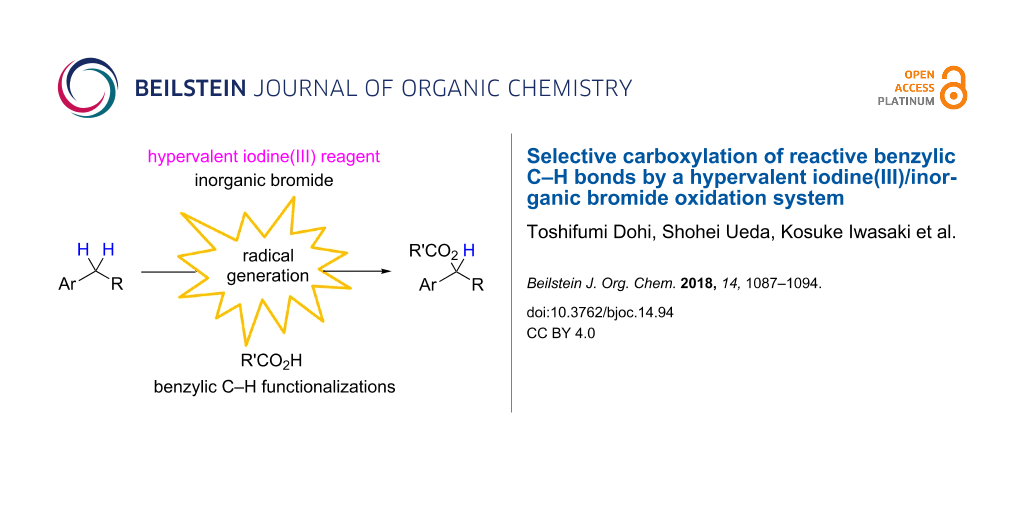
Abstract
An oxidation system comprising phenyliodine(III) diacetate (PIDA) and iodosobenzene with inorganic bromide, i.e., sodium bromide, in an organic solvent led to the direct introduction of carboxylic acids into benzylic C–H bonds under mild conditions. The unique radical species, generated by the homolytic cleavage of the labile I(III)–Br bond of the in situ-formed bromo-λ3-iodane, initiated benzylic carboxylation with a high degree of selectivity for the secondary benzylic position.

Graphical Abstract
Introduction
The oxidative activation of a C(sp3)–H bond in organic molecules to directly install various functional groups and new carbon–carbon networks is a topic of interest for researchers engaged in modern synthetic chemistry [1-8]. Benzylic oxidation is of particular interest because it is a convenient direct approach to arylcarbonyl compounds; it has a long history of research and development, and thus is included among the well-investigated C(sp3)–H transformations [9-12]. To widen the scope, recent studies and reaction systems have been further elaborated to include elegant C–H coupling methodologies. Several important researches that provide a new benzylic C–H coupling strategy have been reported over the past few years, involving the promising catalytic activities of metal complexes [13,14]. On the other hand, reports aimed at realizing efficient and selective metal-free C(sp3)–H transformations are rather limited; however, investigations by several research groups are still ongoing [15-30].
Hypervalent iodine reagents are now widely accepted as a safe replacement for certain heavy-metal oxidizers, such as lead, mercury, and thallium-based salts, due to their low toxicities, high stabilities, operational simplicities, and many other user-friendly characteristics [31,32]. By virtue of their wide array of reactivity patterns, the controllable radical and single-electron-transfer (SET) reactivities [33-37] allow selective activation of the benzylic C(sp3)–H bond for oxidative functionalization and coupling reactions. Initially, the SET oxidation ability of pentavalent iodine reagents, especially o-iodoxybenzoic acid (IBX), in benzylic oxidations was recognized for displaying the new reactivities of hypervalent iodine reagents toward C(sp3)–H bonds [38,39]. By exploiting the radical behavior of trivalent iodine reagents discovered previously [40,41], the activation of trivalent iodine reagents, e.g., phenyliodine(III) diacetate (PIDA), phenyliodine(III) bis(trifluoroacetate) (PIFA), and iodosobenzene, has since become a popular choice for benzylic oxidations, which further expanded the scope and availability of methods for direct C–H functionalization and several coupling reactions [42-50]. As such, we reported aqueous benzylic oxidations using polymeric iodosobenzene in the presence of inorganic bromide and montmorillonite-K10 [51]. In addition, a radical C–H activation strategy, using nonaqueous hypervalent iodine(III)/inorganic bromide systems that can work in organic solvents, was developed for the novel synthesis of lactones via the intramolecular oxidative cyclization of aryl carboxylic acids at the benzyl carbon under transition-metal-free conditions [52]. Based on our previous research and general interest in the unique reactivity of hypervalent iodine(III)–Br bonds [53-56], we report the results of our extensive study and optimization of our radical C–H activation strategy for the intermolecular oxidative coupling between the benzylic secondary C–H bond and the O–H group of carboxylic acids (Scheme 1).
![[1860-5397-14-94-i1]](/bjoc/content/inline/1860-5397-14-94-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Hypervalent iodine(III)-induced benzylic C–H functionalization for oxidative coupling with carboxylic acids.
Scheme 1: Hypervalent iodine(III)-induced benzylic C–H functionalization for oxidative coupling with carboxyl...
Results and Discussion
Benzylic C–H carboxylation can provide a convenient route to benzyl esters from non-functionalized aromatic hydrocarbons, and thus has attracted continuous interest in the community of synthetic chemists. Significant advances for realizing such transformations have been made over the last decade, however, strategies that do not utilize a directing group to facilitate the activation of the benzylic C(sp3)–H bond are rare [21,57-62]. Furthermore, only a limited number of transition-metal-free methods have been reported; successful examples include the Wohl–Ziegler-type conditions [21], the sodium bromate system [57] for the conversion of benzylmethyl groups, and the use of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) [58] or catalytic tetrabutylammonium iodide with tert-butyl hydrogen peroxide for reactions with a large excess of aromatic hydrocarbons [59]. Other than these excellent examples of metal-free methods, two protocols using a hypervalent iodine reagent were reported, both of which include the formation of benzyl radicals during the key initial reaction step. Togo and co-workers developed a reaction system consisting of stoichiometric amounts of PIDA with catalytic amounts of molecular iodine and p-toluenesulfonamide for the benzylic acetoxylation and benzoyloxylation of alkylbenzenes, where an in situ-generated sulfonamidyl radical is the essential radical mediator that effectively abstracts the benzylic hydrogen [49]. More recently, Maruoka et al. succeeded in the photolytic benzylic C–H bond oxygenation of alkylbenzenes initiated by the decomposition of PIFA to form the trifluoroacetoxy radical under visible light irradiation [50].
Our approach for the generation of radical species for the benzylic carboxylation using a hypervalent iodine reagent relies on the unique reactivity of the hypervalent iodine(III)–bromine bond with the following mechanistic principles: As illustrated in Scheme 2, ligand exchange at the iodine(III) center of the phenyliodine(III) dicarboxylate with the bromide ion can gradually produce the corresponding bromo-λ3-iodane in the first step [63,64]. This unstable hypervalent iodine(III) species subsequently decomposes by facile homolytic cleavage of the I(III)–Br bond, generating the iodanyl and bromo radicals [51,52]. It appears that these radicals can then selectively abstract the benzylic hydrogen atom of organic substrates, even in the presence of a wide variety of functional groups, such as electrophilic aromatic rings, non-acidic carbonyl groups, and suitable oxygen, nitrogen, and sulfur functionalities. Carbonyloxy radicals derived from typical hypervalent iodine(III) carboxylates by photolysis and other conditions [65-68] are known to undergo irreversible decarboxylation [69,70]. Therefore, the formation of carbonyloxy radicals must be avoided to prevent non-productive side reactions and achieve the desired benzylic C–H transformations for an extended series of carboxylic acids.
![[1860-5397-14-94-i2]](/bjoc/content/inline/1860-5397-14-94-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Radical reactivities of the I(III)–Br bond generated from PIDA.
