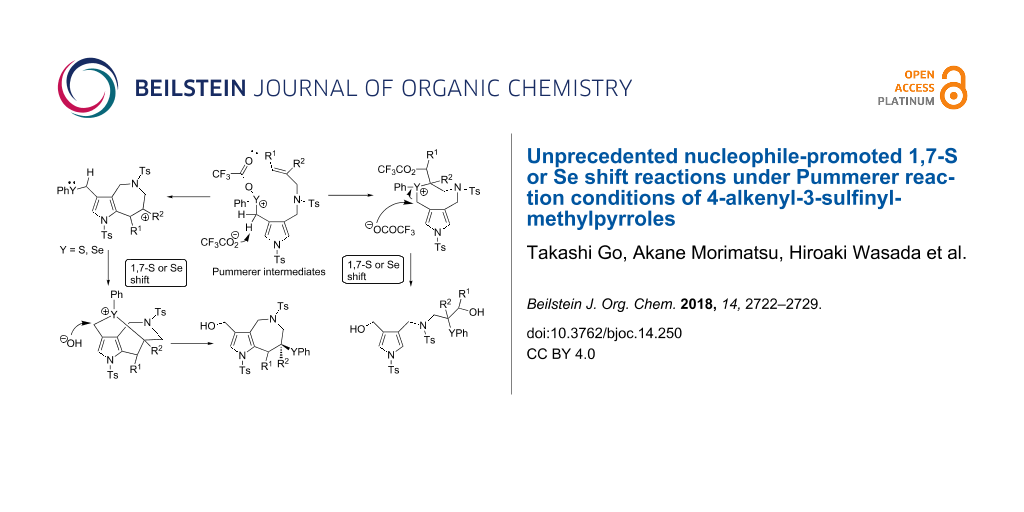
Abstract
A unique 1,7-S- and Se-shift reaction under Pummerer reaction conditions of 4-alkenyl-3-sulfinyl- and seleninylpyrroles was described. The usual Pummerer reaction of 4-(alkenylaminomethyl)-3-phenylsulfinylpyrroles and a successive reaction with tetrabutylammonium hydroxide (TBAH) yielded either pyrrolo[3,2-c]azepines or N-pyrrol-3-ylmethyl-N-(4-hydroxy-3-sulfanylpropyl)-p-toluenesulfonamides (diols). Seleno-Pummerer reactions of 3-selanylmethylpyrroles also proceeded via in situ generation of selenoxides, followed by a treatment with TBAH.

Graphical Abstract
Introduction
Pyrrolo- and indoloazepine skeletons are chemical frameworks present in marine natural products [1,2] such as hymenialdisines [3,4], latonduines [5], paullones, kenpaullones and alsterpaullones [6,7]. These compounds can be used for various pharmaceutical applications [8] such as for the treatment of neurodegenerative and proliferative disorders. These compounds operate by inhibiting proteins or enzymes that regulate cell cycles, including cyclin-dependent kinases [9], tyrosine kinase [10], glycogen-synthase kinase and mitochondrial malate dehydrogenase [11]. These interesting biological activities led us to develop one-pot sequential or cascade protocols for the synthesis of [2,3-b]-, [2,3-c]- and [3,4-b]pyrrolo- and indoloazepines [12-20]. However, the yields of the pyrrolo- and indolo[3,2-c]azepines were relatively low. One of the most efficient procedures for preparing pyrrolo[3,2-c]azepin-4-ones involves the Beckmann rearrangement of 6,7-dihydroindol-4(5H)-ones. However, after implementing this approach, an undesirable isomer was obtained [21]. Although a number of acid- [22,23] and metal-catalysed cascade processes [24,25] have been developed (for example, Pictet–Spengler [26] and Ugi-type reactions [27], the acyl radical cyclisations [28] and the 1,7-electrocyclisation of azomethine ylides or ring expansion sequences [29]), most of these approaches are ineffective for synthesising pyrrolo[3,2-c]azepines. Recently, Echavarren and Beller reported a novel Au- or Pt-catalysed cycloisomerisation reaction of pyrrole-2-carboxamides that produces pyrrolo[2,3-c]- and [2,3-d]azepinones [30,31]. However, this reaction is not suitable for the synthesis of pyrrolo[3,2-c]azepines in viewpoint of the difficulties in preparing the starting materials.
We previously reported that the hydroamination/cyclisation reaction of sulfanyl-1,6-diynes with secondary amines resulted in the formation of 3-sulfanylmethyl-4-aminomethylpyrroles (Scheme 1) [32,33]. The sulfanyl group of pyrroles is easily oxidised to produce their corresponding sulfoxides. When we performed the Pummerer reaction and cyclization of pyrrolesulfoxides to obtain their corresponding pyrroloazepines, we observed unprecedented events, i.e., 1,7-sulfanyl group (1,7-S) shift reactions, on the cation intermediates. Herein, we report the unique 1,7-S and 1,7-Se shift reactions through Pummerer reactions of alkenyl pyrrolomethyl sulfoxides and the corresponding successive cyclisation reactions.
