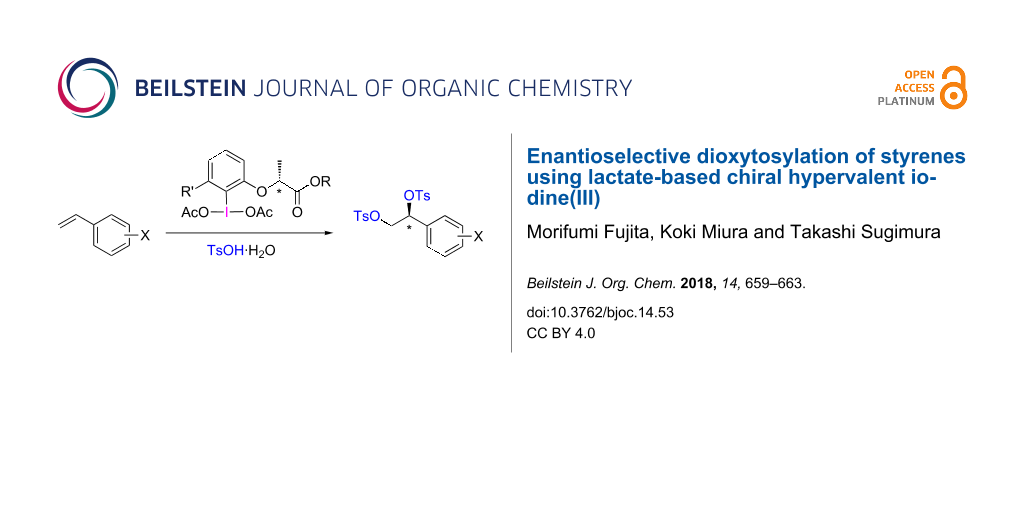
Abstract
A series of optically active hypervalent iodine(III) reagents prepared from the corresponding (R)-2-(2-iodophenoxy)propanoate derivative was employed for the asymmetric dioxytosylation of styrene and its derivatives. The electrophilic addition of the hypervalent iodine(III) compound toward styrene proceeded with high enantioface selectivity to give 1-aryl-1,2-di(tosyloxy)ethane with an enantiomeric excess of 70–96% of the (S)-isomer.

Graphical Abstract
Findings
Hypervalent aryl-λ3-iodanes have been widely used for metal-free oxidation with high selectivity in organic synthesis [1-3]. The reactivity of an aryl-λ3-iodane is controlled by the electronic and steric properties of the aryl group and the heteroatomic ligand coordinated to the iodine atom. Optically active hypervalent iodine compounds contain chiral ligands or chiral aryl groups. Several types of optically active hypervalent iodine reagents and catalysts have been developed for highly stereocontrolled oxidative transformations [4-14]. The enantioselective vicinal difunctionalization of alkenes constitutes one type of attractive transformation achieved by chiral hypervalent iodine compounds. As a seminal example in this field, Wirth et al. [15-17] reported the dioxytosylation of styrene (1a, Scheme 1). Chiral hypervalent iodine reagents 2 bearing a 1-methoxyethyl side chain were used for enantiocontrol of the dioxytosylation, and the maximum enantiomeric excess (ee) of the product 3a reached 65%. Despite recent rapid progress in the field of asymmetric oxidation achieved by chiral hypervalent iodine compounds, there has been no subsequent examination of dioxytosylation, which can be used as a standard reaction for comparing the enantiocontrolling ability of chiral hypervalent iodine reagents.
![[1860-5397-14-53-i1]](/bjoc/content/inline/1860-5397-14-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Enantioselective dioxytosylation of styrene as a seminal example.
Scheme 1: Enantioselective dioxytosylation of styrene as a seminal example.
The design of chiral hypervalent iodine reagents using a lactate motif has been employed for several types of oxidation reaction since we first reported this procedure [18]. Enantioselective oxidative transformations include the dearomatization of phenols [19-24], α-functionalization of carbonyl compounds [25-29], and vicinal difunctionalization of alkenes [18,30-50]. Here, the efficiency of the lactate-based chiral hypervalent iodine reagents 4a–e (Figure 1) was assessed using the dioxytosylation of styrenes as a reference reaction.
