J. N.; Senapati, K.; Kumar, S. J. Org. Chem. 2009, 74, 6287. doi:10.1021/jo9007892 |
| 67. | Yusubov, M. S.; Drygunova, L. A.; Zhdankin, V. V. Synthesis 2004, 2289. doi:10.1055/s-2004-831175 |
| 68. | Yusubov, M. S.; Yusubova, R. J.; Filimonov, V. D.; Chi, K.-W. Synth. Commun. 2004, 34, 443. doi:10.1081/scc-120027283 |
| 69. | Yusubov, M. S.; Yusubova, R. Y.; Filimonov, V. D.; Chi, K.-W. Russ. J. Org. Chem. 2002, 38, 902. doi:10.1023/a:1020320011241 |
| 70. | De Corso, A. R.; Panunzi, B.; Tingoli, M. Tetrahedron Lett. 2001, 42, 7245. doi:10.1016/s0040-4039(01)01509-x |
| 71. | Hokamp, T.; Storm, A. T.; Yusubov, M.; Wirth, T. Synlett 2018, 29, 415. doi:10.1055/s-0036-1589119 |
| 72. | Terent’ev, A. O.; Krylov, I. B.; Borisov, D. A.; Nikishin, G. I. Synthesis 2007, 2979. doi:10.1055/s-2007-990776 |
| 73. | Terent’ev, A. O.; Borisov, A. M.; Platonov, M. M.; Starikova, Z. A.; Chernyshev, V. V.; Nikishin, G. I. Synthesis 2009, 4159. doi:10.1055/s-0029-1217062 |
| 60. | Wirth, T.; Singh, F. V. Synthesis by substitution of other halogens. Science of Synthesis Knowledge Updates: 2015/2; Thieme, 2015; pp 407–414. |
| 61. | Myint, Y. Y.; Pasha, M. A. Synth. Commun. 2004, 34, 4477. doi:10.1081/scc-200043180 |
| 62. | Sanseverino, A. M.; de Mattos, M. C. S. Synthesis 1998, 1584. doi:10.1055/s-1998-2187 |
| 78. | Rafiee, M.; Wang, F.; Hruszkewycz, D. P.; Stahl, S. S. J. Am. Chem. Soc. 2018, 140, 22. doi:10.1021/jacs.7b09744 |
| 79. | Nair, V.; Augustine, A.; George, T. G.; Nair, L. G. Tetrahedron Lett. 2001, 42, 6763. doi:10.1016/s0040-4039(01)01377-6 |
| 80. | Jörissen, J.; Speiser, B. Preparative Electrolysis on the Laboratory Scale. In Organic Electrochemistry, Fifth Edition: Revised and Expanded; Hammerich, O.; Speiser, B., Eds.; CRC Press, 2015; pp 265–329. |
| 34. | Qian, P.-C.; Liu, Y.; Song, R.-J.; Hu, M.; Yang, X.-H.; Xiang, J.-N.; Li, J.-H. Eur. J. Org. Chem. 2015, 1680. doi:10.1002/ejoc.201403616 |
| 55. | Samanta, S.; Donthiri, R. R.; Ravi, C.; Adimurthy, S. J. Org. Chem. 2016, 81, 3457. doi:10.1021/acs.joc.6b00266 |
| 59. | Xia, X.-F.; Gu, Z.; Liu, W.; Wang, H.; Xia, Y.; Gao, H.; Liu, X.; Liang, Y.-M. J. Org. Chem. 2015, 80, 290. doi:10.1021/jo502327r |
| 76. | Xia, X.-F.; Zhu, S.-L.; Zhang, D. Tetrahedron 2015, 71, 8517. doi:10.1016/j.tet.2015.09.040 |
| 77. | Krylov, I. B.; Kompanets, M. O.; Novikova, K. V.; Opeida, I. O.; Kushch, O. V.; Shelimov, B. N.; Nikishin, G. I.; Levitsky, D. O.; Terent'ev, A. O. J. Phys. Chem. A 2016, 120, 68. doi:10.1021/acs.jpca.5b10722 |
| 45. | Bag, R.; De, P. B.; Pradhan, S.; Punniyamurthy, T. Eur. J. Org. Chem. 2017, 5424. doi:10.1002/ejoc.201700512 |
| 74. | Wu, X.-F.; Gong, J.-L.; Qi, X. Org. Biomol. Chem. 2014, 12, 5807. doi:10.1039/c4ob00276h |
| 75. | Thottumkara, A. P.; Bowsher, M. S.; Vinod, T. K. Org. Lett. 2005, 7, 2933. doi:10.1021/ol050875o |
© 2018 Krylov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)