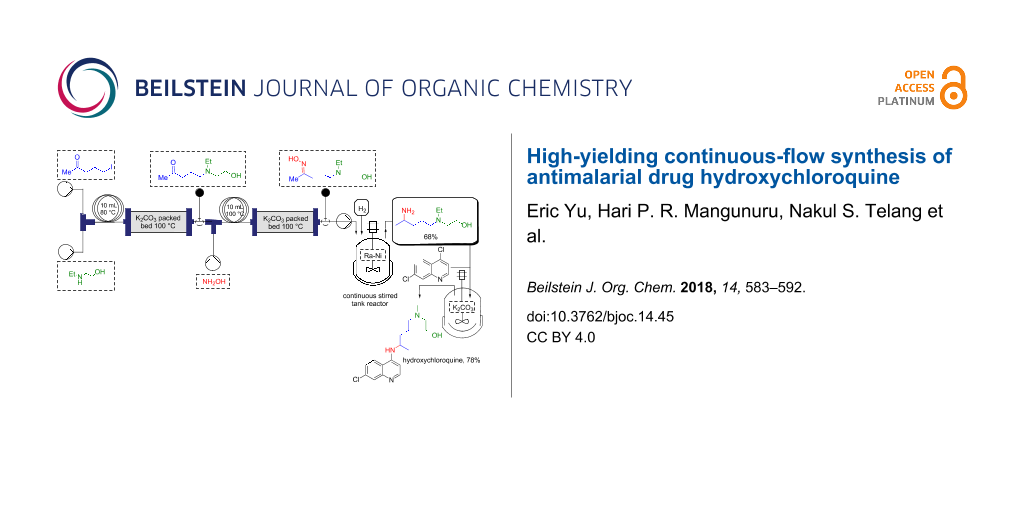
Abstract
Numerous synthetic methods for the continuous preparation of fine chemicals and active pharmaceutical ingredients (API’s) have been reported in recent years resulting in a dramatic improvement in process efficiencies. Herein we report a highly efficient continuous synthesis of the antimalarial drug hydroxychloroquine (HCQ). Key improvements in the new process include the elimination of protecting groups with an overall yield improvement of 52% over the current commercial process. The continuous process employs a combination of packed bed reactors with continuous stirred tank reactors for the direct conversion of the starting materials to the product. This high-yielding, multigram-scale continuous synthesis provides an opportunity to achieve increase global access to hydroxychloroquine for treatment of malaria.

Graphical Abstract
Introduction
Our research group has been focused on the development of new synthetic methods for the preparation of a variety of active pharmaceutical ingredients for global health applications by employing the principles of process intensification [1-3]. In 2016, estimated 212 million cases of malaria, including 429,000 fatalities, were reported worldwide, with the majority of these cases occurring in sub-Saharan Africa and Southern Asia [4]. The malaria epidemic is particularly difficult to control due to the multidrug resistant nature of the malaria parasite Plasmodium falciparum. Hydroxychloroquine (1) is an antimalarial drug developed for both treatment and prevention of the disease in response to the widespread malaria resistance to chloroquine (2, Figure 1) [5,6].
![[1860-5397-14-45-1]](/bjoc/content/figures/1860-5397-14-45-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Commercially available antimalarial drugs.
Figure 1: Commercially available antimalarial drugs.
Additionally, hydroxychloroquine (1, HCQ) is an effective non-steroidal drug with anti-inflammatory activity for the treatment of rheumatoid arthritis (RA) in patients with cardiovascular disease [7-9]. The World Health Organization has identified HCQ (1) as an essential antimalarial medication for a basic healthcare system, but global access to HCQ (1) has been hindered by high manufacturing costs of the API. Thus, the development of cost effective synthetic strategies to increase the global access to this important global health drug is of great importance. Effective strategies for accomplishing such objectives often include identifying more cost effective starting materials and reagents, simplifying the synthetic route in terms of reducing the total number of steps as well as reducing the cost and improving the efficiency of individual steps.
Flow chemistry methodologies have been increasingly investigated in recent years in the pharmaceutical industry for multistep preparations of highly-complex natural products and APIs [10-19]. Advantages include precise control of key reaction parameters such as heat and pressure, improved heat and mass transfer capabilities for better thermal control, enhanced selection of kinetically controlled products to potentially maximize conversion, smaller equipment footprint, and increased safety profiles when working with hazardous materials and reaction conditions [20]. These advantages often result in flow processes being significantly more efficient, as well as less costly, when compared to batch processes.
HCQ (1) is currently produced via the batch method shown in Scheme 1. Therefore, continuous-flow chemistry approaches to synthesizing HCQ (1) offer a great potential to maximize the efficiency, and thus significantly reduces the overall manufacturing costs of this important medicine.
![[1860-5397-14-45-i1]](/bjoc/content/inline/1860-5397-14-45-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Current batch syntheses of the key intermediate 5-(ethyl(2-hydroxyethyl)amino)pentan-2-one (6).
Scheme 1: Current batch syntheses of the key intermediate 5-(ethyl(2-hydroxyethyl)amino)pentan-2-one (6).
The commercial HCQ synthesis employs a key intermediate, 5-(ethyl(2-hydroxyethyl)amino)pentan-2-one (6), which is a major cost driver in the process. The protection–deprotection strategy of chloro-ketone starting material 3 used in the commercial route (Scheme 1a) [21] has been targeted as a significant opportunity for optimization. While the recent improved route (Scheme 1b) by Li and co-workers [21] eliminates the protection–deprotection steps, its use of a complex multi-transition-metal-catalyst system to achieve direct SN2 substitution of the chlorine on 3 by amine 7, is sub-optimal [22,23]. With these issues in mind, we carried out a retrosynthetic analysis (Scheme 2) in which 10, an iodo analogue to the starting material 3, could be generated in a single step via a decarboxylative ring-opening of α-acetyl butyrolactone 8. The iodo analogue 10 could then be used without isolation to prepare compound 6.
![[1860-5397-14-45-i2]](/bjoc/content/inline/1860-5397-14-45-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic strategy to hydroxychloroquine (1).