![[1860-5397-14-53-1]](/bjoc/content/figures/1860-5397-14-53-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Series of lactate-based hypervalent iodine reagents.
Figure 1: Series of lactate-based hypervalent iodine reagents.
A series of lactate-derived aryl-λ3-iodanes 4a–e was used for the oxidation of styrenes 1 in the presence of p-toluenesulfonic acid (TsOH) in dichloromethane. The reaction proceeded at −50 °C to give the 1,2-dioxytosylated product 3 and the rearranged product 5. The yields of 3 and 5 were determined by 1H NMR using an internal standard. The ee of 3 was determined by chiral HPLC analysis. The results for the yields and ee are summarized in Table 1.
Table 1: Enantioselective dioxytosylation of styrenes 1 using aryl-λ3-iodanes 4.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-53-i3.svg?max-width=637&scale=1.0)
|
|||||
| Yield (%)b | |||||
| Entry | Substrate | Reagent | 3 | 5 | ee of 3 (%)c,d |
| 1 | 1a (X = H) | 4a | 53 | 15 | 70 (S) |
| 2 | 1a (X = H) | 4b | 49 | 16 | 80 (S) |
| 3 | 1a (X = H) | 4c | 41 | 14 | 78 (S) |
| 4 | 1a (X = H) | 4d | 41 | 22 | 70 (S) |
| 5 | 1a (X = H) | 4e | 80 | 20 | 92 (S) |
| 6 | 1b (X = p-Cl)e | 4a | 63 | 6 | 70 |
| 7 | 1b (X = p-Cl)e | 4b | 46 | 5 | 76 |
| 8 | 1b (X = p-Cl)e | 4e | 79 | 5 | 90 |
| 9 | 1c (X = o-Me) | 4a | 7 | 34 | 79 |
| 10 | 1c (X = o-Me) | 4e | 10 | 35 | 96 |
aThe reaction was carried out at −50 °C in dichloromethane containing 4 (47 mM), TsOH (86 mM), and 1 (43 mM) for 4 h. bThe yield was determined by 1H NMR using an internal standard. cThe ee was determined by chiral HPLC using a Daicel CHIRALPAK AD column (ø 4.6 mm × 250 mm). dPreferential configuration of product 3. The absolute stereochemistry of 3b and 3c was not determined. eThe reaction was carried out for 20 h.
The reaction of styrene (1a) with 4a gave the 1,2-dioxytosylated product 3a with 70% ee of the (S)-isomer (Table 1, entry 1). An ee of equal to or greater than 70% was also achieved in the reactions with the other lactate-based reagents 4b–e (Table 1, entries 2–5). The reaction with the 2,6-bis(lactate)aryl reagent 4e provided a high ee of 92%. The reactions of p-chlorostyrene (1b) gave 3b with a similar ee, and the ratios of 3 to 5 (3b to 5b) were higher than those in the reaction of 1a (Table 1, entries 6–8). In the reactions of o-methylstyrene (1c), the ee of the 1,2-dioxytosylated product 3c was slightly higher than those of 3a and 3b, but the regioselectivity for 3c over 5c was poor (Table 1, entries 9 and 10).
Scheme 2 illustrates possible reaction pathways that lead to 3 and the achiral byproduct 5. The treatment of (diacetoxyiodo)benzene with TsOH readily gives Koser’s reagent [PhI(OH)OTs] [51], which has a higher electrophilicity toward the carbon–carbon double bond in 1. The dioxytosylation of alkenes with Koser’s reagent was found to proceed via an SN2 reaction of a cyclic intermediate such as I1, judging from the syn selectivity of the dioxytosylation [52,53]. The attack of the tosylate ion on I1 possibly takes place at the benzylic position or at the methylene carbon atom. The positive charge of I1 may be stabilized by the aryl group and localized at the benzylic position. This may allow the preferential formation of I3 from I1. If I2 was the major intermediate in the pathway leading to 3, the stereochemical purity of 3 would have decreased owing to the facile elimination of the iodonium group [54] at the benzylic position of I2 (SN1). The high enantiomeric ratio of 3 can be rationalized via a preference for the I1→I3→3 pathway over the I1→I2→3 pathway. The product ratio of 3 to 5 was affected by the ring substituent in styrenes 1: the electron-withdrawing chloro substituent in 1b increased the amount of 3, whereas the electron-donating methyl substituent in 1c decreased the amount of 3. An electron-donating aryl group increases the rate of participation of the aryl group (I3→I4). In other words, a reaction pathway that bifurcates from I3 to 3 and 5 agrees well with the regioselectivity for 3 over 5 observed for the substituted styrenes. The phenonium cation intermediate I4 contains two reaction sites on the ethylene bridge. Electron donation due to the lone pair on the oxygen atom of the internal tosyloxy group may weaken the bond between the tosyloxy-bonded carbon and the quaternary carbon in I4.