Scheme 2: Radical reactivities of the I(III)–Br bond generated from PIDA.
Based on these considerations, we performed an optimization study using 1-ethyl-4-methoxybenzene (1a) as a model substrate for the oxidative C–H coupling of the benzyl group with acetic acid derived from PIDA (Table 1). Reactions using PIDA with finely powdered inorganic bromide in degassed dichloromethane containing 0.1 M of the substrate were first examined at room temperature. The use of potassium bromide [52] afforded modest yields of the carboxylation product 2a (Table 1, entry 1). Interestingly, a dramatic influence was observed when altering the bromide source to other types; the use of lithium bromide or organic bromides, e.g., bromotrimethylsilane and tetraethylammonium bromide, instead of the potassium salt, were unsuccessful in forming the carboxylate 2a (Table 1, entries 2–4). The reason for this behavior was thought to be because lithium bromide or organic bromides in combination with PIDA generated the electrophilic ‘Br+’ species [71] and molecular bromine [72], or hypobromite and bisacetoxy bromate(I) [73], respectively, rather than the desired bromo radical. As a result, bromination at the aromatic ring of substrate 1a occurred when LiBr was used (Table 1, entry 2), while no reaction was observed in the other two trials using organic bromides (Table 1, entries 3 and 4). At the boiling temperature of dichloromethane, ca. 40 °C, the reaction time was shorter (Table 1, entry 5). Aiming to improve the yield of the reaction, we then examined other inorganic bromides, among which sodium bromide was the most promising, and product 2a was obtained in 76% yield (Table 1, entry 6). By adding extra acetic acid, the benzylic acetoxylation was further improved to provide an 86% yield of product 2a. Since most of the sodium bromide was present as a precipitate in the flask, the reaction was found to work even with catalytic amounts of the bromide activator (Table 1, entry 8). Other reaction factors, such as solvent, concentration, reaction time, and reagent quantities, were screened and, eventually, the reaction conditions of entries 6 and 7, which consisted of 1.2 equiv of PIDA with 2 equiv of sodium bromide in dichloromethane (0.1 M of the reaction substrate) at 40 °C, were determined to be the best in terms of product yield. No reaction was observed in the absence of sodium bromide (Table 1, entry 9) and other representative hypervalent iodine(III) reagents, such as PIFA and PhI(OH)OTs, and pentavalent Dess–Martin periodinane and IBX, were inferior for this carboxylation when compared to PIDA.
Table 1: Reaction optimization of benzylic C–H acetoxylation using PIDA.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-94-i6.svg?max-width=637&scale=1.0)
|
|||
| entry | bromide | conditions | yield of 2a (%) |
| 1 | potassium bromide | rt, 6 h | 55 |
| 2 | lithium bromide | rt, 20 h | 11a |
| 3 | bromotrimethylsilane | rt, 20 h | nd |
| 4 | tetraethylammonium bromide | rt, 20 h | nd |
| 5 | potassium bromide | 40 °C, 4 h | 57 |
| 6 | sodium bromide | 40 °C, 4 h | 76 |
| 7b | sodium bromide | 40 °C, 3 h | 86c |
| 8b | sodium bromided | 40 °C, 3 h | 72 |
| 9 | none | 40 °C, 3 h | nd |
aMainly bromination of the aromatic ring was observed. bAcetic acid (20 equiv) was added. cThe reaction was performed on a 10 mmol scale. dCatalytic amounts of sodium bromide were added (0.5 equiv). nd: not determined.
We then examined the reactivity of different benzyl groups under the optimized reaction conditions for the radical activation system for C–H acetoxylations (Table 2). When the reactions were performed using ethylbenzene (1b) and its derivatives without an electron-donating group (1c–e), the corresponding benzyl bromides were mainly obtained along with a small amount of the target C–H acetoxylation product; this byproduct formation might imply the intermediacy of these organic bromides before the production of benzyl acetates. Hence, the reaction system was modified to include zinc(II) acetate [74] for substrates 1b–d and benzyl acetates 2b–d were obtained in moderate to excellent yields after prolonged reaction times (Table 2, entries 1–4). Furthermore, iterative oxidative coupling at the aromatic and benzylic C–H position using hypervalent iodine chemistry is possible, and 1-ethyl-4-methoxy-3-thiocyanatobenzene (1f), prepared from 1-ethyl-4-methoxybenzene (1a) through the hypervalent iodine(III)-induced aromatic cation radical coupling with thiocyanate [75,76], similarly acetoxylated under the standard reaction conditions without zinc(II) acetate (Table 2, entry 5). The acetoxylation at the activated benzyl carbon adjacent to an oxygen atom proceeded smoothly as shown by the formation of the pseudo acetal 2g (Table 2, entry 6). Diphenylmethane and its derivatives were very good substrates for our system and were less sensitive to the electronic effects of the aromatic ring substituents than in other previously described compounds, showing higher reactivity and chemoselectivity of the benzylic position (see ref. [51]). The reactions of substrates 1h–j proceeded without the use of zinc(II) acetate (see Table 2, entries 7–9 versus entry 2).
Table 2: Substrate screening for benzylic C–H acetoxylation by the PIDA/NaBr system.a
| entry | substrate | product | time | yield of 2 (%) |
![[Graphic 2]](/bjoc/content/inline/1860-5397-14-94-i7.svg?max-width=637&scale=1.0)
1b–e |
![[Graphic 3]](/bjoc/content/inline/1860-5397-14-94-i8.svg?max-width=637&scale=1.0)
2b–e |
|||
| 1 | 1b (R1 = H) | 2b (R1 = H) | 20 h | 55b |
| 2 | 1c (R1 = Ph) | 2c (R1 = Ph) | 20 h | 91b |
| 3 | 1d (R1 = Br) | 2d (R1 = Br) | 20 h | 65b |
| 4 | 1e (R1 = CO2CH3) | 2e (R1 = CO2CH3) | 48 h | ndc |
| 5 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-14-94-i9.svg?max-width=637&scale=1.0)
1f |
![[Graphic 5]](/bjoc/content/inline/1860-5397-14-94-i10.svg?max-width=637&scale=1.0)
2f |
20 h | 62 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-14-94-i11.svg?max-width=637&scale=1.0)
1g |
![[Graphic 7]](/bjoc/content/inline/1860-5397-14-94-i12.svg?max-width=637&scale=1.0)
2g |
4 h | 68 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-14-94-i13.svg?max-width=637&scale=1.0)
1h–j |
![[Graphic 9]](/bjoc/content/inline/1860-5397-14-94-i14.svg?max-width=637&scale=1.0)
2h–j |
|||
| 7 | 1h (R2 = R3 = H) | 2h (R2 = R3 = H) | 4 h | 90 |
| 8 | 1i (R2 = R3 = F) | 2i (R2 = R3 = F) | 4 h | 93 |
| 9 | 1j (R2 = Ph, R3 = H) | 2j (R2 = Ph, R3 = H) | 4 h | 45 |
| 10 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-14-94-i15.svg?max-width=637&scale=1.0)
1k |
![[Graphic 11]](/bjoc/content/inline/1860-5397-14-94-i16.svg?max-width=637&scale=1.0)
2k |
4 h | nd |
aConditions: PIDA (1.2 equiv), sodium bromide (2 equiv), and acetic acid (20 equiv) in dichloromethane (0.1 M of substrate 1) at 40 °C. nd: not determined. bZinc acetate (1 equiv) was added for the conversion of the benzyl bromide byproduct to the desired benzyl acetates 2b–d. cThe corresponding benzyl bromide was obtained in 60% yield.
The installation of other carboxylic acids, such as propionic acid, cyclohexyl carboxylic acid, pivalic acid, and benzoic acid, were also possible by simply replacing PIDA with iodosobenzene (Scheme 3). Here, the addition of 3 Å molecular sieves was essential for removing water derived from the iodosobenzene and to suppress the benzylic oxidation forming aryl ketones [52]. Note that the successful coupling of a range of secondary and tertiary carboxylic acids now supports the direct and selective C–H bond activation at the benzyl position by avoiding the formation of carbonyloxy radicals, which are susceptible to decarboxylation. In addition, it was revealed that more acidic benzoic acids were also suitable substrates for our method. However, even more acidic acids, such as trifluoroacetic acid and methanesulfonic acid, were not effectively introduced by our benzylic C–H carboxylation procedures.