Scheme 2: Retrosynthetic strategy to hydroxychloroquine (1).
It is well known that the direct one-step reductive amination of 6 to give 12 can be accomplished by simple heterogeneous reduction with H2/Raney-nickel [24]. However, THF is employed in all of our prior flow steps and is a poor choice as a solvent for the reductive amination step due to limited solubility of ammonia in THF. H2/Raney-nickel reductions are often carried out in alcoholic media where much higher concentrations of ammonia are achievable but would require a solvent exchange. There are many reports of continuous-flow chemistry methods for reductive amination of ketones [25-31]; however, such processes typically require soluble reductants such as diisobutylaluminium hydride (DIBAL-H), superhydrides, or supported borohydride species [32-36]. Although these approaches are effective, they are significantly more costly than using simple heterogeneous reduction with H2/Raney-nickel. Therefore, we explored an alternate strategy: simple conversion of the ketone group of 6 to oxime 11, followed by reduction to give 5-(ethyl(2-hydroxyethyl)amino)-2-aminopentane (12). We have found H2/Raney-nickel efficiently reduces 11 to 12 with THF as the solvent in a continuous stirred tank reactor (CSTR).
The last step requires the reaction of 12 with 4,7-dichloroquinoline (13) which when used neat takes 24–48 hours at 120–140 °C to give 75–80% yield of HCQ (1) [37]. We have found that this step can be accelerated by employing K2CO3/triethylamine, to facilitate the formation of 1, resulting in a comparable yield in less than 6 hours. Thus, we have integrated the continuous preparation of reaction with our new efficient continuous-flow synthesis of 12 with the final step by using a CSTR to accommodate the longer reaction time required to produce HCQ (1).
Results and Discussion
Initial optimization efforts to prepare 6 (Scheme 1) revealed poor reactivity of starting material 3, so we pursued the iodo analogue of 3, 5-iodopentan-2-one (10) as an alternative. By optimizing the reaction concentration, we have also shown that (see Table S1 in Supporting Information File 1) 10 reacts rapidly and cleanly with 7 under flow conditions to give 6 in high yield (>80%). Furthermore, we have developed and optimized a continuous synthesis of 10 (Table 1), wherein hydroiodic acid is reacted with neat 3-acetyldihydrofuran-2(3H)-one (8) to provide a rapid route to 10 which is significantly higher in yield than in previously reported syntheses [38,39]. Initial results using diluted hydroiodic acid (20–40%) provided only modest conversion to product over a range of temperatures (Table 1, entries 1–5); however, the use of 55% hydroiodic acid (Table 1, entries 6–8) was found to give near quantitative conversion. The reaction profile was monitored using GC–MS and 1H NMR – no intermediates were observed under these conditions. Optimization of the flow rate with 55% hydroiodic acid (Table 1, entries 6–8) revealed that a flow rate of 1.0 mL min−1 (tR = 5 min) gave an isolated yield of 89%.
Table 1: Optimization of the flow process for the synthesis of 10.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-45-i5.svg?max-width=637&scale=1.0)
|
|||||
| entry | HI [aqueous %] | temp (°C) | tR = min | pressure (bar) | conv.a (%) |
|---|---|---|---|---|---|
| 1 | 20 | r.t. | 5 | 1.5 | 5 |
| 2 | 20 | 40 | 5 | 2.0 | 31 |
| 3 | 20 | 80 | 5 | 2.0 | 34 |
| 4 | 40 | 80 | 5 | 2.0 | 43 |
| 5 | 40 | 80 | 5 | 2.5 | 46 |
| 6 | 55 | 80 | 5 | 3.0 | 98 (89%)b |
| 7 | 55 | 80 | 2.5 | 3.0 | 91 |
| 8 | 55 | 80 | 10 | 3.0 | 92 |
aConversion determined by GC–MS and 1H NMR. bIsolated yield.
Due to the need to use an excess of hydroiodic acid it is important to remove its excess from the eluting reaction stream before telescoping into the next step in flow. The product stream containing crude 10 was mixed in-line with methyl tert-butyl ether (MTBE) and saturated NaHCO3 before phase separation using a hydrophobic, membrane-based separator (Zaiput) [40] (Scheme 3) to afford purified 10 in the organic phase. A loss of 5–10% of product to the water layer was observed, however, this was deemed adequate as it prevented the need for a complete work-up step in batch.
![[1860-5397-14-45-i3]](/bjoc/content/inline/1860-5397-14-45-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Schematic representation for continuous in-line extraction of 10.
Scheme 3: Schematic representation for continuous in-line extraction of 10.
In the next step 6 was reacted with hydroxylamine, which was facilitated by passing through a packed-bed of K2CO3 to give oxime 11 (Table 2). As was seen with the reaction to produce 6 (Table S1 in Supporting Information File 1), reactant concentrations also had a dramatic effect on the oxime formation. A series of experiments were conducted to optimize the continuous formation of 11. Reaction yields were modest at lower reactant concentrations across several temperatures and residence times (Table 2). Conversion to 11 increased when reactant concentrations were increased (9% at 0.1 M to 72% at 1 M, Table 2, entries 1–6). Optimization of the flow rate with 1 M concentrations of each reactant (Table 2, entries 6–8) showed that a flow rate of 1.0 mL min−1 (tR = 20 min) was optimal, giving an isolated yield of 78% (Table 2, entry 7).
Table 2: Schematic representation for the continuous telescoped process to synthesize 11.
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-45-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | concentrationa | temp (°C) | tR = min | conv. of 11 (%)b |
|---|---|---|---|---|
| 1 | 0.1 M | 100 | 10 | 9 |
| 2 | 0.2 M | 100 | 10 | 16 |
| 3 | 0.4 M | 100 | 10 | 34 |
| 4 | 0.6 M | 100 | 10 | 37 |
| 5 | 0.8 M | 100 | 10 | 62 |
| 6 | 1.0 M | 100 | 10 | 72 |
| 7 | 1.0 M | 100 | 20 | 85 (78)c |
| 8 | 1.0 M | 100 | 40 | 76 |
aConcentration of 10 , 7 and hydroxylamine. bConversion determined by GC–MS and 1H NMR. cIsolated yield.