![[1860-5397-14-53-i2]](/bjoc/content/inline/1860-5397-14-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible pathways in dioxytosylation of styrenes.
Scheme 2: Plausible pathways in dioxytosylation of styrenes.
The reaction of styrene with 4a–e preferentially gave (S)-3, which forms via an electrophilic addition of the iodane toward the Si face of styrene, followed by an SN2 reaction with the tosylate ion. If an SN1 mechanism were involved in the oxytosylation of I1, the enantiomeric ratio of 3 would decrease owing to the planar structure of the benzylic cation. Thus, the tosylate ion may act as an effective nucleophile for the SN2 reaction of I1. The stereoface-differentiation in the dioxytosylation reaction using the lactate-derived aryl-λ3-iodanes is similar to that in preceding reactions [14], which include the diacetoxylation [38,39,50] and diamination [30,49] of styrene.
In summary, the reaction of styrenes with lactate-derived aryl-λ3-iodanes gave the dioxytosylated product with an ee of 70–96%.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra are available. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Zhdankin, V. V. Hypervalent Iodine Chemistry; John Wiley & Sons: Chichester, U.K., 2014.
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Wirth, T., Ed. Hypervalent Iodine Chemistry; Springer: Basel, Switzerland, 2016. doi:10.1007/978-3-319-33733-3
Return to citation in text: [1] -
Ngatimin, M.; Lupton, D. W. Aust. J. Chem. 2010, 63, 653–658. doi:10.1071/CH09625
Return to citation in text: [1] -
Liang, H.; Ciufolini, M. A. Angew. Chem., Int. Ed. 2011, 50, 11849–11851. doi:10.1002/anie.201106127
Angew. Chem. 2011, 123, 12051–12053. doi:10.1002/ange.201106127
Return to citation in text: [1] -
Uyanik, M.; Ishihara, K. J. Synth. Org. Chem., Jpn. 2012, 70, 1116–1122. doi:10.5059/yukigoseikyokaishi.70.1116
Return to citation in text: [1] -
Parra, A.; Reboredo, S. Chem. – Eur. J. 2013, 19, 17244–17260. doi:10.1002/chem.201302220
Return to citation in text: [1] -
Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950–971. doi:10.1002/asia.201301582
Return to citation in text: [1] -
Romero, R. M.; Wöste, T. H.; Muñiz, K. Chem. – Asian J. 2014, 9, 972–983. doi:10.1002/asia.201301637
Return to citation in text: [1] -
Harned, A. M. Tetrahedron Lett. 2014, 55, 4681–4689. doi:10.1016/j.tetlet.2014.06.051
Return to citation in text: [1] -
Zheng, Z. S.; Zhang-Negrerie, D.; Du, Y. F.; Zhao, K. Sci. China: Chem. 2014, 57, 189–214. doi:10.1007/s11426-013-5043-1
Return to citation in text: [1] -
Berthiol, F. Synthesis 2015, 47, 587–603. doi:10.1055/s-0034-1379892
Return to citation in text: [1] -
Basdevant, B.; Guilbault, A.-A.; Beaulieu, S.; Lauriers, A. J.-D.; Legault, C. Y. Pure Appl. Chem. 2017, 89, 781–789. doi:10.1515/pac-2016-1212
Return to citation in text: [1] -
Fujita, M. Tetrahedron Lett. 2017, 58, 4409–4419. doi:10.1016/j.tetlet.2017.10.019