![[1860-5397-14-94-i3]](/bjoc/content/inline/1860-5397-14-94-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Benzylic C–H carboxylations by the iodosobenzene/NaBr system.
Scheme 3: Benzylic C–H carboxylations by the iodosobenzene/NaBr system.
We believe that the reaction mechanism involves a benzyl radical formation as the initiating step (Scheme 4). Either the iodanyl radical or the bromo radical may cause the H-atom abstraction generating the benzyl radical, and the resulting benzyl radical is trapped by the persistent bromo radical, giving rise to the observed benzyl bromide intermediate (path A). Alternatively, the benzyl radical formed in situ couples with the iodanyl radical to give the pseudo halide, benzyl-λ3-iodane, which is more reactive to nucleophilic substitution by a carboxylic acid [77,78]. Another possible course of the reaction mechanism assumes the formation of a benzyl cation, resulting from the SET oxidation of the benzyl radical, probably by the iodanyl radicals (path B).
![[1860-5397-14-94-i4]](/bjoc/content/inline/1860-5397-14-94-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Outline of the proposed reaction mechanism for the PIDA/NaBr system.
Scheme 4: Outline of the proposed reaction mechanism for the PIDA/NaBr system.
A significant point of interest in this study is understanding the relative reactivity of the iodanyl radical versus the bromo radical. To gain more information about this, we confirmed the chemoselectivity of our reaction system towards different classes of benzyl carbon atoms. To this end, the clear direction to the secondary benzylic position in substrates 1a–j [51], and the lack of benzylic acetoxylation of the tertiary carbon in substrate 1k (see Table 2) are noteworthy. As bromo radicals are known to readily abstract hydrogen atoms at the tertiary carbon centers [79], the results suggest a predominant participation of the iodanyl radical as the hydrogen atom abstractor for the selective secondary benzylic C–H bond activation in our reaction. It seems that the iodanyl radical is more reactive than the bromo radical and a large-size effect of the hypervalent iodine species establishes the selectivity over the electronically favored tertiary radical formation. A similar trend in the secondary-preferential hydrogen abstraction at the sp3 carbons was recently reported by Maruoka and co-workers for the C–H oxidations of unreactive alkanes by iodanyl radicals [80]. Based on these observations, the reaction mechanism via path A that involves the formation of benzyl bromides (X = Br) seems to be more reasonable for our benzylic C–H carboxylation system based on the hypervalent iodine(III) reagent/inorganic bromide combination. This mechanistic course was also partially supported by the control experiment, whereby one of the separately prepared bromides, 2h’, was gradually transformed into the corresponding acetate 2h in good yield under the radical C–H acetoxylation reaction conditions (Scheme 5).
![[1860-5397-14-94-i5]](/bjoc/content/inline/1860-5397-14-94-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of benzyl bromide 2h’ under radical C–H acetoxylation conditions.
Scheme 5: Reaction of benzyl bromide 2h’ under radical C–H acetoxylation conditions.
Conclusion
In conclusion, we have described the optimization and scope of an oxidation system for benzyl C–H carboxylation that utilizes the radical reactivity of a hypervalent iodine(III) reagent produced under suitable conditions. The mechanistic information obtained in this study indicates that iodanyl radicals, generated by the homolytic cleavage of the labile I(III)–Br bond of in situ formed bromo-λ3-iodane, are the key initiators for this benzylic carboxylation, and they show a high degree of selectivity towards the secondary benzylic position under mild reaction conditions.
Experimental
Representative experimental procedure for the benzylic C–H acetoxylation by the PIDA/NaBr system
In a flame-dried two-necked round-bottomed flask, under nitrogen, phenyliodine(III) diacetate (PIDA, 193 mg, 0.6 mmol) and finely powdered sodium bromide (103 mg, 1.0 mmol) were subsequently added to a stirred solution of arylalkane 1 (0.50 mmol) and acetic acid (0.57 mL, ca. 10 mmol) in dry dichloromethane (5 mL). Then the mixture was vigorously stirred at 40 °C with or without zinc acetate (92 mg, 0.5 mmol). After checking the reaction completion by TLC, saturated aqueous sodium carbonate was added to the mixture and the resulting solution was stirred for an additional 5 min. The organic layer was separated, washed again with saturated aqueous sodium carbonate, then with dilute aqueous sodium thiosulfate, and was dried over anhydrous sodium sulfate. After removal of the solvents, the residue was subjected to column chromatography on silica gel (eluents: n-hexane/ethyl acetate) to give the benzyl acetate 2 in the indicated yield; the physical and spectral data of the reported compounds (2a–d, g–j) matched those of the authentic samples (see Supporting Information File 1).
1-(4-Methoxy-3-thiocyanatophenyl)ethyl acetate (2f). Yield 62% (156 mg). Obtained as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 1.51 (d, J = 6.8 Hz, 3H), 2.05 (s, 3H), 3.89 (s, 3H), 5.80 (q, J = 6.8 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H), 7.32 (dd, J = 8.3, 1.9 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H) ppm; 13C NMR (100 MHz, CDCl3) δ 21.3, 22.1, 56.3, 71.3, 110.3, 111.2, 113.3, 127.6, 128.7, 135.8, 156.0, 170.2 ppm; IR (KBr): 2984, 2158, 1741, 1603, 1499, 1370, 1291, 1242, 1208, 1062, 1023 cm−1; HRMS (MALDI): [M + Na]+ calcd for C12H13NO3SNa, 274.0507; found, 274.0508.
Representative experimental procedure for the benzylic C–H carboxylation by the iodosobenzene/NaBr system
In a flame-dried two-necked round-bottomed flask, under nitrogen, iodosobenzene (132 mg, 0.6 mmol) and finely powdered sodium bromide (103 mg, 1.0 mmol) were subsequently added to a stirred solution of diphenylmethane (1h, 84 mg, 0.50 mmol) and the carboxylic acid (ca. 2.5 mmol) with freshly-dried molecular sieves 3 Å (ca. 300 mg) in dry dichloromethane (5 mL). Then the mixture was vigorously stirred at 40 °C. After checking the reaction completion by TLC, saturated aqueous sodium carbonate was added to the mixture and the resulting solution was stirred for an additional 5 min. The organic layer was separated, washed again with saturated aqueous sodium carbonate, then with dilute aqueous sodium thiosulfate, and was dried over anhydrous sodium sulfate. After removal of the solvents, the residue was subjected to column chromatography on silica gel (eluents: n-hexane/ethyl acetate) to give the benzylic C–H carboxylation product in the indicated yield; the physical and spectral data of the carboxylation products (3h, 4h, 5h, 6h) matched those previously reported (see Supporting Information File 1).
Supporting Information
| Supporting Information File 1: Starting materials and Copies of 1H and 13C NMR spectra of all products. | ||
| Format: PDF | Size: 624.5 KB | Download |
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (A) to Y.K. from JSPS and a Grant-in-Aid for Encouragement of Young Scientists (A) and Scientific Research (C) to T.D. from JSPS, and Ritsumeikan Global Innovation Research Organization (R-GIRO) project. T.D. thanks the research fund of the Asahi Glass Foundation.
References
-
Gini, A.; Brandhofer, T.; Mancheño, O. G. Org. Biomol. Chem. 2017, 15, 1294. doi:10.1039/C6OB02474B
For other recent summarizations, see refs. [2-8].