The reductive amination of 11 performed in the first generation batch process was carried out using Raney-nickel at 80 °C and 10 bar hydrogen pressure for 4–6 h [21-24]. In order to perform this step in a continuous fashion, a continuous stirred tank reactor [25,41] was employed (Table 3). Materials were delivered to the CSTR vessel through an HPLC pump and were reacted under hydrogen pressure with mechanical stirring. The dip tube in the CSTR was outfitted with a fritted metal filter, allowing for retention of the heterogeneous catalyst within the CSTR vessel. Optimization of this CSTR-based flow process (Table 3) showed near quantitative yields of 12 over a broad range of oxime 11 reactant concentrations. An optimum residence time was determined to be 4 hours.
Table 3: Optimization of the flow process for the reductive amination of 12 using a CSTR.
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-45-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | oxime [concentration] | temp. (°C) |
pressure
(bar) |
tR = hours | conv. of 12 (%)a |
|---|---|---|---|---|---|
| 1 | 0.05 M | 80 | 10 | 4 | 94% |
| 2 | 0.25 M | 80 | 10 | 4 | 96% |
| 3 | 0.5 M | 80 | 10 | 4 | 97% |
| 4 | 2.0 M | 80 | 10 | 4 | 98% (89%)b |
| 5 | 2.0 M | 80 | 10 | 2 | 56% |
| 6 | 2.0 M | 80 | 10 | 1 | 46% |
aConversion determined by GC–MS and 1H NMR. bIsolated yield.
After optimizing the individual steps up to compound 12 the entire reaction was telescoped into a continuous reaction process that convert 10 and 6 into 12 (Scheme 4) with an overall isolated yield of 68% for compound 12.
![[1860-5397-14-45-i4]](/bjoc/content/inline/1860-5397-14-45-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Optimization of the flow process for the synthesis of 12.
Scheme 4: Optimization of the flow process for the synthesis of 12.
With an optimized continuous process for producing the key intermediate 12 in-hand the reaction conditions for the conversion of 12 to HCQ (1) were examined. In the commercial process this step is carried out in batch under neat reactant conditions and requires a relatively long reaction time of 24–48 h [42-44]. In order to convert this step to a flow chemistry method, we selected to employ a CSTR (Table 4). This final step, transforming 13 and 12 into 1, was first investigated in batch to optimize the conditions before implemented in a CSTR.
Table 4: Optimization of the reaction conditions for the preparation of hydroxychloroquine (1).a
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-45-i8.svg?max-width=637&scale=1.0)
|
||||
| entry | base | solvent | temp. (°C) | conv. to 1 (%)b |
|---|---|---|---|---|
| 1 | NaOH | EtOH | 125 | 30 |
| 2 | KOH | EtOH | 125 | 35 |
| 3 | K2CO3 | EtOH | 125 | 82 |
| 4 | Et3N | EtOH | 125 | 61 |
| 5 | DIPEA | EtOH | 125 | 55 |
| 6 | K2CO3/Et3N | EtOH | 125 | 88 (78)c |
aReaction conditions: 4,7-dichloroquinoline (13, 1.0 equiv), base (1.0 equiv), amine 12 (1.2 equiv). bConversion determined by HPLC and 1H NMR, cIsolated yield.
Process optimization for the final step started with the screening of the effect of solvents and base(s) on the yield of HCQ (1). Screening of different polar–protic and non-protic solvents (see Table S2 in Supporting Information File 1) demonstrated that ethanol is the most effective for this transformation. During the screening of bases, the pKa of the amine and alcohol groups present in compound 12 were given careful consideration in order to minimize C–O bond formation (Table 4). NaOH or KOH in ethanol gave low (<40%) conversion, whereas using K2CO3 in ethanol gave 82% conversion to HCQ (Table 4, entry 3). Attempts with organic bases (Table 4, entries 5 and 6) resulted in only moderate conversions to the desired product; however, using a 1:1 mixture of K2CO3/Et3N (1:1) resulted in 88% conversion (Table 4, entry 6) to 1, with corresponds to an isolated yield of 78%.
Conclusion
In summary, we have developed a high-yielding continuous-flow process for the synthesis of hydroxychloroquine (1, HCQ) by optimizing continuous-flow methods for the synthesis of key intermediates 6 and 12. Additionally, we have developed and optimized flow-chemistry conditions for performing reductive amination of 11 using Raney-nickel as catalyst in a continuous stirring tank reactor (CSTR) for the synthesis of compound 12, and have incorporated it into a fully continuous telescoped process for the synthesis of 12 from lactone 8 and aminoethanol 7. Feeding the output stream containing 12 from the above CSTR into a second CSTR in which 12 is converted to HCQ (1) provides a completely continuous-flow process for producing HCQ (1) from readily available starting materials. This efficient process has the potential to increase the global access to this strategically important antimalarial drug. We are currently working to demonstrate that this fully integrated continuous-flow process for the synthesis of HCQ (1) can be scaled to commercial operations.
Experimental
General information
All reactions for the preparation of substrates were performed in standard, dry glassware under an inert atmosphere of nitrogen or argon unless otherwise described. All starting materials and reagents were purchased from commercial sources and were used as received unless otherwise noted. 1H and 13C NMR spectra were recorded using 600 MHz spectrometers. Chemical shift (δ) values are given in ppm, and coupling constants (J) are given in Hz. The 7.26 ppm resonance of residual CHCl3 (or 0 ppm of TMS) for proton spectra and the 77.23 ppm resonance of CDCl3 for carbon spectra were used as internal references. Continuous-flow experiments were carried out using the E-series flow reactor instrument purchased from Vapourtec Ltd. PFA tubing (1/16 OD × 1 mm ID) was used for all reactor coils in flow experiments. Most of the reagents and starting materials were purchased from commercial sources and used as received. All HPLC chromatograms were recorded on an Agilent Technologies 1260 Infinity instrument with a Poroshell 120 EC-C18 column (4.6 × 50 mm, 2.7 micron). Continuous flow hydrogenation was performed using a FlowCAT instrument.