Return to citation in text: [1] [2] -
Wirth, T.; Hirt, U. H. Tetrahedron: Asymmetry 1997, 8, 23–26. doi:10.1016/S0957-4166(96)00469-7
Return to citation in text: [1] -
Hirt, U. H.; Spingler, B.; Wirth, T. J. Org. Chem. 1998, 63, 7674–7679. doi:10.1021/jo980475x
Return to citation in text: [1] -
Hirt, U. H.; Schuster, M. F. H.; French, A. N.; Wiest, O. G.; Wirth, T. Eur. J. Org. Chem. 2001, 1569–1579. doi:10.1002/1099-0690(200104)2001:8<1569::AID-EJOC1569>3.0.CO;2-T
Return to citation in text: [1] -
Fujita, M.; Okuno, S.; Lee, H. J.; Sugimura, T.; Okuyama, T. Tetrahedron Lett. 2007, 48, 8691–8694. doi:10.1016/j.tetlet.2007.10.015
Return to citation in text: [1] [2] -
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2010, 49, 2175–2177. doi:10.1002/anie.200907352
Angew. Chem. 2010, 122, 2221–2223. doi:10.1002/ange.200907352
Return to citation in text: [1] -
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2013, 52, 9215–9218. doi:10.1002/anie.201303559
Angew. Chem. 2013, 125, 9385–9388. doi:10.1002/ange.201303559
Return to citation in text: [1] -
Zhang, D.-Y.; Xu, L.; Wu, H.; Gong, L.-Z. Chem. – Eur. J. 2015, 21, 10314–10317. doi:10.1002/chem.201501583
Return to citation in text: [1] -
Yoshida, Y.; Magara, A.; Mino, T.; Sakamoto, M. Tetrahedron Lett. 2016, 57, 5103–5107. doi:10.1016/j.tetlet.2016.10.016
Return to citation in text: [1] -
Uyanik, M.; Sasakura, N.; Mizuno, M.; Ishihara, K. ACS Catal. 2017, 7, 872–876. doi:10.1021/acscatal.6b03380
Return to citation in text: [1] -
Jain, N.; Xu, S.; Ciufolini, M. A. Chem. – Eur. J. 2017, 23, 4542–4546. doi:10.1002/chem.201700667
Return to citation in text: [1] -
Mizar, P.; Wirth, T. Angew. Chem., Int. Ed. 2014, 53, 5993–5997. doi:10.1002/anie.201400405
Angew. Chem. 2014, 126, 6103–6107. doi:10.1002/ange.201400405
Return to citation in text: [1] -
Wu, H.; He, Y.-P.; Xu, L.; Zhang, D.-Y.; Gong, L.-Z. Angew. Chem., Int. Ed. 2014, 53, 3466–3469. doi:10.1002/anie.201309967
Angew. Chem. 2014, 126, 3534–3537. doi:10.1002/ange.201309967
Return to citation in text: [1] -
Basdevant, B.; Legault, C. Y. Org. Lett. 2015, 17, 4918–4921. doi:10.1021/acs.orglett.5b02501
Return to citation in text: [1] -
Feng, Y.; Huang, R.; Hu, L.; Xiong, Y.; Coeffard, V. Synthesis 2016, 48, 2637–2644. doi:10.1055/s-0035-1561442
Return to citation in text: [1] -
Cao, Y.; Zhang, X.; Lin, G.; Zhang-Negrerie, D.; Du, Y. Org. Lett. 2016, 18, 5580–5583. doi:10.1021/acs.orglett.6b02816
Return to citation in text: [1] -
Muñiz, K.; Barreiro, L.; Romero, R. M.; Martínez, C. J. Am. Chem. Soc. 2017, 139, 4354–4357. doi:10.1021/jacs.7b01443
Return to citation in text: [1] [2] -
Gelis, C.; Dumoulin, A.; Bekkaye, M.; Neuville, L.; Masson, G. Org. Lett. 2017, 19, 278–281. doi:10.1021/acs.orglett.6b03631
Return to citation in text: [1] -
Qurban, J.; Elsherbini, M.; Wirth, T. J. Org. Chem. 2017, 82, 11872–11876. doi:10.1021/acs.joc.7b01571