Return to citation in text: [1] -
Dailler, D.; Danoun, G.; Baudoin, O. Top. Organomet. Chem. 2016, 56, 133. doi:10.1007/3418_2015_122
Return to citation in text: [1] [2] -
Chu, J. C. K.; Rovis, T. Angew. Chem., Int. Ed. 2018, 57, 62. doi:10.1002/anie.201703743
Return to citation in text: [1] [2] -
Roslin, S.; Odell, L. R. Eur. J. Org. Chem. 2017, 1993. doi:10.1002/ejoc.201601479
Return to citation in text: [1] [2] -
Guo, S.-r.; Kumar, P. S.; Yang, M. Adv. Synth. Catal. 2017, 359, 2. doi:10.1002/adsc.201600467
Return to citation in text: [1] [2] -
Rit, R. K.; Shankar, M.; Sahoo, A. K. Org. Biomol. Chem. 2017, 15, 1282. doi:10.1039/C6OB02162J
Return to citation in text: [1] [2] -
Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74. doi:10.1002/anie.201304268
Return to citation in text: [1] [2] -
Narayan, R.; Matcha, K.; Antonchick, A. P. Chem. – Eur. J. 2015, 21, 14678. doi:10.1002/chem.201502005
Return to citation in text: [1] [2] -
Andrus, M. B. Allylic and benzylic oxidation. In Science of Synthesis, Stereoselective Synthesis, 1st ed.; De Vries, J. G.; Molander, G. A.; Evans, P. A., Eds.; Thieme Verlag: Stuttgart, 2011; Vol. 3, pp 469–482. doi:10.1055/sos-SD-203-00294
Return to citation in text: [1] -
Sheldon, R. A. Allylic and benzylic oxidation. In Fine Chemicals through Heterogeneous Catalysis; Sheldon, R. A.; van Bekkum, H., Eds.; Wiley-VCH: Weinheim, 2001; pp 519–526.
Return to citation in text: [1] -
Hudlicky, M. Oxidations in Organic Chemistry; ACS Monograph, Vol. 186; American Chemical Society: Washington DC, 1990.
Return to citation in text: [1] -
Liu, J.; Hu, K.-F.; Qu, J.-P.; Kang, Y.-B. Org. Lett. 2017, 19, 5593. doi:10.1021/acs.orglett.7b02731
See for a recent study.
Return to citation in text: [1] -
Deng, G.-J.; Xiao, F.; Yang, L. Cross-dehydrogenative-coupling reactions involving allyl, benzyl and alkyl C–H bonds. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; RSC Green Chemistry Series, 2015; Vol. 26, pp 93–113.
See also refs. [14-20] for recent new developments.
Return to citation in text: [1] -
Pandey, G.; Laha, R.; Singh, D. J. Org. Chem. 2016, 81, 7161. doi:10.1021/acs.joc.6b00970
and references cited therein.
Return to citation in text: [1] [2] -
Chen, K.; Zhang, P.; Wang, Y.; Li, H. Green Chem. 2014, 16, 2344. doi:10.1039/C3GC42135J
Return to citation in text: [1] [2] -
Zhang, B.; Cui, Y.; Jiao, N. Chem. Commun. 2012, 48, 4498. doi:10.1039/C2CC30684K
See for 2;2;6;6-tetramethylpiperidinyloxyl, radical (TEMPO).
Return to citation in text: [1] [2] -
Neel, A. J.; Hehn, J. P.; Tripet, P. F.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14044. doi:10.1021/ja407410b
See for oxoammonium salt.
Return to citation in text: [1] [2] -
Ramesh, D.; Ramulu, U.; Mukkanti, K.; Venkateswarlu, Y. Tetrahedron Lett. 2012, 53, 2904. doi:10.1016/j.tetlet.2012.03.137
See for DDQ.
Return to citation in text: [1] [2] -
Kong, S.; Zhang, L.; Dai, X.; Tao, L.; Xie, C.; Shi, L.; Wang, M. Adv. Synth. Catal. 2015, 357, 2453. doi:10.1002/adsc.201500096
and references therein. For other recent metal-free methods, see refs. [20-30].
Return to citation in text: [1] [2] -
Liu, W.; Liu, C.; Zhang, Y.; Sun, Y.; Abdukadera, A.; Wang, B.; Li, H.; Ma, X.; Zhang, Z. Org. Biomol. Chem. 2015, 13, 7154. doi:10.1039/C5OB00781J
Return to citation in text: [1] [2] [3] -
Shimojo, H.; Moriyama, K.; Togo, H. Synthesis 2015, 47, 1280. doi:10.1055/s-0034-1380069
Return to citation in text: [1] [2] [3] [4] -
Oss, G.; de Vos, S. D.; Luc, K. N. H.; Harper, J. B.; Nguyen, T. V. J. Org. Chem. 2018, 83, 1000. doi:10.1021/acs.joc.7b02584
Return to citation in text: [1] [2] -
Yi, X.; Jiao, L.; Xi, C. Org. Biomol. Chem. 2016, 14, 9912. doi:10.1039/C6OB01827K
Return to citation in text: [1] [2] -
Zhang, H.; Muñiz, K. ACS Catal. 2017, 7, 4122. doi:10.1021/acscatal.7b00928
Return to citation in text: [1] [2] -
Zhao, D.; Wang, T.; Li, J.-X. Chem. Commun. 2014, 50, 6471. doi:10.1039/C4CC02648A
See for di-tert-butyl peroxide.
Return to citation in text: [1] [2] -
Wu, X.-F.; Gong, J.-L.; Qi, X. Org. Biomol. Chem. 2014, 12, 5807. doi:10.1039/C4OB00276H
See for cat. tetraalkylammmonium iodide/tert-butyl hydrogen peroxide system.
Return to citation in text: [1] [2] -
Chu, X.; Duan, T.; Liu, X.; Feng, L.; Jia, J.; Ma, C. Org. Biomol. Chem. 2017, 15, 1606. doi:10.1039/C6OB02731H
and references cited therein.
Return to citation in text: [1] [2] -
Pandey, G.; Laha, R. Angew. Chem., Int. Ed. 2015, 54, 14875. doi:10.1002/anie.201506990
See for a light-harvesting green method.
Return to citation in text: [1] [2] -
Zhang, L.; Yi, H.; Wang, J.; Lei, A. Green Chem. 2016, 18, 5122. doi:10.1039/C6GC01880G
Return to citation in text: [1] [2] -
Xiang, M.; Xin, Z.-K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Org. Lett. 2017, 19, 3009. doi:10.1021/acs.orglett.7b01270
Return to citation in text: [1] [2] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Wirth, T., Ed. Hypervalent Iodine Chemistry; Springer: Switzerland, 2016. doi:10.1007/978-3-319-33733-3
Return to citation in text: [1] -
Togo, H.; Katohgi, M. Synlett 2001, 565. doi:10.1055/s-2001-13349
For other recent review and account, see refs. [34] and [35].
Return to citation in text: [1] -
Dohi, T.; Kita, Y. ChemCatChem 2014, 6, 76. doi:10.1002/cctc.201300666
Return to citation in text: [1] [2] -
Wang, X.; Studer, A. Acc. Chem. Res. 2017, 50, 1712. doi:10.1021/acs.accounts.7b00148
Return to citation in text: [1] [2] -
Dohi, T.; Kita, Y. Top. Curr. Chem. 2016, 373, 1. doi:10.1007/128_2016_667
See for oxidative couplings in natural product synthesis.
Return to citation in text: [1] -
Kita, Y.; Dohi, T. Chem. Rec. 2015, 15, 886. doi:10.1002/tcr.201500020
See for our pioneering studies in this area.
Return to citation in text: [1] -
Nicolaou, K. C.; Baran, P. S.; Zhong, Y.-L. J. Am. Chem. Soc. 2001, 123, 3183. doi:10.1021/ja004218x
See for a leading report for benzylic oxidation using IBX.
Return to citation in text: [1] -
Ojha, L. R.; Kudugunti, S.; Maddukuri, P. P.; Kommareddy, A.; Gunna, M. R.; Dokuparthi, P.; Gottam, H. B.; Botha, K. K.; Parapati, D. R.; Vinod, T. K. Synlett 2009, 117. doi:10.1055/s-0028-1087384
See for the catalytic use of hypervalent iodine(V) reagents in benzylic oxidations.