Synthesis of 5-iodopentan-2-one (10)
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-45-i9.svg?max-width=637&scale=1.18182)
Two solutions, 2-acetylbutyrolactone (8, 1.176 mL, 10.35 mmol, 1.0 equiv) and hydroiodic acid (55% aqueous solution) were pumped at 1.0 mL min−1 using peristaltic pumps through a 10 mL coil (residence time, tR = 5 min) at 80 °C. The completion of the reaction was monitored using GC–MS. Complete consumption of starting material was observed. The reaction mixture was cooled to room temperature and sodium hydrogencarbonate was added until neutralized at pH 7. The crude mixture was extracted with hexanes/MTBE and the combined organic phase was dried over anhydrous sodium sulfate and evaporated in vacuo to dryness yielding the desired product as a light brown liquid (14.72 g, 89%). 1H NMR (600 MHz, CDCl3) δ 3.22 (t, J = 6.9 Hz, 2H), 2.59 (t, J = 6.9 Hz, 2H), 2.17 (s, 3H), 2.06 (quin, J = 7.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 207.4, 44.0, 30.3, 27.2, 6.7. Spectra were obtained in accordance with those previously reported [3].
Synthesis of 5-(ethyl(2-hydroxyethyl)amino)pentan-2-one (6)
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-45-i10.svg?max-width=637&scale=1.18182)
Telescope of compound 6: Prior to the start of the experiment, the flow reactor unit was rinsed with dry THF and flushed with nitrogen gas. At room temperature, the stock solutions of 5-iodopentan-2-one (10, 1.0 M) and 2-(ethylamino)ethan-1-ol (7) in THF solution (1.0 M) were streamed in at 0.5 mL min−1 via a T-piece into a 10 mL reactor coil (tR = 10 min) and passed through a packed bed reactor of potassium carbonate at 100 °C. The output solution was collected and quenched with a saturated solution of ammonium chloride. The aqueous phase was extracted by DCM (3 × 50 mL) and the organic layers were combined, dried over sodium sulfate, and evaporated in vacuo to give a light brown liquid (14.05 g, 86%). 1H NMR (600 MHz, CDCl3) δ 3.53 (t, J = 5.2 Hz, 2H), 2.58 (m, 3H), 2.53 (m, 2H), 2.45 (t, J = 6.7 Hz, 4H), 2.59 (t, J = 6.9 Hz, 2H), 2.17 (s, 3H), 2.07 (quin, J = 7.0 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 208.9, 58.6, 55.0, 52.4, 47.2, 41.3, 30.0, 21.2, 11.7. Spectra were obtained in accordance with those previously reported [38,39].
Synthesis of (E)-5-(ethyl(2-hydroxyethyl)amino)pentan-2-one oxime (11)
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-45-i11.svg?max-width=637&scale=1.18182)
Flow: Prior to the start of the experiment, the flow reactor unit was rinsed with dry THF and flushed with nitrogen gas. At room temperature, the stock solutions of 5-iodopentan-2-one (10, 1.0 M) and 2-(ethylamino)ethan-1-ol (7) in THF solution (1.0 M) were streamed in at 0.5 mL min−1 via a T-piece into a 10 mL reactor coil (tR =10 min) and passed through a packed bed reactor of potassium carbonate. The output solution was streamlined with hydroxylamine (1.0 M) at 1.0 mL min−1 via a T-piece into a 10 mL reactor coil (tR =10 min) and passed through a packed bed reactor of potassium carbonate at 100 °C. The reaction mixture was then concentrated in vacuo, taken up in dichloromethane (3 × 20 mL) and concentrated under reduced pressure to yield 11 as light brown liquid. The crude product was used in the next step without further purification.
Synthesis of 2-((4-aminopentyl)(ethyl)amino)ethan-1-ol (12)
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-45-i12.svg?max-width=637&scale=1.18182)
Flow: The synthesis of compound 12 was performed in a HEL continuous stirred tank reactor (CSTR) with a reaction volume of 150 mL. The reaction vessel was first charged with Raney-nickel (1.0 g). The Raney-nickel catalyst was retained in the CSTR by the 2 µm metal filter frit on the dip tube of the exit stream. The reaction mixture, consisting of compound 11 (0.05–2.0 M) in THF, was pumped by a HPLC pump set at a flow rate of 0.6–2.5 mL min−1 into the reaction vessel. The reaction pressure was set to 10 bar of hydrogen supplied by hydrogen gas (ultrahigh purity) at a flow rate of 0.5 mL min−1. The reaction temperature was set to 80 °C which was controlled by a thermocouple positioned in the reaction mixture. The reaction was stirred with mechanical stirring (750 rpm) to provide proper mixing. Two thermocouples were used to control the reaction volume in the reactor by setting a level control of −3 °C. The lower thermocouple constantly measured and controlled the reaction temperature and the upper thermocouple measured the temperature at approximately 150 mL reactor volume. When the two thermocouples were within 3 °C, the level control ‘opened’ the exit stream dip tube to allow products to exit the reactor, or ‘closed’ the exit stream dip tube to allow the reactor to fill when the temperature difference between the two thermocouples was greater than 3 °C. The product was collected after a full reaction volume of material (150 mL) had passed through the CSTR indicating that steady-state was reached. The reaction was monitored by liquid chromatography and 1H NMR. The reaction mixture was filtered through a celite pad and dried under reduced pressure. The solution was extracted with water (10 mL) and dichloromethane (3 × 20 mL). The organic layers were combined, washed with brine and dried over sodium sulfate and evaporated in vacuo. The resulting oil was fractionally distilled to give a colorless liquid (16.83 g, 84%). 1H NMR (600 MHz, CDCl3) δ 3.53 (t, J =5.3 Hz, 2H), 2.89 (sx, J = 6.4 Hz, 1H), 2.57 (t, J = 5.5 Hz, 2H), 2.55 (t, J = 7.0 Hz, 2H), 2.45 (t, J = 7.0 Hz, 2H), 1.55–1.44 (m, 2H), 1.36–1.27 (m, 2H), 1.22 (t, J = 7.1 Hz, 2H), 1.07 (d, J = 7.1 Hz, 2H), 1.00 (t, J = 7.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 58.2, 54.9, 53.2, 46.9, 46.7, 36.6, 23.8, 22.4, 10.6. Spectra were obtained in accordance with those previously reported [38,39].