![[1860-5397-14-250-i1]](/bjoc/content/inline/1860-5397-14-250-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Our synthetic plan for pyrrolo[3,2-c]azepines.
Scheme 1: Our synthetic plan for pyrrolo[3,2-c]azepines.
Results and Discussion
We prepared sulfoxides as substrates for the Pummerer reactions according to the aforementioned method [32,33]; subsequently, oxidation with mCPBA was performed (Scheme 2). Both the allylic sulfides 4a–f and their sulfoxides 5a–f were obtained in good to high yields. Corresponding pyrroloselenides 6a–f were prepared by applying a similar synthetic sequence; however, the selenoxides could not be obtained by implementing the usual reaction conditions. The structure of the products derived from the amination–cyclisation reaction of sulfanyl 1,6-diyne 1a was determined by analysing the single-crystal X-ray diffraction spectrum of the corresponding sulfone of 4a; the structure was a pyrrole bearing both 3-sulfanylmethyl and 4-allylaminomethyl groups (see Supporting Information File 1).
![[1860-5397-14-250-i2]](/bjoc/content/inline/1860-5397-14-250-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of precursors for the Pummerer reactions.
Scheme 2: Preparation of precursors for the Pummerer reactions.
To develop a useful Pummerer reaction procedure, we selected allylamine derivative 5a as representative substrate and performed a screening to find suitable conditions for its reaction with anhydrides (Table 1). Since the reaction of 5a with acetic anhydride/acetic acid completely stagnated at room temperature, we performed the same reaction at 120 °C. However, the reaction conditions did not yield the desired cyclic products and ester 7a was instead produced in good yield (entry 1 in Table 1). We next examined the reaction of 5a with 1.5 equiv of trifluoroacetic anhydride (TFAA) in dichloromethane at 0 °C; however, the reaction yielded a complex mixture (Table 1, entry 2). Since the reaction with trifluoromethanesulfonic anhydride resulted in the recovery of 5a (Table 1, entry 3), we reinvestigated the reaction of 5a with TFAA in detail. The use of an excess of TFAA, followed by the treatment with triethylamine yielded aldehyde 8a instead of alcohol 7a (Table 1, entry 4). These results show that the reaction conducted in the presence of an excess of TFAA was indeed successful and the Pummerer reaction products such as α-trifluoroacetoxysulfide underwent hydrolysis to yield aldehyde 8a. In entries 5 and 6, we present the results from performing the reactions at lower temperatures. Fortunately, we found the formation of unprecedented product 9a, which was contaminated with unknown products at both −20 °C and −40 °C. The Pummerer rearrangement and successive cyclisation with either the intramolecular C–C double bond or the pyrrole were unsuccessful; however, N-(3-hydroxy-2-(phenylthio)propyl)-N-((4-(hydroxymethyl)-1-tosyl-1H-pyrrol-3-yl)methyl)-4-methylbenzenesulfonamide (9a) was apparently obtained via the sulfanyl group migration (S-shift reaction) (refer the NMR studies of 9a in Supporting Information File 1). To clarify the details of the S-shift reactions, we performed the usual Pummerer reaction, followed by treatment with bases at −20 °C (Table 1, entries 8–10). We determined that the S-shift reaction was strongly affected by the base. Tetrabutylammonium hydroxide was found to be the best base for the formation of 9a (Table 1, entry 10). Neither trimethylsilyl trifluoromethanesulfonate nor oxallyl chloride/triethylamine proved effective for the formation of 9a (Table 1, entries 11 and 12).
Table 1: Screening the reaction conditions for the Pummerer reactions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-250-i8.svg?max-width=637&scale=1.0)
|
||||||||
| entry | anhydride [equiv] | solvent | T [°C] | time [h] | 5a | yields [%] 7a | 8a | 9a |
|---|---|---|---|---|---|---|---|---|
| 1 | Ac2O (5) | AcOH | 120 | 6 | 86 | – | – | |
| 2 | TFAA (1.5) | DCM | 0 | 2 | – | – | – | – |
| 3 | Tf2O (1.5) | MeCN | 0 | 1 | – | – | – | |
| 4 | TFAA (8), Et3N (3) | DCM | 0 | 1.3 | – | 36 | – | |
| 5 | TFAA (5) | DCM | −20 | 0.5 | – | – | –a | |
| 6 | TFAA (5) | DCE | −40 | 0.3 | – | – | –a | |
| 7 | TFAA (5) | THF | −20 | 0.5 | – | 86 | – | |
| 8 | TFAA (5), Bu4NOH aq | DCM | −20 | 2 | – | – | 67 | |
| 9 | TFAA (5), K2CO3 aq | DCM | −20 | 1.5 | – | – | 46 | |
| 10 | TFAA (5), Bu4NOH aq | DCM | −20 | 0.5 | – | – | 93 | |
| 11 | TMSOTf (3) | DCE | −20 | 0.7 | – | – | – | |
| 12 | (COCl)2 (5), Et3N (5) | DCE | 0 | 0.3 | – | 74 | 18 | |
a9a was obtained contaminated with traces of other unknown compounds.