Return to citation in text: [1] -
Shimogaki, M.; Fujita, M.; Sugimura, T. J. Org. Chem. 2017, 82, 11836–11840. doi:10.1021/acs.joc.7b01141
Return to citation in text: [1] -
Banik, S. M.; Medley, J. W.; Jacobsen, E. N. Science 2016, 353, 51–54. doi:10.1126/science.aaf8078
Return to citation in text: [1] -
Banik, S. M.; Medley, J. W.; Jacobsen, E. N. J. Am. Chem. Soc. 2016, 138, 5000–5003. doi:10.1021/jacs.6b02391
Return to citation in text: [1] -
Woerly, E.; Banik, S. M.; Jacobsen, E. N. J. Am. Chem. Soc. 2016, 138, 13858–13861. doi:10.1021/jacs.6b09499
Return to citation in text: [1] -
Ahmad, A.; Silva, L. F., Jr. J. Org. Chem. 2016, 81, 2174–2181. doi:10.1021/acs.joc.5b02803
Return to citation in text: [1] -
Haubenreisser, S.; Wöste, T. H.; Martínez, C.; Ishihara, K.; Muñiz, K. Angew. Chem., Int. Ed. 2016, 55, 413–417. doi:10.1002/anie.201507180
Angew. Chem. 2016, 128, 422–426. doi:10.1002/ange.201507180
Return to citation in text: [1] [2] -
Wöste, T. H.; Muñiz, K. Synthesis 2016, 48, 816–827. doi:10.1055/s-0035-1561313
Return to citation in text: [1] [2] -
Mizar, P.; Niebuhr, R.; Hutchings, M.; Farooq, U.; Wirth, T. Chem. – Eur. J. 2016, 22, 1614–1617. doi:10.1002/chem.201504636
Return to citation in text: [1] -
Brown, M.; Kumar, R.; Rehbein, J.; Wirth, T. Chem. – Eur. J. 2016, 22, 4030–4035. doi:10.1002/chem.201504844
Return to citation in text: [1] -
Shimogaki, M.; Fujita, M.; Sugimura, T. Angew. Chem., Int. Ed. 2016, 55, 15797–15801. doi:10.1002/anie.201609110
Angew. Chem. 2016, 128, 16029–16033. doi:10.1002/ange.201609110
Return to citation in text: [1] -
Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453–1456. doi:10.1021/acs.orglett.5b00333
Return to citation in text: [1] -
Takesue, T.; Fujita, M.; Sugimura, T.; Akutsu, H. Org. Lett. 2014, 16, 4634–4637. doi:10.1021/ol502225p
Return to citation in text: [1] -
Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Angew. Chem., Int. Ed. 2013, 52, 2469–2473. doi:10.1002/anie.201208471
Angew. Chem. 2013, 125, 2529–2533. doi:10.1002/ange.201208471
Return to citation in text: [1] -
Farid, U.; Malmedy, F.; Claveau, R.; Albers, L.; Wirth, T. Angew. Chem., Int. Ed. 2013, 52, 7018–7022. doi:10.1002/anie.201302358
Angew. Chem. 2013, 125, 7156–7160. doi:10.1002/ange.201302358
Return to citation in text: [1] -
Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. RSC Adv. 2013, 3, 17717–17725. doi:10.1039/c3ra43230k
Return to citation in text: [1] -
Farid, U.; Wirth, T. Angew. Chem., Int. Ed. 2012, 51, 3462–3465. doi:10.1002/anie.201107703
Angew. Chem. 2012, 124, 3518–3522. doi:10.1002/ange.201107703
Return to citation in text: [1] -
Röben, C.; Souto, J. A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Angew. Chem., Int. Ed. 2011, 50, 9478–9482. doi:10.1002/anie.201103077
Angew. Chem. 2011, 123, 9650–9654. doi:10.1002/ange.201103077