Synthesis of 2-((4-((7-chloroquinolin-4-yl)amino)pentyl)(ethyl)amino)ethan-1-ol (1)
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-45-i13.svg?max-width=637&scale=1.18182)
Batch: In a CSTR reactor, to a mixture of 4,7-dichloroquinoline (200 mg, 1.0 mmol), compound 12 (208 mg, 1.2 mmol), triethylamine (0.069 mL, 0.5 mmol, 0.5 equiv) and potassium carbonate (69 mg, 0.5 mmol, 0.5 equiv) was added ethanol (1.0 mL). The ethanol was distilled off from the reaction mixture and kept under nitrogen atmosphere (15 psi). The reaction was left at 125 °C in the nitrogen atmosphere for 6 h. After cooling, the mixture was transferred into a separatory funnel using 1 M aqueous sodium hydroxide (5 mL) and dichloromethane (2 × 20 mL). The organic phases were separated and the aqueous phase was re-extracted with dichloromethane (2 × 10 mL). The organic layers were combined and dried over sodium sulfate and evaporated in vacuo. The crude material was purified using flash chromatography with DCM/Et3N/MeOH 95:3:2 to give a white solid (0.263 g, 78%). 1H NMR (600 MHz, CDCl3) δ 8.48 (d, J = 5.4 Hz, 1H), 7.93 (d, J = 5.4 Hz, 1H), 7.70 (d, J = 9.2 Hz, 1H), 7.34 (dd, J = 8.8, 7.3 Hz, 1H), 6.39 (d, J = 5.4 Hz, 1H), 4.96 (d, J = 7.5 Hz, 1H), 3.70 (sx, J = 6.8 Hz, 1H), 3.55 (m, 2H), 2.57 (m, 5H), 2.49 (m, 2H), 1.74–1.62 (m, 1H), 1.65–1.53 (m, 3H), 1.31 (d, J = 6.9 Hz, 3H), 1.24 (d, J = 7.2 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 152.2, 149.5, 149.2, 135.0, 129.0, 125.4, 121.2, 117.4, 99.4, 58.6, 54.9, 53.18, 48.5, 47.9, 34.5, 24.1, 20.6, 11.9. Spectra were obtained in accordance with those previously reported [38,39].
Supporting Information
| Supporting Information File 1: Additional experimental descriptions and NMR spectra. | ||
| Format: PDF | Size: 920.8 KB | Download |
References
-
Verghese, J.; Kong, C. J.; Rivalti, D.; Yu, E. C.; Krack, R.; Alcázar, J.; Manley, J. B.; McQuade, D. T.; Ahmad, S.; Belecki, K.; Gupton, B. F. Green Chem. 2017, 19, 2986–2991. doi:10.1039/C7GC00937B
Return to citation in text: [1] -
Kong, C. J.; Fisher, D.; Desai, B. K.; Yang, Y.; Ahmad, S.; Belecki, K.; Gupton, B. F. Bioorg. Med. Chem. 2017, 25, 6203–6208. doi:10.1016/j.bmc.2017.07.004
Return to citation in text: [1] -
Martin, A. D.; Siamaki, A. R.; Belecki, K.; Gupton, B. F. J. Flow Chem. 2015, 5, 145–147. doi:10.1556/JFC-D-15-00002
Return to citation in text: [1] [2] -
World Health Organization. WHO Malaria Report 2016. Geneva, 2016; http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/.
Return to citation in text: [1] -
Surrey, A.-R.; Hammer, H. F. J. Am. Chem. Soc. 1950, 72, 1814–1815. doi:10.1021/ja01160a116
Return to citation in text: [1] -
Bailey, D. M. J. Med. Chem. 1969, 12, 184–185. doi:10.1021/jm00301a055
Return to citation in text: [1] -
Pavelka, K., Jr.; Sen, K. P.; Pelísková, Z.; Vácha, J.; Trnavský, K. Ann. Rheum. Dis. 1989, 48, 542–546. doi:10.1136/ard.48.7.542
Return to citation in text: [1] -
Hage, M. P.; Al-Badri, M. R.; Azar, S. T. Ther. Adv. Endocrinol. Metab. 2014, 5, 77–85. doi:10.1177/2042018814547204
Return to citation in text: [1] -
Poorvashree, J.; Suneela, D. Drug Delivery Transl. Res. 2017, 7, 709–730. doi:10.1007/s13346-017-0420-5
Return to citation in text: [1] -
Belecki, K.; Gupton, B. F. Continuous Processing in Drug Discovery. Green Chemistry Strategies for Drug Discovery; The Royal Society of Chemistry, 2015; pp 127–150. doi:10.1039/9781782622659-00127
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2015, 11, 1194–1219. doi:10.3762/bjoc.11.134
Return to citation in text: [1] -
Baxendale, I. R.; Brocken, L.; Mallia, C. J. Green Process. Synth. 2013, 2, 211–230. doi:10.1515/gps-2013-0029
Return to citation in text: [1] -
Opalka, S. M.; Park, J. K.; Longstreet, A. R.; McQuade, D. T. Org. Lett. 2013, 15, 996–999. doi:10.1021/ol303442m
Return to citation in text: [1] -
Alonso, N.; Miller, L. Z.; Muñoz, J. d. M.; Alcázar, J.; McQuade, D. T. Adv. Synth. Catal. 2014, 356, 3737–3741. doi:10.1002/adsc.201400243
Return to citation in text: [1] -
McQuade, D. T.; Seeberger, P. H. J. Org. Chem. 2013, 78, 6384–6389. doi:10.1021/jo400583m
Return to citation in text: [1] -
Poechlauer, P.; Colberg, J.; Fisher, E.; Jansen, M.; Johnson, M. D.; Koenig, S. G.; Lawler, M.; Laporte, T.; Manley, J.; Martin, B.; Kearney-McMullan, A. Org. Process Res. Dev. 2013, 17, 1472–1478. doi:10.1021/op400245s
Return to citation in text: [1] -
Poechlauer, P.; Manley, J.; Broxterman, R.; Gregertsen, B.; Ridemark, M. Org. Process Res. Dev. 2012, 16, 1586–1590. doi:10.1021/op300159y
Return to citation in text: [1] -
Britton, J.; Raston, C. L. Chem. Soc. Rev. 2017, 46, 1250–2171. doi:10.1039/C6CS00830E
Return to citation in text: [1] -
Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183
Return to citation in text: [1] -
Gutmann, B.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed. 2015, 54, 6688–6728. doi:10.1002/anie.201409318
Return to citation in text: [1] -
You, H.; Liu, Y.; Ning, F.; Zheng, Z.; Yu, Q.; Niu, X.; Li, C. CN104803859A, 2015.