Although the synthesis of the desired pyrrolo[3,2-c]azepines was unsuccessful, we discovered the unique 1,7-S shift reactions. We next examined the scope of 1,7-S- and 1,7-Se-shift reactions under Pummerer reaction conditions of sulfoxides and the results are shown in Scheme 3. We first examined the following one-pot sequential reactions of the selenium analog: the mCPBA oxidation of N-allylpyrrolomethylselenide 6a, the following Pummerer reaction, and final treatment with TBAH. The stepwise procedure succeeded to give 7-phenylselenodiol 10a in 91% yield. Next, we performed the Pummerer reaction of N-β-methallyl sulfoxide 5b in order to clarify the substituent effects on the N-alkenyl groups. Surprisingly, the reaction of 5b afforded the intramolecular cyclised pyrroloazepine 11b. The N-methallylselenopyrrole 6b also gave the pyrroloazepine 10b, which was formed via the 1,7-Se shift reaction. While N-3-methylbut-2-enyl derivatives 4c and 6c yielded 1,7-S- and Se-shifted diols 9c and 10c, respectively. Interestingly, the reaction of N-cinnamylpyrroles 4d and 6d exclusively afforded trans-azepines 11d and 12d via intramolecular cyclization. The same procedure applied to the bulky N-(3-methylbut-2-enyl) derivatives 4e and 6e yielded the corresponding diols 9e and 10e. Whether the Pummerer reactions yielded cyclised products or not was strongly affected by the substituent patterns. If the azepinium cation intermediates were stabilised by either α-methyl or β-phenyl substituents, the intramolecular cyclisation with the pyrrole ring proceeded to afford azepinoindoles. If both substituents stabilised the intermediates, N-allyl (n = 1; R1 = R2 = H), N-2-butenyl (n = 1; R1 = H, R2 = Me) and N-3-methyl-2-butenyl derivatives (n = 1; R1 = R2 = Me) exclusively gave the diols 9a, 9c, 9e, 10a, 10c and 10e. In particular, all products were obtained through 1,7-S- or 1,7-Se shift reactions. The yields of the Pummerer reactions of both N-3-butenyl derivatives 9f and 10f were very low. These products were confirmed by X-ray spectra and/or single-crystal X-ray diffraction patterns.
![[1860-5397-14-250-i3]](/bjoc/content/inline/1860-5397-14-250-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of 1,7-S and 1,7-Se shift reactions.
Scheme 3: Substrate scope of 1,7-S and 1,7-Se shift reactions.
The unique S-shift reactions in the formation of either pyrrolo[3,2-c]azepines or diols proceeded according to the mechanism we proposed, which is depicted in Scheme 4. Sulfoxide 13 is activated by TFAA to form sulfonium salt 14, which is characterised by an intramolecular double bond [34,35]. A less substituted double bond easily approaches the sulfonium ion and undergoes an addition reaction to produce intermediate 15. The trifluoroacetate anion attacks the posterior side of the α-carbon of the sulfur atom. Bis(trifluoro)acetate 16 resulted from the opening of the ring is hydrolysed by TBAH to yield diol 17. Conversely, sterically hindered double bonds of 14 are not easily accessible to the sulfonium ion; therefore, the Pummerer intermediate α-thio carbenium ion 18 is generated through detrifluoroacetoxylation of 14 (path b: normal Pummerer reactions) and undergoes intramolecular cyclisation on the pyrrole ring of 19. Aromatisation of dihydropyrrole intermediate 20 leads to the formation of pyrroloazepinic cation 21, which is stabilised by the phenylsulfanyl group at its 3-position. The S-shift reaction involves the transannular sulfonium intermediate 22, which is attacked by a hydroxide anion from the anti-direction. Both products, diols and azepinopyrroles, are obtained in a stereoselective manner. In order to confirm the reaction mechanism, we performed crossover experiments with arylthiol (Scheme 5). First, we examined the Pummerer reactions in the presence of p-methoxybenzenethiol or p-chlorobenzenethiol; however, the reactions yielded the reductive sulfide 4a. We next examined the stepwise process: i) the Pummerer reaction of 5a with TFAA, followed by ii) the usual work-up and successive treatment with p-methoxybenzenethiol/TBAH, which exclusively produced 9a (the diol 24a was not detected by ESI mass spectroscopy). A similar result was obtained from the reaction of 5a with p-chlorobenzenethiol/TBAH. These results support the hypothesis that the reaction proceeds via the initial formation of the key intermediate 15, of which the sulfanyl group would intramolecularly migrate to produce 16 stereoselectively (path c, not d). The hydrolysis of 16 using TBAH yields diol 17. We further confirmed the mechanism of the 1,7-S shift reaction by performing density functional theory (DFT)-based calculations (Table 2) [36]. Whether the reactions lead to the formation of pyrroloazepines or diols clearly depends on the substituents in the alkenyl group. We performed DFT calculations for the reaction pathways starting from the key intermediate 14 bearing methyl and phenyl groups. The DFT-calculated ΔG values of 15a is by 24 kcal/mol lower than that of 21a (R1 = R2 = H), which is in accordance with the experimental result that the reaction of 4a leads to the formation of diol 9a. The calculated results related to 15b,c were also found to match the experimental data. The S-shift process of pyrrolo[3,2-c]azepinium cation 21x enables the reaction to proceed via an intramolecular 1,7-S shift, followed by a hydroxide attack on 22 (relevant details are reported in Supporting Information File 1).