Return to citation in text: [1] [2] -
Fujita, M.; Wakita, M.; Sugimura, T. Chem. Commun. 2011, 47, 3983–3985. doi:10.1039/c1cc10129c
Return to citation in text: [1] [2] -
Koser, G. F.; Wettach, R. H. J. Org. Chem. 1977, 42, 1476–1478. doi:10.1021/jo00428a052
Return to citation in text: [1] -
Koser, G. F.; Rebrovic, L.; Wettach, R. H. J. Org. Chem. 1981, 46, 4324–4326. doi:10.1021/jo00334a057
Return to citation in text: [1] -
Rebrovic, L.; Koser, G. F. J. Org. Chem. 1984, 49, 2462–2472. doi:10.1021/jo00187a032
Return to citation in text: [1] -
Okuyama, T.; Takino, T.; Sueda, T.; Ochiai, M. J. Am. Chem. Soc. 1995, 117, 3360–3367. doi:10.1021/ja00117a006
Return to citation in text: [1]
| 1. | Zhdankin, V. V. Hypervalent Iodine Chemistry; John Wiley & Sons: Chichester, U.K., 2014. |
| 2. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 3. | Wirth, T., Ed. Hypervalent Iodine Chemistry; Springer: Basel, Switzerland, 2016. doi:10.1007/978-3-319-33733-3 |
| 19. |
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2010, 49, 2175–2177. doi:10.1002/anie.200907352
Angew. Chem. 2010, 122, 2221–2223. doi:10.1002/ange.200907352 |
| 20. |
Uyanik, M.; Yasui, T.; Ishihara, K. Angew. Chem., Int. Ed. 2013, 52, 9215–9218. doi:10.1002/anie.201303559
Angew. Chem. 2013, 125, 9385–9388. doi:10.1002/ange.201303559 |
| 21. | Zhang, D.-Y.; Xu, L.; Wu, H.; Gong, L.-Z. Chem. – Eur. J. 2015, 21, 10314–10317. doi:10.1002/chem.201501583 |
| 22. | Yoshida, Y.; Magara, A.; Mino, T.; Sakamoto, M. Tetrahedron Lett. 2016, 57, 5103–5107. doi:10.1016/j.tetlet.2016.10.016 |
| 23. | Uyanik, M.; Sasakura, N.; Mizuno, M.; Ishihara, K. ACS Catal. 2017, 7, 872–876. doi:10.1021/acscatal.6b03380 |
| 24. | Jain, N.; Xu, S.; Ciufolini, M. A. Chem. – Eur. J. 2017, 23, 4542–4546. doi:10.1002/chem.201700667 |
| 18. | Fujita, M.; Okuno, S.; Lee, H. J.; Sugimura, T.; Okuyama, T. Tetrahedron Lett. 2007, 48, 8691–8694. doi:10.1016/j.tetlet.2007.10.015 |
| 15. | Wirth, T.; Hirt, U. H. Tetrahedron: Asymmetry 1997, 8, 23–26. doi:10.1016/S0957-4166(96)00469-7 |
| 16. | Hirt, U. H.; Spingler, B.; Wirth, T. J. Org. Chem. 1998, 63, 7674–7679. doi:10.1021/jo980475x |
| 17. | Hirt, U. H.; Schuster, M. F. H.; French, A. N.; Wiest, O. G.; Wirth, T. Eur. J. Org. Chem. 2001, 1569–1579. doi:10.1002/1099-0690(200104)2001:8<1569::AID-EJOC1569>3.0.CO;2-T |
| 30. | Muñiz, K.; Barreiro, L.; Romero, R. M.; Martínez, C. J. Am. Chem. Soc. 2017, 139, 4354–4357. doi:10.1021/jacs.7b01443 |
| 49. |
Röben, C.; Souto, J. A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Angew. Chem., Int. Ed. 2011, 50, 9478–9482. doi:10.1002/anie.201103077
Angew. Chem. 2011, 123, 9650–9654. doi:10.1002/ange.201103077 |
| 4. | Ngatimin, M.; Lupton, D. W. Aust. J. Chem. 2010, 63, 653–658. doi:10.1071/CH09625 |
| 5. |
Liang, H.; Ciufolini, M. A. Angew. Chem., Int. Ed. 2011, 50, 11849–11851. doi:10.1002/anie.201106127