Return to citation in text: [1] -
Ochiai, M.; Ito, T.; Takahashi, H.; Nakanishi, A.; Toyonari, M.; Sueda, T.; Goto, S.; Shiro, M. J. Am. Chem. Soc. 1996, 118, 7716. doi:10.1021/ja9610287
See for a case of trivalent hypervalent iodine.
Return to citation in text: [1] -
Kita, Y.; Tohma, H.; Takada, T.; Mitoh, S.; Fujita, S.; Gyoten, M. Synlett 1994, 427. doi:10.1055/s-1994-22875
Return to citation in text: [1] -
Pedersen, C. M.; Marinescu, L. G.; Bols, M. Org. Biomol. Chem. 2005, 3, 816. doi:10.1039/B500037H
Return to citation in text: [1] -
Zhou, W.; Zhang, L.; Jiao, N. Angew. Chem., Int. Ed. 2009, 48, 7094. doi:10.1002/anie.200903838
See for a recent application of our method in ref. [41].
Return to citation in text: [1] -
Fan, R.; Li, W.; Pu, D.; Zhang, L. Org. Lett. 2009, 11, 1425. doi:10.1021/ol900090f
For other methods introducing nitrogen nucleophiles, see refs. [45-47].
Return to citation in text: [1] -
Lamar, A. A.; Nicholas, K. M. J. Org. Chem. 2010, 75, 7644. doi:10.1021/jo1015213
Return to citation in text: [1] [2] -
Kim, H. J.; Kim, J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2011, 133, 16382. doi:10.1021/ja207296y
Return to citation in text: [1] [2] -
Zhu, C.; Liang, Y.; Hong, X.; Sun, H.; Sun, W.-Y.; Houk, K. N.; Shi, Z. J. Am. Chem. Soc. 2015, 137, 7564. doi:10.1021/jacs.5b03488
Return to citation in text: [1] [2] -
Qian, P.-C.; Liu, Y.; Song, R.-J.; Hu, M.; Yang, X.-H.; Xiang, J.-N.; Li, J.-H. Eur. J. Org. Chem. 2015, 1680. doi:10.1002/ejoc.201403616
Return to citation in text: [1] -
Baba, H.; Moriyama, K.; Togo, H. Tetrahedron Lett. 2011, 52, 4303. doi:10.1016/j.tetlet.2011.06.036
Return to citation in text: [1] [2] -
Sakamoto, R.; Inada, T.; Selvakumar, S.; Moteki, S. A.; Maruoka, K. Chem. Commun. 2016, 52, 3758. doi:10.1039/C5CC07647A
Return to citation in text: [1] [2] -
Dohi, T.; Takenaga, N.; Goto, A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365. doi:10.1021/jo8012435
Return to citation in text: [1] [2] [3] [4] -
Dohi, T.; Takenaga, N.; Goto, A.; Maruyama, A.; Kita, Y. Org. Lett. 2007, 9, 3129. doi:10.1021/ol071315n
Return to citation in text: [1] [2] [3] [4] -
Tohma, H.; Takizawa, S.; Maegawa, T.; Kita, Y. Angew. Chem., Int. Ed. 2000, 39, 1306. doi:10.1002/(SICI)1521-3773(20000403)39:7<1306::AID-ANIE1306>3.0.CO;2-J
Return to citation in text: [1] -
Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328. doi:10.1002/1615-4169(200206)344:3/4<328::AID-ADSC328>3.0.CO;2-S
Return to citation in text: [1] -
Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668. doi:10.1021/ja808940n
Return to citation in text: [1] -
Dohi, T.; Ito, M.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Angew. Chem., Int. Ed. 2010, 49, 3334. doi:10.1002/anie.200907281
Return to citation in text: [1] -
Khan, K. M.; Maharvi, G. M.; Hayat, S.; Zia-Ullah; Choudhary, M. I.; Atta-ur-Rahman. Tetrahedron 2003, 59, 5549. doi:10.1016/S0040-4020(03)00812-3
Return to citation in text: [1] [2] -
Yi, H.; Liu, Q.; Liu, J.; Zeng, Z.; Yang, Y.; Lei, A. ChemSusChem 2012, 5, 2143. doi:10.1002/cssc.201200458
Return to citation in text: [1] [2] -
Feng, J.; Liang, S.; Chen, S.-Y.; Zhang, J.; Fu, S.-S.; Yu, X.-Q. Adv. Synth. Catal. 2012, 354, 1287. doi:10.1002/adsc.201100920
Return to citation in text: [1] [2] -
Liu, H.; Shi, G.; Pan, S.; Jiang, Y.; Zhang, Y. Org. Lett. 2013, 15, 4098. doi:10.1021/ol401687f
Return to citation in text: [1] -
Lu, B.; Zhu, F.; Sun, H.-M.; Shen, Q. Org. Lett. 2017, 19, 1132. doi:10.1021/acs.orglett.7b00148
Return to citation in text: [1] -
Ren, T.-L.; Xu, B.-H.; Mahmood, S.; Sun, M.-X.; Zhang, S.-J. Tetrahedron 2017, 73, 2943. doi:10.1016/j.tet.2017.04.002
Return to citation in text: [1] -
Banks, D. F.; Huyser, E. S.; Kleinberg, J. J. Org. Chem. 1964, 29, 3692. doi:10.1021/jo01035a504
Return to citation in text: [1] -
Amey, R. L.; Martin, J. C. J. Am. Chem. Soc. 1979, 101, 3060. doi:10.1021/ja00505a038
Return to citation in text: [1] -
Moriarty, R. M.; Khosrowshahi, J. S.; Dalecki, T. M. J. Chem. Soc., Chem. Commun. 1987, 675. doi:10.1039/c39870000675
Return to citation in text: [1] -
Togo, H.; Aoki, M.; Kuramochi, T.; Yokoyama, M. J. Chem. Soc., Perkin Trans. 1 1993, 2417. doi:10.1039/P19930002417
Return to citation in text: [1] -
Kiyokawa, K.; Watanabe, T.; Fra, L.; Kojima, T.; Minakata, S. J. Org. Chem. 2017, 82, 11711. doi:10.1021/acs.joc.7b01202
Return to citation in text: [1] -
Sakamoto, R.; Kashiwagi, H.; Maruoka, K. Org. Lett. 2017, 19, 5126. doi:10.1021/acs.orglett.7b02416
Return to citation in text: [1] -
Li, Y.; Ge, L.; Muhammad, M. T.; Bao, H. Synthesis 2017, 49, 5263. doi:10.1055/s-0036-1590935
Return to citation in text: [1] -
Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 15632. doi:10.1002/anie.201505731
Return to citation in text: [1] -
Braddock, D. C.; Cansell, G.; Hermitage, S. A. Synlett 2004, 461. doi:10.1055/s-2004-815410
Return to citation in text: [1] -
Evans, P. A.; Brandt, T. A. Tetrahedron Lett. 1996, 37, 6443. doi:10.1016/0040-4039(96)01427-X
Return to citation in text: [1] -
Hashem, M. A.; Jung, A.; Ries, M.; Kirschning, A. Synlett 1998, 195. doi:10.1055/s-1998-1596
Return to citation in text: [1] -
Gurudutt, K. N.; Ravindranath, B.; Srinivas, P. Tetrahedron 1982, 38, 1843. doi:10.1016/0040-4020(82)80261-5
See for the role of zinc acetate in this transformation.