![[1860-5397-14-250-i4]](/bjoc/content/inline/1860-5397-14-250-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-14-250-i5]](/bjoc/content/inline/1860-5397-14-250-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-250-i9.svg?max-width=637&scale=1.0)
We further investigated the cyclisation of diols to synthesise pyrrolo[3,2-c]azepines (Scheme 6). However, the products obtained were not the desired pyrrolo[3,2-c]azepines but were 2,6,7,8,9,10-hexahydro-4H-pyrrolo[3,4-g][1,5]oxazonines 25a–e, which were formed via a reaction route that involved the intramolecular dehydration of diols. To confirm the structure of 4H-pyrrolo[3,4-g]oxazines, we prepared an N-bromobenzenesulfonyl derivative, which was expected to act as a good substrate for the isolation of crystals, and performed the same sequential process of sulfanyl-1,6-diynes to the oxazine derivatives (Scheme 7). Hydroamination–cyclisation of diyne 1 with N-allyl-p-bromobenzenesulfonamide 3g and the successive oxidation proceeded to give the corresponding sulfoxide 5g in high yield. The sequential process, Pummerer reaction of 5g with TFAA, followed by treatment with TBAH to afford the 1,7-sulfur-shifted product 9g as crystals; however, the yield was relatively low. The final ytterbium-catalysed intramolecular cyclisation afforded 25g with a 56% yield. We obtained the 4H-pyrrolo[3,4-g]oxazine derivative as crystals. The structure of these products was determined through NMR spectroscopy experiments (COSY and HMBC data are reported and discussed in Supporting Information File 1).
![[1860-5397-14-250-i6]](/bjoc/content/inline/1860-5397-14-250-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Lewis acid-catalysed cyclization of diols.
Scheme 6: Lewis acid-catalysed cyclization of diols.
![[1860-5397-14-250-i7]](/bjoc/content/inline/1860-5397-14-250-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Sequential process of sulfanyl-1,6-diyne 1 to 4H-pyrrolo[3,4-g]oxazine 25g.
Scheme 7: Sequential process of sulfanyl-1,6-diyne 1 to 4H-pyrrolo[3,4-g]oxazine 25g.
Conclusion
We found unique 1,7-S and 1,7-Se shift reactions by investigating the synthesis of pyrroloazepines through amination–cyclisation of 1,6-diynes, followed by the Pummerer reaction and TBAH hydrolysis. We are now investigating the syntheses of pyrroloazepines using 4-heteroarylmethyl-3-sulfanylpyrroles, which could be easily obtained by hydroamination–cyclization of sulfanyl-1,6-diynes/the successive cyclization. The results of this investigation will be reported elsewhere.
Supporting Information
For crystallographic data see also CCDC 1824587 and 1824588.
| Supporting Information File 1: Schemes S1 and S2 on the syntheses of compounds 1, 2, and 3a–g; the NMR study for the structure determinations, further DFT calculations, the ORTEP drawing of both sulfone of 5a, 11d and the 1H and 13C NMR charts. | ||
| Format: PDF | Size: 5.1 MB | Download |
| Supporting Information File 2: Crystallographic information of compound 11d. | ||
| Format: TXT | Size: 39.8 KB | Download |
| Supporting Information File 3: Crystallographic information of the sulfone of compound 5a. | ||
| Format: TXT | Size: 37.2 KB | Download |
References
-
Kunick, C.; Lemcke, T.; Meijer, L. Development, Selectivity, and Application of Paullones, a Family of CDK Inhibitors. In Inhibitors of Cyclin-dependent Kinases as Anti-tumor Agents; Smith, P. J.; Yue, E. W., Eds.; Enzyme Inhibitors Series; CRC Press: Boca Raton, FL, U.S.A., 2006; pp 227–249. doi:10.1201/9781420005400.ch10
Return to citation in text: [1] -
Gussio, R.; Zaharevitz, D. W.; McGrath, C. F.; Pattabiraman, N.; Kellog, G. E.; Schultz, C.; Link, A.; Kunick, C.; Leost, M.; Meijer, L.; Sausville, E. A. Anti-Cancer Drug Des. 2000, 15, 53–66.