Return to citation in text: [1] [2] [3] -
Glenn, J. S.; Pham, E. A. Use of chloroquine and clemizole compounds for treatment of inflammatory and cancerous conditions PCT. Int. Appl. WO2017004454 A1, Jan 5, 2017.
Return to citation in text: [1] [2] -
Min, Y. S.; Cho, H.-S.; Mo, K. W. New preparation of hydroxychloroquine. PCT Int. Appl. WO 2010027150 A2, March 11, 2010.
Return to citation in text: [1] [2] -
Ashok, K.; Dharmendra, S.; Snajay, N.; Sanjay, B.; Atul, J. An improved process for the preparation of 7-chloro-4-(5-N-ethyl-N-2-hydroxyethylamine)-2-pentyl]aminoquinoline and its intermediates. WO2005/062723, July 14, 2005.
Return to citation in text: [1] [2] -
Continuous flow chemistry catalytic reactions with FlowCAT. http://www.helgroup.com/reactor-systems/hydrogenation-catalysis/flowcat/.
Return to citation in text: [1] [2] -
Jensen, R. K.; Thykier, N.; Enevoldsen, M. V.; Lindhardt, A. T. Org. Process Res. Dev. 2017, 21, 370–376. doi:10.1021/acs.oprd.6b00441
Return to citation in text: [1] -
Falus, P.; Boros, Z.; Hornyánszky, G.; Nagy, J.; Darvas, F.; Ürge, L.; Poppe, L. Tetrahedron Lett. 2011, 52, 1310–1312. doi:10.1016/j.tetlet.2011.01.062
Return to citation in text: [1] -
Chi, Y.; Zhou, Y.-G.; Zhang, X. J. Org. Chem. 2003, 68, 4120–4122. doi:10.1021/jo026856z
Return to citation in text: [1] -
Hoffmann, S.; Seayad, A. M.; List, B. Angew. Chem., Int. Ed. 2005, 44, 7424–7427. doi:10.1002/anie.200503062
Return to citation in text: [1] -
Kitamura, M.; Lee, D.; Hayashi, S.; Tanaka, S.; Yoshimura, M. J. Org. Chem. 2002, 67, 8685–8687. doi:10.1021/jo0203701
Return to citation in text: [1] -
Kadyrov, R.; Riermeier, T. H. Angew. Chem., Int. Ed. 2003, 42, 5472–5474. doi:10.1002/anie.200352503
Return to citation in text: [1] -
Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.; Shah, R. D. J. Org. Chem. 1996, 61, 3849–3862. doi:10.1021/jo960057x
Return to citation in text: [1] -
Gilmore, K.; Vukelić, S.; McQuade, D. T.; Koksch, B.; Seeberger, P. H. Org. Process Res. Dev. 2014, 18, 1771–1776. doi:10.1021/op500310s
Return to citation in text: [1] -
Webb, D.; Jamison, T. F. Org. Lett. 2012, 14, 568–571. doi:10.1021/ol2031872
Return to citation in text: [1] -
Fan, X.; Sans, V.; Yaseneva, P.; Plaza, D. D.; Williams, J.; Lapkin, A. Org. Process Res. Dev. 2012, 16, 1039–1042. doi:10.1021/op200373m
Return to citation in text: [1] -
Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem., Int. Ed. 2001, 40, 650–679. doi:10.1002/1521-3773(20010216)40:4<650::AID-ANIE6500>3.0.CO;2-C
Return to citation in text: [1] -
Blaney, P. M.; Byard, S. J.; Carr, G.; Ellames, G. J.; Herbert, J. M.; Peace, J. E.; Smith, D. I.; Michne, W. F.; Sanner, M. S. Tetrahedron: Asymmetry 1994, 5, 1815–1822. doi:10.1016/0957-4166(94)80090-1
Return to citation in text: [1] -
Cornish, C. A.; Warren, S. J. Chem. Soc., Perkin Trans. 1 1985, 2585–2598. doi:10.1039/P19850002585
Return to citation in text: [1] [2] [3] [4] -
Münstedt, R.; Wannagat, U.; Wrobel, D. J. Organomet. Chem. 1984, 264, 135–148. doi:10.1016/0022-328X(84)85139-6
Return to citation in text: [1] [2] [3] [4] -
Hamlin, T. A.; Lazarus, G. M. L.; Kelly, C. B.; Leadbeater, N. E. Org. Process Res. Dev. 2014, 18, 1253–1258. doi:10.1021/op500190j
Return to citation in text: [1] -
Chapman, M. R.; Kwan, M. H. T.; King, G.; Jolley, K. E.; Hussain, M.; Hussain, S.; Salama, I. E.; Niño, C. G.; Thompson, L. A.; Bayana, M. E.; Clayton, A. D.; Nguyen, B. N.; Turner, N. J.; Kapur, N.; Blacker, A. J. Org. Process Res. Dev. 2017, 21, 1294–1301. doi:10.1021/acs.oprd.7b00173
Return to citation in text: [1] -
Xiao, H.; Tong, R.; Liao, Z.; Chuan, J.; Zhang, L.; Zhang, Y.; Bian, Y. Hydroxychloroquine linolenate and synthesis method thereof. CN103772277 A, May 7, 2014.