Return to citation in text: [1] -
Kita, Y.; Tohma, H.; Hatanaka, K.; Takada, T.; Fujita, S.; Mitoh, S.; Sakurai, H.; Oka, S. J. Am. Chem. Soc. 1994, 116, 3684. doi:10.1021/ja00088a003
Return to citation in text: [1] -
Kita, Y.; Takada, T.; Mihara, S.; Whelan, B. A.; Tohma, H. J. Org. Chem. 1995, 60, 7144. doi:10.1021/jo00127a018
Return to citation in text: [1] -
Davidson, R. I.; Kropp, P. J. J. Org. Chem. 1982, 47, 1904. doi:10.1021/jo00349a016
Return to citation in text: [1] -
Morris, D. G.; Shepherd, A. G. J. Chem. Soc., Chem. Commun. 1981, 1250. doi:10.1039/c39810001250
Return to citation in text: [1] -
Russell, G. A.; Brown, H. C. J. Am. Chem. Soc. 1955, 77, 4025. doi:10.1021/ja01620a020
Return to citation in text: [1] -
Moteki, S. A.; Usui, A.; Zhang, T.; Solorio, A.; Alvarado, C. R. S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 8657. doi:10.1002/anie.201304359
Return to citation in text: [1]
| 74. |
Gurudutt, K. N.; Ravindranath, B.; Srinivas, P. Tetrahedron 1982, 38, 1843. doi:10.1016/0040-4020(82)80261-5
See for the role of zinc acetate in this transformation. |
| 75. | Kita, Y.; Tohma, H.; Hatanaka, K.; Takada, T.; Fujita, S.; Mitoh, S.; Sakurai, H.; Oka, S. J. Am. Chem. Soc. 1994, 116, 3684. doi:10.1021/ja00088a003 |
| 76. | Kita, Y.; Takada, T.; Mihara, S.; Whelan, B. A.; Tohma, H. J. Org. Chem. 1995, 60, 7144. doi:10.1021/jo00127a018 |
| 51. | Dohi, T.; Takenaga, N.; Goto, A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365. doi:10.1021/jo8012435 |
| 1. |
Gini, A.; Brandhofer, T.; Mancheño, O. G. Org. Biomol. Chem. 2017, 15, 1294. doi:10.1039/C6OB02474B
For other recent summarizations, see refs. [2-8]. |
| 2. | Dailler, D.; Danoun, G.; Baudoin, O. Top. Organomet. Chem. 2016, 56, 133. doi:10.1007/3418_2015_122 |
| 3. | Chu, J. C. K.; Rovis, T. Angew. Chem., Int. Ed. 2018, 57, 62. doi:10.1002/anie.201703743 |
| 4. | Roslin, S.; Odell, L. R. Eur. J. Org. Chem. 2017, 1993. doi:10.1002/ejoc.201601479 |
| 5. | Guo, S.-r.; Kumar, P. S.; Yang, M. Adv. Synth. Catal. 2017, 359, 2. doi:10.1002/adsc.201600467 |
| 6. | Rit, R. K.; Shankar, M.; Sahoo, A. K. Org. Biomol. Chem. 2017, 15, 1282. doi:10.1039/C6OB02162J |
| 7. | Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74. doi:10.1002/anie.201304268 |
| 8. | Narayan, R.; Matcha, K.; Antonchick, A. P. Chem. – Eur. J. 2015, 21, 14678. doi:10.1002/chem.201502005 |
| 31. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328. doi:10.1021/acs.chemrev.5b00547 |
| 32. | Wirth, T., Ed. Hypervalent Iodine Chemistry; Springer: Switzerland, 2016. doi:10.1007/978-3-319-33733-3 |
| 57. | Khan, K. M.; Maharvi, G. M.; Hayat, S.; Zia-Ullah; Choudhary, M. I.; Atta-ur-Rahman. Tetrahedron 2003, 59, 5549. doi:10.1016/S0040-4020(03)00812-3 |
| 14. |
Pandey, G.; Laha, R.; Singh, D. J. Org. Chem. 2016, 81, 7161. doi:10.1021/acs.joc.6b00970
and references cited therein. |
| 15. | Chen, K.; Zhang, P.; Wang, Y.; Li, H. Green Chem. 2014, 16, 2344. doi:10.1039/C3GC42135J |
| 16. |
Zhang, B.; Cui, Y.; Jiao, N. Chem. Commun. 2012, 48, 4498. doi:10.1039/C2CC30684K
See for 2;2;6;6-tetramethylpiperidinyloxyl, radical (TEMPO). |
| 17. |
Neel, A. J.; Hehn, J. P.; Tripet, P. F.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14044. doi:10.1021/ja407410b
See for oxoammonium salt. |
| 18. |
Ramesh, D.; Ramulu, U.; Mukkanti, K.; Venkateswarlu, Y. Tetrahedron Lett. 2012, 53, 2904. doi:10.1016/j.tetlet.2012.03.137
See for DDQ. |
| 19. |
Kong, S.; Zhang, L.; Dai, X.; Tao, L.; Xie, C.; Shi, L.; Wang, M. Adv. Synth. Catal. 2015, 357, 2453. doi:10.1002/adsc.201500096
and references therein. For other recent metal-free methods, see refs. [20-30]. |
| 20. | Liu, W.; Liu, C.; Zhang, Y.; Sun, Y.; Abdukadera, A.; Wang, B.; Li, H.; Ma, X.; Zhang, Z. Org. Biomol. Chem. 2015, 13, 7154. doi:10.1039/C5OB00781J |
| 15. | Chen, K.; Zhang, P.; Wang, Y.; Li, H. Green Chem. 2014, 16, 2344. doi:10.1039/C3GC42135J |
| 16. |
Zhang, B.; Cui, Y.; Jiao, N. Chem. Commun. 2012, 48, 4498. doi:10.1039/C2CC30684K
See for 2;2;6;6-tetramethylpiperidinyloxyl, radical (TEMPO). |
| 17. |
Neel, A. J.; Hehn, J. P.; Tripet, P. F.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 14044. doi:10.1021/ja407410b
See for oxoammonium salt. |
| 18. |
Ramesh, D.; Ramulu, U.; Mukkanti, K.; Venkateswarlu, Y. Tetrahedron Lett. 2012, 53, 2904. doi:10.1016/j.tetlet.2012.03.137
See for DDQ. |
| 19. |
Kong, S.; Zhang, L.; Dai, X.; Tao, L.; Xie, C.; Shi, L.; Wang, M. Adv. Synth. Catal. 2015, 357, 2453. doi:10.1002/adsc.201500096
and references therein. For other recent metal-free methods, see refs. [20-30]. |
| 20. | Liu, W.; Liu, C.; Zhang, Y.; Sun, Y.; Abdukadera, A.; Wang, B.; Li, H.; Ma, X.; Zhang, Z. Org. Biomol. Chem. 2015, 13, 7154. doi:10.1039/C5OB00781J |
| 21. | Shimojo, H.; Moriyama, K.; Togo, H. Synthesis 2015, 47, 1280. doi:10.1055/s-0034-1380069 |
| 22. | Oss, G.; de Vos, S. D.; Luc, K. N. H.; Harper, J. B.; Nguyen, T. V. J. Org. Chem. 2018, 83, 1000. doi:10.1021/acs.joc.7b02584 |
| 23. | Yi, X.; Jiao, L.; Xi, C. Org. Biomol. Chem. 2016, 14, 9912. doi:10.1039/C6OB01827K |
| 24. | Zhang, H.; Muñiz, K. ACS Catal. 2017, 7, 4122. doi:10.1021/acscatal.7b00928 |
| 25. |
Zhao, D.; Wang, T.; Li, J.-X. Chem. Commun. 2014, 50, 6471. doi:10.1039/C4CC02648A
See for di-tert-butyl peroxide. |
| 26. |
Wu, X.-F.; Gong, J.-L.; Qi, X. Org. Biomol. Chem. 2014, 12, 5807. doi:10.1039/C4OB00276H
See for cat. tetraalkylammmonium iodide/tert-butyl hydrogen peroxide system. |