Return to citation in text: [1] -
Forte, B.; Malgesini, B.; Piutti, C.; Quartieri, F.; Scolaro, A.; Papeo, G. Mar. Drugs 2009, 7, 705–753. doi:10.3390/md7040705
Return to citation in text: [1] -
Nguyen, T. N. T.; Tepe, J. J. Curr. Med. Chem. 2009, 16, 3122–3143. doi:10.2174/092986709788803015
Return to citation in text: [1] -
Linington, R. G.; Williams, D. E.; Tahir, A.; van Soest, R.; Andersen, R. J. Org. Lett. 2003, 5, 2735–2738. doi:10.1021/ol034950b
Return to citation in text: [1] -
Schultz, C.; Link, A.; Leost, M.; Zaharevitz, D. W.; Gussio, R.; Sausville, E. A.; Meijer, L.; Kunick, C. J. Med. Chem. 1999, 42, 2909–2919. doi:10.1021/jm9900570
Return to citation in text: [1] -
Tolle, N.; Kunick, C. Curr. Top. Med. Chem. 2011, 11, 1320–1332. doi:10.2174/156802611795589601
Return to citation in text: [1] -
Bridges, A. J. Chem. Rev. 2001, 101, 2541–2571. doi:10.1021/cr000250y
Return to citation in text: [1] -
Brault, L.; Migianu, E.; Neguesque, A.; Battaglia, E.; Bagrel, D.; Kirsch, G. Eur. J. Med. Chem. 2005, 40, 757–763. doi:10.1016/j.ejmech.2005.02.010
Return to citation in text: [1] -
Tang, P. C.; Su, Y. D.; Feng, J.; Fu, J. H.; Yang, J. L.; Xiao, L.; Peng, J. H.; Li, Y. L.; Zhang, L.; Hu, B.; Zhou, Y.; Li, F. Q.; Fu, B. B.; Lou, L. G.; Gong, A. S.; She, G. H.; Sun, W. H.; Mong, X. T. J. Med. Chem. 2010, 53, 8140–8149. doi:10.1021/jm101036c
Return to citation in text: [1] -
Meijer, L.; Thunnissen, A.-M.; White, A. W.; Garnier, M.; Nikolic, M.; Tsai, L.-H.; Walter, J.; Cleverley, K. E.; Salinas, P. C.; Wu, Y. Z.; Biernat, J.; Mandelkow, E.-M.; Kim, S.-H.; Pettit, G. R. Chem. Biol. 2000, 7, 51–63. doi:10.1016/s1074-5521(00)00063-6
Return to citation in text: [1] -
Albrecht, B. K.; Gehling, V. S.; Hewitt, M. C.; Vaswani, R. G.; Côté, A.; Leblanc, Y.; Nasveschuk, C. G.; Bellon, S.; Bergeron, L.; Campbell, R.; Cantone, N.; Cooper, M. R.; Cummings, R. T.; Jayaram, H.; Joshi, S.; Mertz, J. A.; Neiss, A.; Normant, E.; O’Meara, M.; Pardo, E.; Poy, F.; Sandy, P.; Supko, J.; Sims, R. J., III; Harmange, J.-C.; Taylor, A. M.; Audia, J. E. J. Med. Chem. 2016, 59, 1330–1339. doi:10.1021/acs.jmedchem.5b01882
Return to citation in text: [1] -
Kumar, A.; Vachhani, D. D.; Modha, S. G.; Sharma, S. K.; Parmar, V. S.; Van der Eycken, E. V. Eur. J. Org. Chem. 2013, 2288–2292. doi:10.1002/ejoc.201300132
Return to citation in text: [1] -
Haibach, M. C.; Deb, I.; De, C. K.; Seidel, D. J. Am. Chem. Soc. 2011, 133, 2100–2103. doi:10.1021/ja110713k
Return to citation in text: [1] -
Modha, S. G.; Kumar, A.; Vachhani, D. D.; Sharma, S. K.; Parmar, V. S.; Van der Eycken, E. V. Chem. Commun. 2012, 48, 10916–10918. doi:10.1039/c2cc35900f
Return to citation in text: [1] -
Yokosaka, T.; Nemoto, T.; Hamada, Y. Chem. Commun. 2012, 48, 5431–5433. doi:10.1039/c2cc31699d
Return to citation in text: [1] -
Zhu, W.; Zhao, L.; Wang, M.-X. J. Org. Chem. 2015, 80, 12047–12057. doi:10.1021/acs.joc.5b02021
Return to citation in text: [1] -
Garcia, S.; Schulz, J.; Pellet-Rostaing, S.; Lemaire, M. Synlett 2008, 1852–1856. doi:10.1055/s-2008-1078567