Angew. Chem. 2011, 123, 12051–12053. doi:10.1002/ange.201106127 |
| 6. | Uyanik, M.; Ishihara, K. J. Synth. Org. Chem., Jpn. 2012, 70, 1116–1122. doi:10.5059/yukigoseikyokaishi.70.1116 |
| 7. | Parra, A.; Reboredo, S. Chem. – Eur. J. 2013, 19, 17244–17260. doi:10.1002/chem.201302220 |
| 8. | Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950–971. doi:10.1002/asia.201301582 |
| 9. | Romero, R. M.; Wöste, T. H.; Muñiz, K. Chem. – Asian J. 2014, 9, 972–983. doi:10.1002/asia.201301637 |
| 10. | Harned, A. M. Tetrahedron Lett. 2014, 55, 4681–4689. doi:10.1016/j.tetlet.2014.06.051 |
| 11. | Zheng, Z. S.; Zhang-Negrerie, D.; Du, Y. F.; Zhao, K. Sci. China: Chem. 2014, 57, 189–214. doi:10.1007/s11426-013-5043-1 |
| 12. | Berthiol, F. Synthesis 2015, 47, 587–603. doi:10.1055/s-0034-1379892 |
| 13. | Basdevant, B.; Guilbault, A.-A.; Beaulieu, S.; Lauriers, A. J.-D.; Legault, C. Y. Pure Appl. Chem. 2017, 89, 781–789. doi:10.1515/pac-2016-1212 |
| 14. | Fujita, M. Tetrahedron Lett. 2017, 58, 4409–4419. doi:10.1016/j.tetlet.2017.10.019 |
| 52. | Koser, G. F.; Rebrovic, L.; Wettach, R. H. J. Org. Chem. 1981, 46, 4324–4326. doi:10.1021/jo00334a057 |
| 53. | Rebrovic, L.; Koser, G. F. J. Org. Chem. 1984, 49, 2462–2472. doi:10.1021/jo00187a032 |
| 14. | Fujita, M. Tetrahedron Lett. 2017, 58, 4409–4419. doi:10.1016/j.tetlet.2017.10.019 |
| 51. | Koser, G. F.; Wettach, R. H. J. Org. Chem. 1977, 42, 1476–1478. doi:10.1021/jo00428a052 |
| 38. |
Haubenreisser, S.; Wöste, T. H.; Martínez, C.; Ishihara, K.; Muñiz, K. Angew. Chem., Int. Ed. 2016, 55, 413–417. doi:10.1002/anie.201507180
Angew. Chem. 2016, 128, 422–426. doi:10.1002/ange.201507180 |
| 39. | Wöste, T. H.; Muñiz, K. Synthesis 2016, 48, 816–827. doi:10.1055/s-0035-1561313 |
| 50. | Fujita, M.; Wakita, M.; Sugimura, T. Chem. Commun. 2011, 47, 3983–3985. doi:10.1039/c1cc10129c |
| 18. | Fujita, M.; Okuno, S.; Lee, H. J.; Sugimura, T.; Okuyama, T. Tetrahedron Lett. 2007, 48, 8691–8694. doi:10.1016/j.tetlet.2007.10.015 |
| 30. | Muñiz, K.; Barreiro, L.; Romero, R. M.; Martínez, C. J. Am. Chem. Soc. 2017, 139, 4354–4357. doi:10.1021/jacs.7b01443 |
| 31. | Gelis, C.; Dumoulin, A.; Bekkaye, M.; Neuville, L.; Masson, G. Org. Lett. 2017, 19, 278–281. doi:10.1021/acs.orglett.6b03631 |
| 32. | Qurban, J.; Elsherbini, M.; Wirth, T. J. Org. Chem. 2017, 82, 11872–11876. doi:10.1021/acs.joc.7b01571 |
| 33. | Shimogaki, M.; Fujita, M.; Sugimura, T. J. Org. Chem. 2017, 82, 11836–11840. doi:10.1021/acs.joc.7b01141 |
| 34. | Banik, S. M.; Medley, J. W.; Jacobsen, E. N. Science 2016, 353, 51–54. doi:10.1126/science.aaf8078 |
| 35. | Banik, S. M.; Medley, J. W.; Jacobsen, E. N. J. Am. Chem. Soc. 2016, 138, 5000–5003. doi:10.1021/jacs.6b02391 |
| 36. | Woerly, E.; Banik, S. M.; Jacobsen, E. N. J. Am. Chem. Soc. 2016, 138, 13858–13861. doi:10.1021/jacs.6b09499 |
| 37. | Ahmad, A.; Silva, L. F., Jr. J. Org. Chem. 2016, 81, 2174–2181. doi:10.1021/acs.joc.5b02803 |