| 27. |
Chu, X.; Duan, T.; Liu, X.; Feng, L.; Jia, J.; Ma, C. Org. Biomol. Chem. 2017, 15, 1606. doi:10.1039/C6OB02731H
and references cited therein. |
| 28. |
Pandey, G.; Laha, R. Angew. Chem., Int. Ed. 2015, 54, 14875. doi:10.1002/anie.201506990
See for a light-harvesting green method. |
| 29. | Zhang, L.; Yi, H.; Wang, J.; Lei, A. Green Chem. 2016, 18, 5122. doi:10.1039/C6GC01880G |
| 30. | Xiang, M.; Xin, Z.-K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Org. Lett. 2017, 19, 3009. doi:10.1021/acs.orglett.7b01270 |
| 58. | Yi, H.; Liu, Q.; Liu, J.; Zeng, Z.; Yang, Y.; Lei, A. ChemSusChem 2012, 5, 2143. doi:10.1002/cssc.201200458 |
| 20. | Liu, W.; Liu, C.; Zhang, Y.; Sun, Y.; Abdukadera, A.; Wang, B.; Li, H.; Ma, X.; Zhang, Z. Org. Biomol. Chem. 2015, 13, 7154. doi:10.1039/C5OB00781J |
| 21. | Shimojo, H.; Moriyama, K.; Togo, H. Synthesis 2015, 47, 1280. doi:10.1055/s-0034-1380069 |
| 22. | Oss, G.; de Vos, S. D.; Luc, K. N. H.; Harper, J. B.; Nguyen, T. V. J. Org. Chem. 2018, 83, 1000. doi:10.1021/acs.joc.7b02584 |
| 23. | Yi, X.; Jiao, L.; Xi, C. Org. Biomol. Chem. 2016, 14, 9912. doi:10.1039/C6OB01827K |
| 24. | Zhang, H.; Muñiz, K. ACS Catal. 2017, 7, 4122. doi:10.1021/acscatal.7b00928 |
| 25. |
Zhao, D.; Wang, T.; Li, J.-X. Chem. Commun. 2014, 50, 6471. doi:10.1039/C4CC02648A
See for di-tert-butyl peroxide. |
| 26. |
Wu, X.-F.; Gong, J.-L.; Qi, X. Org. Biomol. Chem. 2014, 12, 5807. doi:10.1039/C4OB00276H
See for cat. tetraalkylammmonium iodide/tert-butyl hydrogen peroxide system. |
| 27. |
Chu, X.; Duan, T.; Liu, X.; Feng, L.; Jia, J.; Ma, C. Org. Biomol. Chem. 2017, 15, 1606. doi:10.1039/C6OB02731H
and references cited therein. |
| 28. |
Pandey, G.; Laha, R. Angew. Chem., Int. Ed. 2015, 54, 14875. doi:10.1002/anie.201506990
See for a light-harvesting green method. |
| 29. | Zhang, L.; Yi, H.; Wang, J.; Lei, A. Green Chem. 2016, 18, 5122. doi:10.1039/C6GC01880G |
| 30. | Xiang, M.; Xin, Z.-K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Org. Lett. 2017, 19, 3009. doi:10.1021/acs.orglett.7b01270 |
| 13. |
Deng, G.-J.; Xiao, F.; Yang, L. Cross-dehydrogenative-coupling reactions involving allyl, benzyl and alkyl C–H bonds. In From C–H to C–C Bonds: Cross-Dehydrogenative-Coupling; Li, C.-J., Ed.; RSC Green Chemistry Series, 2015; Vol. 26, pp 93–113.
See also refs. [14-20] for recent new developments. |
| 14. |
Pandey, G.; Laha, R.; Singh, D. J. Org. Chem. 2016, 81, 7161. doi:10.1021/acs.joc.6b00970
and references cited therein. |
| 21. | Shimojo, H.; Moriyama, K.; Togo, H. Synthesis 2015, 47, 1280. doi:10.1055/s-0034-1380069 |
| 57. | Khan, K. M.; Maharvi, G. M.; Hayat, S.; Zia-Ullah; Choudhary, M. I.; Atta-ur-Rahman. Tetrahedron 2003, 59, 5549. doi:10.1016/S0040-4020(03)00812-3 |
| 58. | Yi, H.; Liu, Q.; Liu, J.; Zeng, Z.; Yang, Y.; Lei, A. ChemSusChem 2012, 5, 2143. doi:10.1002/cssc.201200458 |
| 59. | Feng, J.; Liang, S.; Chen, S.-Y.; Zhang, J.; Fu, S.-S.; Yu, X.-Q. Adv. Synth. Catal. 2012, 354, 1287. doi:10.1002/adsc.201100920 |
| 60. | Liu, H.; Shi, G.; Pan, S.; Jiang, Y.; Zhang, Y. Org. Lett. 2013, 15, 4098. doi:10.1021/ol401687f |
| 61. | Lu, B.; Zhu, F.; Sun, H.-M.; Shen, Q. Org. Lett. 2017, 19, 1132. doi:10.1021/acs.orglett.7b00148 |
| 62. | Ren, T.-L.; Xu, B.-H.; Mahmood, S.; Sun, M.-X.; Zhang, S.-J. Tetrahedron 2017, 73, 2943. doi:10.1016/j.tet.2017.04.002 |
| 80. | Moteki, S. A.; Usui, A.; Zhang, T.; Solorio, A.; Alvarado, C. R. S.; Maruoka, K. Angew. Chem., Int. Ed. 2013, 52, 8657. doi:10.1002/anie.201304359 |
| 9. | Andrus, M. B. Allylic and benzylic oxidation. In Science of Synthesis, Stereoselective Synthesis, 1st ed.; De Vries, J. G.; Molander, G. A.; Evans, P. A., Eds.; Thieme Verlag: Stuttgart, 2011; Vol. 3, pp 469–482. doi:10.1055/sos-SD-203-00294 |
| 10. | Sheldon, R. A. Allylic and benzylic oxidation. In Fine Chemicals through Heterogeneous Catalysis; Sheldon, R. A.; van Bekkum, H., Eds.; Wiley-VCH: Weinheim, 2001; pp 519–526. |
| 11. | Hudlicky, M. Oxidations in Organic Chemistry; ACS Monograph, Vol. 186; American Chemical Society: Washington DC, 1990. |
| 12. |
Liu, J.; Hu, K.-F.; Qu, J.-P.; Kang, Y.-B. Org. Lett. 2017, 19, 5593. doi:10.1021/acs.orglett.7b02731
See for a recent study. |
| 21. | Shimojo, H.; Moriyama, K.; Togo, H. Synthesis 2015, 47, 1280. doi:10.1055/s-0034-1380069 |
| 2. | Dailler, D.; Danoun, G.; Baudoin, O. Top. Organomet. Chem. 2016, 56, 133. doi:10.1007/3418_2015_122 |
| 3. | Chu, J. C. K.; Rovis, T. Angew. Chem., Int. Ed. 2018, 57, 62. doi:10.1002/anie.201703743 |
| 4. | Roslin, S.; Odell, L. R. Eur. J. Org. Chem. 2017, 1993. doi:10.1002/ejoc.201601479 |
| 5. | Guo, S.-r.; Kumar, P. S.; Yang, M. Adv. Synth. Catal. 2017, 359, 2. doi:10.1002/adsc.201600467 |
| 6. | Rit, R. K.; Shankar, M.; Sahoo, A. K. Org. Biomol. Chem. 2017, 15, 1282. doi:10.1039/C6OB02162J |
| 7. | Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74. doi:10.1002/anie.201304268 |
| 8. | Narayan, R.; Matcha, K.; Antonchick, A. P. Chem. – Eur. J. 2015, 21, 14678. doi:10.1002/chem.201502005 |
| 42. | Pedersen, C. M.; Marinescu, L. G.; Bols, M. Org. Biomol. Chem. 2005, 3, 816. doi:10.1039/B500037H |
| 43. |
Zhou, W.; Zhang, L.; Jiao, N. Angew. Chem., Int. Ed. 2009, 48, 7094. doi:10.1002/anie.200903838
See for a recent application of our method in ref. [41]. |
| 44. |
Fan, R.; Li, W.; Pu, D.; Zhang, L. Org. Lett. 2009, 11, 1425. doi:10.1021/ol900090f