Return to citation in text: [1] -
Feng, J.-J.; Lin, T.-Y.; Wu, H.-H.; Zhang, J. Angew. Chem., Int. Ed. 2015, 54, 15854–15858. doi:10.1002/anie.201509185
Return to citation in text: [1] -
Feng, J.-J.; Lin, T.-Y.; Wu, H.-H.; Zhang, J. J. Am. Chem. Soc. 2015, 137, 3787–3790. doi:10.1021/jacs.5b01305
Return to citation in text: [1] -
Martı́nez, L. R.; Gustavo Avila Zarraga, J.; Duran, M. E.; Ramı́rez Apam, M. T.; Cañas, R. Bioorg. Med. Chem. Lett. 2002, 12, 1675–1677. doi:10.1016/s0960-894x(02)00232-9
Return to citation in text: [1] -
Mizuno, A.; Ogata, A.; Kamei, T.; Shibata, M.; Shimamoto, T.; Hayashi, Y.; Nakanishi, K.; Takiguchi, C.; Oka, N.; Inomata, N. Chem. Pharm. Bull. 2000, 48, 623–635. doi:10.1248/cpb.48.623
Return to citation in text: [1] -
Li, X.; Chen, D.; Gu, H.; Lin, X. Chem. Commun. 2014, 50, 7538–7541. doi:10.1039/c4cc02295e
Return to citation in text: [1] -
Grigg, R.; Sridharan, V.; Sykes, D. A. Tetrahedron 2008, 64, 8952–8962. doi:10.1016/j.tet.2008.06.044
Return to citation in text: [1] -
Shi, Z.; Ren, Y.; Li, B.; Lu, S.; Zhang, W. Chem. Commun. 2010, 46, 3973–3975. doi:10.1039/c001715a
Return to citation in text: [1] -
Sharma, S. K.; Sharma, S.; Agarwal, P. K.; Kundu, B. Eur. J. Org. Chem. 2009, 1309–1312. doi:10.1002/ejoc.200801201
Return to citation in text: [1] -
Beaumont, S.; Retailleau, P.; Dauban, P.; Dodd, R. H. Eur. J. Org. Chem. 2008, 5162–5175. doi:10.1002/ejoc.200800643
Return to citation in text: [1] -
Bennasar, M.-L.; Roca, T.; García-Díaz, D. J. Org. Chem. 2007, 72, 4562–4565. doi:10.1021/jo070322m
Return to citation in text: [1] -
Nyerges, M.; Pintér, Á.; Virányi, A.; Bitter, I.; Tőke, L. Tetrahedron Lett. 2005, 46, 377–380. doi:10.1016/j.tetlet.2004.11.127
Return to citation in text: [1] -
Ferrer, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2006, 45, 1105–1109. doi:10.1002/anie.200503484
Return to citation in text: [1] -
Gruit, M.; Michalik, D.; Tillack, A.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 7212–7216. doi:10.1002/anie.200902937
Return to citation in text: [1] -
Nagata, H.; Sugimoto, Y.; Ito, Y.; Tanaka, M.; Yoshimatsu, M. Tetrahedron 2014, 70, 1306–1316. doi:10.1016/j.tet.2013.12.049
Return to citation in text: [1] [2] -
Kobayashi, Y.; Tanahashi, R.; Yamaguchi, Y.; Hatae, N.; Kobayashi, M.; Ueno, Y.; Yoshimatsu, M. J. Org. Chem. 2017, 82, 2436–2449. doi:10.1021/acs.joc.6b02841
Return to citation in text: [1] [2] -
Kennedy, M.; McKervey, M. A. Oxidation adjacent to Sulfur. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I.; Ley, S. V., Eds.; Pergamon Press: Oxford, United Kingdom, 1991; Vol. 7, pp 193–216. doi:10.1016/b978-0-08-052349-1.00187-6
Return to citation in text: [1] -
Akai, S.; Kita, Y. Recent Advances in Pummerer Reactions. In Sulfur-Mediated Rearrangements I; Schaumann, E., Ed.; Topics in Current Chemistry, Vol. 274; Springer: Berlin, Germany, 2006; pp 35–76. doi:10.1007/128_073
Return to citation in text: [1] -
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian, Inc., Wallingford, CT, U.S.A., 2016.