| 38. |
Haubenreisser, S.; Wöste, T. H.; Martínez, C.; Ishihara, K.; Muñiz, K. Angew. Chem., Int. Ed. 2016, 55, 413–417. doi:10.1002/anie.201507180
Angew. Chem. 2016, 128, 422–426. doi:10.1002/ange.201507180 |
| 39. | Wöste, T. H.; Muñiz, K. Synthesis 2016, 48, 816–827. doi:10.1055/s-0035-1561313 |
| 40. | Mizar, P.; Niebuhr, R.; Hutchings, M.; Farooq, U.; Wirth, T. Chem. – Eur. J. 2016, 22, 1614–1617. doi:10.1002/chem.201504636 |
| 41. | Brown, M.; Kumar, R.; Rehbein, J.; Wirth, T. Chem. – Eur. J. 2016, 22, 4030–4035. doi:10.1002/chem.201504844 |
| 42. |
Shimogaki, M.; Fujita, M.; Sugimura, T. Angew. Chem., Int. Ed. 2016, 55, 15797–15801. doi:10.1002/anie.201609110
Angew. Chem. 2016, 128, 16029–16033. doi:10.1002/ange.201609110 |
| 43. | Alhalib, A.; Kamouka, S.; Moran, W. J. Org. Lett. 2015, 17, 1453–1456. doi:10.1021/acs.orglett.5b00333 |
| 44. | Takesue, T.; Fujita, M.; Sugimura, T.; Akutsu, H. Org. Lett. 2014, 16, 4634–4637. doi:10.1021/ol502225p |
| 45. |
Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Angew. Chem., Int. Ed. 2013, 52, 2469–2473. doi:10.1002/anie.201208471
Angew. Chem. 2013, 125, 2529–2533. doi:10.1002/ange.201208471 |
| 46. |
Farid, U.; Malmedy, F.; Claveau, R.; Albers, L.; Wirth, T. Angew. Chem., Int. Ed. 2013, 52, 7018–7022. doi:10.1002/anie.201302358
Angew. Chem. 2013, 125, 7156–7160. doi:10.1002/ange.201302358 |
| 47. | Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. RSC Adv. 2013, 3, 17717–17725. doi:10.1039/c3ra43230k |
| 48. |
Farid, U.; Wirth, T. Angew. Chem., Int. Ed. 2012, 51, 3462–3465. doi:10.1002/anie.201107703
Angew. Chem. 2012, 124, 3518–3522. doi:10.1002/ange.201107703 |
| 49. |
Röben, C.; Souto, J. A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Angew. Chem., Int. Ed. 2011, 50, 9478–9482. doi:10.1002/anie.201103077
Angew. Chem. 2011, 123, 9650–9654. doi:10.1002/ange.201103077 |
| 50. | Fujita, M.; Wakita, M.; Sugimura, T. Chem. Commun. 2011, 47, 3983–3985. doi:10.1039/c1cc10129c |
| 25. |
Mizar, P.; Wirth, T. Angew. Chem., Int. Ed. 2014, 53, 5993–5997. doi:10.1002/anie.201400405
Angew. Chem. 2014, 126, 6103–6107. doi:10.1002/ange.201400405 |
| 26. |
Wu, H.; He, Y.-P.; Xu, L.; Zhang, D.-Y.; Gong, L.-Z. Angew. Chem., Int. Ed. 2014, 53, 3466–3469. doi:10.1002/anie.201309967
Angew. Chem. 2014, 126, 3534–3537. doi:10.1002/ange.201309967 |
| 27. | Basdevant, B.; Legault, C. Y. Org. Lett. 2015, 17, 4918–4921. doi:10.1021/acs.orglett.5b02501 |
| 28. | Feng, Y.; Huang, R.; Hu, L.; Xiong, Y.; Coeffard, V. Synthesis 2016, 48, 2637–2644. doi:10.1055/s-0035-1561442 |
| 29. | Cao, Y.; Zhang, X.; Lin, G.; Zhang-Negrerie, D.; Du, Y. Org. Lett. 2016, 18, 5580–5583. doi:10.1021/acs.orglett.6b02816 |
| 54. | Okuyama, T.; Takino, T.; Sueda, T.; Ochiai, M. J. Am. Chem. Soc. 1995, 117, 3360–3367. doi:10.1021/ja00117a006 |
© 2018 Fujita et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)