Return to citation in text: [1] -
Sleightholm, R.; Yang, B.; Yu, F.; Xie, Y.; Oupický, D. Biomacromolecules 2017, 18, 2247–2257. doi:10.1021/acs.biomac.7b00023
Return to citation in text: [1] -
Ansari, A. M.; Craig, J. C. J. Chem. Soc., Perkin Trans. 2 1994, 1731–1733. doi:10.1039/P29940001731
Return to citation in text: [1]
| 3. | Martin, A. D.; Siamaki, A. R.; Belecki, K.; Gupton, B. F. J. Flow Chem. 2015, 5, 145–147. doi:10.1556/JFC-D-15-00002 |
| 25. | Continuous flow chemistry catalytic reactions with FlowCAT. http://www.helgroup.com/reactor-systems/hydrogenation-catalysis/flowcat/. |
| 41. | Chapman, M. R.; Kwan, M. H. T.; King, G.; Jolley, K. E.; Hussain, M.; Hussain, S.; Salama, I. E.; Niño, C. G.; Thompson, L. A.; Bayana, M. E.; Clayton, A. D.; Nguyen, B. N.; Turner, N. J.; Kapur, N.; Blacker, A. J. Org. Process Res. Dev. 2017, 21, 1294–1301. doi:10.1021/acs.oprd.7b00173 |
| 42. | Xiao, H.; Tong, R.; Liao, Z.; Chuan, J.; Zhang, L.; Zhang, Y.; Bian, Y. Hydroxychloroquine linolenate and synthesis method thereof. CN103772277 A, May 7, 2014. |
| 43. | Sleightholm, R.; Yang, B.; Yu, F.; Xie, Y.; Oupický, D. Biomacromolecules 2017, 18, 2247–2257. doi:10.1021/acs.biomac.7b00023 |
| 44. | Ansari, A. M.; Craig, J. C. J. Chem. Soc., Perkin Trans. 2 1994, 1731–1733. doi:10.1039/P29940001731 |
| 1. | Verghese, J.; Kong, C. J.; Rivalti, D.; Yu, E. C.; Krack, R.; Alcázar, J.; Manley, J. B.; McQuade, D. T.; Ahmad, S.; Belecki, K.; Gupton, B. F. Green Chem. 2017, 19, 2986–2991. doi:10.1039/C7GC00937B |
| 2. | Kong, C. J.; Fisher, D.; Desai, B. K.; Yang, Y.; Ahmad, S.; Belecki, K.; Gupton, B. F. Bioorg. Med. Chem. 2017, 25, 6203–6208. doi:10.1016/j.bmc.2017.07.004 |
| 3. | Martin, A. D.; Siamaki, A. R.; Belecki, K.; Gupton, B. F. J. Flow Chem. 2015, 5, 145–147. doi:10.1556/JFC-D-15-00002 |
| 10. | Belecki, K.; Gupton, B. F. Continuous Processing in Drug Discovery. Green Chemistry Strategies for Drug Discovery; The Royal Society of Chemistry, 2015; pp 127–150. doi:10.1039/9781782622659-00127 |
| 11. | Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2015, 11, 1194–1219. doi:10.3762/bjoc.11.134 |
| 12. | Baxendale, I. R.; Brocken, L.; Mallia, C. J. Green Process. Synth. 2013, 2, 211–230. doi:10.1515/gps-2013-0029 |
| 13. | Opalka, S. M.; Park, J. K.; Longstreet, A. R.; McQuade, D. T. Org. Lett. 2013, 15, 996–999. doi:10.1021/ol303442m |
| 14. | Alonso, N.; Miller, L. Z.; Muñoz, J. d. M.; Alcázar, J.; McQuade, D. T. Adv. Synth. Catal. 2014, 356, 3737–3741. doi:10.1002/adsc.201400243 |
| 15. | McQuade, D. T.; Seeberger, P. H. J. Org. Chem. 2013, 78, 6384–6389. doi:10.1021/jo400583m |
| 16. | Poechlauer, P.; Colberg, J.; Fisher, E.; Jansen, M.; Johnson, M. D.; Koenig, S. G.; Lawler, M.; Laporte, T.; Manley, J.; Martin, B.; Kearney-McMullan, A. Org. Process Res. Dev. 2013, 17, 1472–1478. doi:10.1021/op400245s |
| 17. | Poechlauer, P.; Manley, J.; Broxterman, R.; Gregertsen, B.; Ridemark, M. Org. Process Res. Dev. 2012, 16, 1586–1590. doi:10.1021/op300159y |
| 18. | Britton, J.; Raston, C. L. Chem. Soc. Rev. 2017, 46, 1250–2171. doi:10.1039/C6CS00830E |
| 19. | Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183 |
| 40. | Hamlin, T. A.; Lazarus, G. M. L.; Kelly, C. B.; Leadbeater, N. E. Org. Process Res. Dev. 2014, 18, 1253–1258. doi:10.1021/op500190j |
| 7. | Pavelka, K., Jr.; Sen, K. P.; Pelísková, Z.; Vácha, J.; Trnavský, K. Ann. Rheum. Dis. 1989, 48, 542–546. doi:10.1136/ard.48.7.542 |
| 8. | Hage, M. P.; Al-Badri, M. R.; Azar, S. T. Ther. Adv. Endocrinol. Metab. 2014, 5, 77–85. doi:10.1177/2042018814547204 |
| 9. | Poorvashree, J.; Suneela, D. Drug Delivery Transl. Res. 2017, 7, 709–730. doi:10.1007/s13346-017-0420-5 |
| 21. | You, H.; Liu, Y.; Ning, F.; Zheng, Z.; Yu, Q.; Niu, X.; Li, C. CN104803859A, 2015. |
| 22. | Glenn, J. S.; Pham, E. A. Use of chloroquine and clemizole compounds for treatment of inflammatory and cancerous conditions PCT. Int. Appl. WO2017004454 A1, Jan 5, 2017. |
| 23. | Min, Y. S.; Cho, H.-S.; Mo, K. W. New preparation of hydroxychloroquine. PCT Int. Appl. WO 2010027150 A2, March 11, 2010. |