Return to citation in text: [1]
| 32. | Nagata, H.; Sugimoto, Y.; Ito, Y.; Tanaka, M.; Yoshimatsu, M. Tetrahedron 2014, 70, 1306–1316. doi:10.1016/j.tet.2013.12.049 |
| 33. | Kobayashi, Y.; Tanahashi, R.; Yamaguchi, Y.; Hatae, N.; Kobayashi, M.; Ueno, Y.; Yoshimatsu, M. J. Org. Chem. 2017, 82, 2436–2449. doi:10.1021/acs.joc.6b02841 |
| 30. | Ferrer, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2006, 45, 1105–1109. doi:10.1002/anie.200503484 |
| 31. | Gruit, M.; Michalik, D.; Tillack, A.; Beller, M. Angew. Chem., Int. Ed. 2009, 48, 7212–7216. doi:10.1002/anie.200902937 |
| 32. | Nagata, H.; Sugimoto, Y.; Ito, Y.; Tanaka, M.; Yoshimatsu, M. Tetrahedron 2014, 70, 1306–1316. doi:10.1016/j.tet.2013.12.049 |
| 33. | Kobayashi, Y.; Tanahashi, R.; Yamaguchi, Y.; Hatae, N.; Kobayashi, M.; Ueno, Y.; Yoshimatsu, M. J. Org. Chem. 2017, 82, 2436–2449. doi:10.1021/acs.joc.6b02841 |
| 1. | Kunick, C.; Lemcke, T.; Meijer, L. Development, Selectivity, and Application of Paullones, a Family of CDK Inhibitors. In Inhibitors of Cyclin-dependent Kinases as Anti-tumor Agents; Smith, P. J.; Yue, E. W., Eds.; Enzyme Inhibitors Series; CRC Press: Boca Raton, FL, U.S.A., 2006; pp 227–249. doi:10.1201/9781420005400.ch10 |
| 2. | Gussio, R.; Zaharevitz, D. W.; McGrath, C. F.; Pattabiraman, N.; Kellog, G. E.; Schultz, C.; Link, A.; Kunick, C.; Leost, M.; Meijer, L.; Sausville, E. A. Anti-Cancer Drug Des. 2000, 15, 53–66. |
| 28. | Bennasar, M.-L.; Roca, T.; García-Díaz, D. J. Org. Chem. 2007, 72, 4562–4565. doi:10.1021/jo070322m |
| 6. | Schultz, C.; Link, A.; Leost, M.; Zaharevitz, D. W.; Gussio, R.; Sausville, E. A.; Meijer, L.; Kunick, C. J. Med. Chem. 1999, 42, 2909–2919. doi:10.1021/jm9900570 |
| 7. | Tolle, N.; Kunick, C. Curr. Top. Med. Chem. 2011, 11, 1320–1332. doi:10.2174/156802611795589601 |
| 29. | Nyerges, M.; Pintér, Á.; Virányi, A.; Bitter, I.; Tőke, L. Tetrahedron Lett. 2005, 46, 377–380. doi:10.1016/j.tetlet.2004.11.127 |
| 5. | Linington, R. G.; Williams, D. E.; Tahir, A.; van Soest, R.; Andersen, R. J. Org. Lett. 2003, 5, 2735–2738. doi:10.1021/ol034950b |
| 26. | Sharma, S. K.; Sharma, S.; Agarwal, P. K.; Kundu, B. Eur. J. Org. Chem. 2009, 1309–1312. doi:10.1002/ejoc.200801201 |
| 3. | Forte, B.; Malgesini, B.; Piutti, C.; Quartieri, F.; Scolaro, A.; Papeo, G. Mar. Drugs 2009, 7, 705–753. doi:10.3390/md7040705 |
| 4. | Nguyen, T. N. T.; Tepe, J. J. Curr. Med. Chem. 2009, 16, 3122–3143. doi:10.2174/092986709788803015 |
| 27. | Beaumont, S.; Retailleau, P.; Dauban, P.; Dodd, R. H. Eur. J. Org. Chem. 2008, 5162–5175. doi:10.1002/ejoc.200800643 |
| 12. | Albrecht, B. K.; Gehling, V. S.; Hewitt, M. C.; Vaswani, R. G.; Côté, A.; Leblanc, Y.; Nasveschuk, C. G.; Bellon, S.; Bergeron, L.; Campbell, R.; Cantone, N.; Cooper, M. R.; Cummings, R. T.; Jayaram, H.; Joshi, S.; Mertz, J. A.; Neiss, A.; Normant, E.; O’Meara, M.; Pardo, E.; Poy, F.; Sandy, P.; Supko, J.; Sims, R. J., III; Harmange, J.-C.; Taylor, A. M.; Audia, J. E. J. Med. Chem. 2016, 59, 1330–1339. doi:10.1021/acs.jmedchem.5b01882 |
| 13. | Kumar, A.; Vachhani, D. D.; Modha, S. G.; Sharma, S. K.; Parmar, V. S.; Van der Eycken, E. V. Eur. J. Org. Chem. 2013, 2288–2292. doi:10.1002/ejoc.201300132 |
| 14. | Haibach, M. C.; Deb, I.; De, C. K.; Seidel, D. J. Am. Chem. Soc. 2011, 133, 2100–2103. doi:10.1021/ja110713k |
| 15. | Modha, S. G.; Kumar, A.; Vachhani, D. D.; Sharma, S. K.; Parmar, V. S.; Van der Eycken, E. V. Chem. Commun. 2012, 48, 10916–10918. doi:10.1039/c2cc35900f |
| 16. | Yokosaka, T.; Nemoto, T.; Hamada, Y. Chem. Commun. 2012, 48, 5431–5433. doi:10.1039/c2cc31699d |
| 17. | Zhu, W.; Zhao, L.; Wang, M.-X. J. Org. Chem. 2015, 80, 12047–12057. doi:10.1021/acs.joc.5b02021 |
| 18. | Garcia, S.; Schulz, J.; Pellet-Rostaing, S.; Lemaire, M. Synlett 2008, 1852–1856. doi:10.1055/s-2008-1078567 |
| 19. | Feng, J.-J.; Lin, T.-Y.; Wu, H.-H.; Zhang, J. Angew. Chem., Int. Ed. 2015, 54, 15854–15858. doi:10.1002/anie.201509185 |
| 20. | Feng, J.-J.; Lin, T.-Y.; Wu, H.-H.; Zhang, J. J. Am. Chem. Soc. 2015, 137, 3787–3790. doi:10.1021/jacs.5b01305 |
| 22. | Mizuno, A.; Ogata, A.; Kamei, T.; Shibata, M.; Shimamoto, T.; Hayashi, Y.; Nakanishi, K.; Takiguchi, C.; Oka, N.; Inomata, N. Chem. Pharm. Bull. 2000, 48, 623–635. doi:10.1248/cpb.48.623 |
| 23. | Li, X.; Chen, D.; Gu, H.; Lin, X. Chem. Commun. 2014, 50, 7538–7541. doi:10.1039/c4cc02295e |
| 11. | Meijer, L.; Thunnissen, A.-M.; White, A. W.; Garnier, M.; Nikolic, M.; Tsai, L.-H.; Walter, J.; Cleverley, K. E.; Salinas, P. C.; Wu, Y. Z.; Biernat, J.; Mandelkow, E.-M.; Kim, S.-H.; Pettit, G. R. Chem. Biol. 2000, 7, 51–63. doi:10.1016/s1074-5521(00)00063-6 |
| 24. | Grigg, R.; Sridharan, V.; Sykes, D. A. Tetrahedron 2008, 64, 8952–8962. doi:10.1016/j.tet.2008.06.044 |
| 25. | Shi, Z.; Ren, Y.; Li, B.; Lu, S.; Zhang, W. Chem. Commun. 2010, 46, 3973–3975. doi:10.1039/c001715a |
| 10. | Tang, P. C.; Su, Y. D.; Feng, J.; Fu, J. H.; Yang, J. L.; Xiao, L.; Peng, J. H.; Li, Y. L.; Zhang, L.; Hu, B.; Zhou, Y.; Li, F. Q.; Fu, B. B.; Lou, L. G.; Gong, A. S.; She, G. H.; Sun, W. H.; Mong, X. T. J. Med. Chem. 2010, 53, 8140–8149. doi:10.1021/jm101036c |
| 34. | Kennedy, M.; McKervey, M. A. Oxidation adjacent to Sulfur. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I.; Ley, S. V., Eds.; Pergamon Press: Oxford, United Kingdom, 1991; Vol. 7, pp 193–216. doi:10.1016/b978-0-08-052349-1.00187-6 |
| 35. | Akai, S.; Kita, Y. Recent Advances in Pummerer Reactions. In Sulfur-Mediated Rearrangements I; Schaumann, E., Ed.; Topics in Current Chemistry, Vol. 274; Springer: Berlin, Germany, 2006; pp 35–76. doi:10.1007/128_073 |
| 9. | Brault, L.; Migianu, E.; Neguesque, A.; Battaglia, E.; Bagrel, D.; Kirsch, G. Eur. J. Med. Chem. 2005, 40, 757–763. doi:10.1016/j.ejmech.2005.02.010 |
| 21. | Martı́nez, L. R.; Gustavo Avila Zarraga, J.; Duran, M. E.; Ramı́rez Apam, M. T.; Cañas, R. Bioorg. Med. Chem. Lett. 2002, 12, 1675–1677. doi:10.1016/s0960-894x(02)00232-9 |
| 36. | Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian, Inc., Wallingford, CT, U.S.A., 2016. |
© 2018 Go et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)