| 24. | Ashok, K.; Dharmendra, S.; Snajay, N.; Sanjay, B.; Atul, J. An improved process for the preparation of 7-chloro-4-(5-N-ethyl-N-2-hydroxyethylamine)-2-pentyl]aminoquinoline and its intermediates. WO2005/062723, July 14, 2005. |
| 5. | Surrey, A.-R.; Hammer, H. F. J. Am. Chem. Soc. 1950, 72, 1814–1815. doi:10.1021/ja01160a116 |
| 6. | Bailey, D. M. J. Med. Chem. 1969, 12, 184–185. doi:10.1021/jm00301a055 |
| 37. | Blaney, P. M.; Byard, S. J.; Carr, G.; Ellames, G. J.; Herbert, J. M.; Peace, J. E.; Smith, D. I.; Michne, W. F.; Sanner, M. S. Tetrahedron: Asymmetry 1994, 5, 1815–1822. doi:10.1016/0957-4166(94)80090-1 |
| 4. | World Health Organization. WHO Malaria Report 2016. Geneva, 2016; http://www.who.int/malaria/publications/world-malaria-report-2016/report/en/. |
| 38. | Cornish, C. A.; Warren, S. J. Chem. Soc., Perkin Trans. 1 1985, 2585–2598. doi:10.1039/P19850002585 |
| 39. | Münstedt, R.; Wannagat, U.; Wrobel, D. J. Organomet. Chem. 1984, 264, 135–148. doi:10.1016/0022-328X(84)85139-6 |
| 22. | Glenn, J. S.; Pham, E. A. Use of chloroquine and clemizole compounds for treatment of inflammatory and cancerous conditions PCT. Int. Appl. WO2017004454 A1, Jan 5, 2017. |
| 23. | Min, Y. S.; Cho, H.-S.; Mo, K. W. New preparation of hydroxychloroquine. PCT Int. Appl. WO 2010027150 A2, March 11, 2010. |
| 25. | Continuous flow chemistry catalytic reactions with FlowCAT. http://www.helgroup.com/reactor-systems/hydrogenation-catalysis/flowcat/. |
| 26. | Jensen, R. K.; Thykier, N.; Enevoldsen, M. V.; Lindhardt, A. T. Org. Process Res. Dev. 2017, 21, 370–376. doi:10.1021/acs.oprd.6b00441 |
| 27. | Falus, P.; Boros, Z.; Hornyánszky, G.; Nagy, J.; Darvas, F.; Ürge, L.; Poppe, L. Tetrahedron Lett. 2011, 52, 1310–1312. doi:10.1016/j.tetlet.2011.01.062 |
| 28. | Chi, Y.; Zhou, Y.-G.; Zhang, X. J. Org. Chem. 2003, 68, 4120–4122. doi:10.1021/jo026856z |
| 29. | Hoffmann, S.; Seayad, A. M.; List, B. Angew. Chem., Int. Ed. 2005, 44, 7424–7427. doi:10.1002/anie.200503062 |
| 30. | Kitamura, M.; Lee, D.; Hayashi, S.; Tanaka, S.; Yoshimura, M. J. Org. Chem. 2002, 67, 8685–8687. doi:10.1021/jo0203701 |
| 31. | Kadyrov, R.; Riermeier, T. H. Angew. Chem., Int. Ed. 2003, 42, 5472–5474. doi:10.1002/anie.200352503 |
| 38. | Cornish, C. A.; Warren, S. J. Chem. Soc., Perkin Trans. 1 1985, 2585–2598. doi:10.1039/P19850002585 |
| 39. | Münstedt, R.; Wannagat, U.; Wrobel, D. J. Organomet. Chem. 1984, 264, 135–148. doi:10.1016/0022-328X(84)85139-6 |
| 21. | You, H.; Liu, Y.; Ning, F.; Zheng, Z.; Yu, Q.; Niu, X.; Li, C. CN104803859A, 2015. |
| 32. | Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.; Shah, R. D. J. Org. Chem. 1996, 61, 3849–3862. doi:10.1021/jo960057x |
| 33. | Gilmore, K.; Vukelić, S.; McQuade, D. T.; Koksch, B.; Seeberger, P. H. Org. Process Res. Dev. 2014, 18, 1771–1776. doi:10.1021/op500310s |
| 34. | Webb, D.; Jamison, T. F. Org. Lett. 2012, 14, 568–571. doi:10.1021/ol2031872 |
| 35. | Fan, X.; Sans, V.; Yaseneva, P.; Plaza, D. D.; Williams, J.; Lapkin, A. Org. Process Res. Dev. 2012, 16, 1039–1042. doi:10.1021/op200373m |
| 36. | Kirschning, A.; Monenschein, H.; Wittenberg, R. Angew. Chem., Int. Ed. 2001, 40, 650–679. doi:10.1002/1521-3773(20010216)40:4<650::AID-ANIE6500>3.0.CO;2-C |
| 21. | You, H.; Liu, Y.; Ning, F.; Zheng, Z.; Yu, Q.; Niu, X.; Li, C. CN104803859A, 2015. |
| 38. | Cornish, C. A.; Warren, S. J. Chem. Soc., Perkin Trans. 1 1985, 2585–2598. doi:10.1039/P19850002585 |
| 39. | Münstedt, R.; Wannagat, U.; Wrobel, D. J. Organomet. Chem. 1984, 264, 135–148. doi:10.1016/0022-328X(84)85139-6 |
| 20. | Gutmann, B.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed. 2015, 54, 6688–6728. doi:10.1002/anie.201409318 |
| 24. | Ashok, K.; Dharmendra, S.; Snajay, N.; Sanjay, B.; Atul, J. An improved process for the preparation of 7-chloro-4-(5-N-ethyl-N-2-hydroxyethylamine)-2-pentyl]aminoquinoline and its intermediates. WO2005/062723, July 14, 2005. |
| 38. | Cornish, C. A.; Warren, S. J. Chem. Soc., Perkin Trans. 1 1985, 2585–2598. doi:10.1039/P19850002585 |
| 39. | Münstedt, R.; Wannagat, U.; Wrobel, D. J. Organomet. Chem. 1984, 264, 135–148. doi:10.1016/0022-328X(84)85139-6 |
© 2018 Yu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)