For other methods introducing nitrogen nucleophiles, see refs. [45-47]. |
| 45. | Lamar, A. A.; Nicholas, K. M. J. Org. Chem. 2010, 75, 7644. doi:10.1021/jo1015213 |
| 46. | Kim, H. J.; Kim, J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2011, 133, 16382. doi:10.1021/ja207296y |
| 47. | Zhu, C.; Liang, Y.; Hong, X.; Sun, H.; Sun, W.-Y.; Houk, K. N.; Shi, Z. J. Am. Chem. Soc. 2015, 137, 7564. doi:10.1021/jacs.5b03488 |
| 48. | Qian, P.-C.; Liu, Y.; Song, R.-J.; Hu, M.; Yang, X.-H.; Xiang, J.-N.; Li, J.-H. Eur. J. Org. Chem. 2015, 1680. doi:10.1002/ejoc.201403616 |
| 49. | Baba, H.; Moriyama, K.; Togo, H. Tetrahedron Lett. 2011, 52, 4303. doi:10.1016/j.tetlet.2011.06.036 |
| 50. | Sakamoto, R.; Inada, T.; Selvakumar, S.; Moteki, S. A.; Maruoka, K. Chem. Commun. 2016, 52, 3758. doi:10.1039/C5CC07647A |
| 52. | Dohi, T.; Takenaga, N.; Goto, A.; Maruyama, A.; Kita, Y. Org. Lett. 2007, 9, 3129. doi:10.1021/ol071315n |
| 51. | Dohi, T.; Takenaga, N.; Goto, A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365. doi:10.1021/jo8012435 |
| 40. |
Ochiai, M.; Ito, T.; Takahashi, H.; Nakanishi, A.; Toyonari, M.; Sueda, T.; Goto, S.; Shiro, M. J. Am. Chem. Soc. 1996, 118, 7716. doi:10.1021/ja9610287
See for a case of trivalent hypervalent iodine. |
| 41. | Kita, Y.; Tohma, H.; Takada, T.; Mitoh, S.; Fujita, S.; Gyoten, M. Synlett 1994, 427. doi:10.1055/s-1994-22875 |
| 53. | Tohma, H.; Takizawa, S.; Maegawa, T.; Kita, Y. Angew. Chem., Int. Ed. 2000, 39, 1306. doi:10.1002/(SICI)1521-3773(20000403)39:7<1306::AID-ANIE1306>3.0.CO;2-J |
| 54. | Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328. doi:10.1002/1615-4169(200206)344:3/4<328::AID-ADSC328>3.0.CO;2-S |
| 55. | Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. J. Am. Chem. Soc. 2009, 131, 1668. doi:10.1021/ja808940n |
| 56. | Dohi, T.; Ito, M.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Angew. Chem., Int. Ed. 2010, 49, 3334. doi:10.1002/anie.200907281 |
| 79. | Russell, G. A.; Brown, H. C. J. Am. Chem. Soc. 1955, 77, 4025. doi:10.1021/ja01620a020 |
| 38. |
Nicolaou, K. C.; Baran, P. S.; Zhong, Y.-L. J. Am. Chem. Soc. 2001, 123, 3183. doi:10.1021/ja004218x
See for a leading report for benzylic oxidation using IBX. |
| 39. |
Ojha, L. R.; Kudugunti, S.; Maddukuri, P. P.; Kommareddy, A.; Gunna, M. R.; Dokuparthi, P.; Gottam, H. B.; Botha, K. K.; Parapati, D. R.; Vinod, T. K. Synlett 2009, 117. doi:10.1055/s-0028-1087384
See for the catalytic use of hypervalent iodine(V) reagents in benzylic oxidations. |
| 52. | Dohi, T.; Takenaga, N.; Goto, A.; Maruyama, A.; Kita, Y. Org. Lett. 2007, 9, 3129. doi:10.1021/ol071315n |
| 33. |
Togo, H.; Katohgi, M. Synlett 2001, 565. doi:10.1055/s-2001-13349
For other recent review and account, see refs. [34] and [35]. |
| 34. | Dohi, T.; Kita, Y. ChemCatChem 2014, 6, 76. doi:10.1002/cctc.201300666 |
| 35. | Wang, X.; Studer, A. Acc. Chem. Res. 2017, 50, 1712. doi:10.1021/acs.accounts.7b00148 |
| 36. |
Dohi, T.; Kita, Y. Top. Curr. Chem. 2016, 373, 1. doi:10.1007/128_2016_667
See for oxidative couplings in natural product synthesis. |
| 37. |
Kita, Y.; Dohi, T. Chem. Rec. 2015, 15, 886. doi:10.1002/tcr.201500020
See for our pioneering studies in this area. |
| 51. | Dohi, T.; Takenaga, N.; Goto, A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365. doi:10.1021/jo8012435 |
| 77. | Davidson, R. I.; Kropp, P. J. J. Org. Chem. 1982, 47, 1904. doi:10.1021/jo00349a016 |
| 78. | Morris, D. G.; Shepherd, A. G. J. Chem. Soc., Chem. Commun. 1981, 1250. doi:10.1039/c39810001250 |
| 50. | Sakamoto, R.; Inada, T.; Selvakumar, S.; Moteki, S. A.; Maruoka, K. Chem. Commun. 2016, 52, 3758. doi:10.1039/C5CC07647A |
| 59. | Feng, J.; Liang, S.; Chen, S.-Y.; Zhang, J.; Fu, S.-S.; Yu, X.-Q. Adv. Synth. Catal. 2012, 354, 1287. doi:10.1002/adsc.201100920 |
| 49. | Baba, H.; Moriyama, K.; Togo, H. Tetrahedron Lett. 2011, 52, 4303. doi:10.1016/j.tetlet.2011.06.036 |
| 35. | Wang, X.; Studer, A. Acc. Chem. Res. 2017, 50, 1712. doi:10.1021/acs.accounts.7b00148 |
| 41. | Kita, Y.; Tohma, H.; Takada, T.; Mitoh, S.; Fujita, S.; Gyoten, M. Synlett 1994, 427. doi:10.1055/s-1994-22875 |
| 72. | Evans, P. A.; Brandt, T. A. Tetrahedron Lett. 1996, 37, 6443. doi:10.1016/0040-4039(96)01427-X |
| 73. | Hashem, M. A.; Jung, A.; Ries, M.; Kirschning, A. Synlett 1998, 195. doi:10.1055/s-1998-1596 |
| 52. | Dohi, T.; Takenaga, N.; Goto, A.; Maruyama, A.; Kita, Y. Org. Lett. 2007, 9, 3129. doi:10.1021/ol071315n |
| 71. | Braddock, D. C.; Cansell, G.; Hermitage, S. A. Synlett 2004, 461. doi:10.1055/s-2004-815410 |
| 65. | Moriarty, R. M.; Khosrowshahi, J. S.; Dalecki, T. M. J. Chem. Soc., Chem. Commun. 1987, 675. doi:10.1039/c39870000675 |
| 66. | Togo, H.; Aoki, M.; Kuramochi, T.; Yokoyama, M. J. Chem. Soc., Perkin Trans. 1 1993, 2417. doi:10.1039/P19930002417 |
| 67. | Kiyokawa, K.; Watanabe, T.; Fra, L.; Kojima, T.; Minakata, S. J. Org. Chem. 2017, 82, 11711. doi:10.1021/acs.joc.7b01202 |
| 68. | Sakamoto, R.; Kashiwagi, H.; Maruoka, K. Org. Lett. 2017, 19, 5126. doi:10.1021/acs.orglett.7b02416 |
| 69. | Li, Y.; Ge, L.; Muhammad, M. T.; Bao, H. Synthesis 2017, 49, 5263. doi:10.1055/s-0036-1590935 |
| 70. | Xuan, J.; Zhang, Z.-G.; Xiao, W.-J. Angew. Chem., Int. Ed. 2015, 54, 15632. doi:10.1002/anie.201505731 |
| 63. | Banks, D. F.; Huyser, E. S.; Kleinberg, J. J. Org. Chem. 1964, 29, 3692. doi:10.1021/jo01035a504 |
| 64. | Amey, R. L.; Martin, J. C. J. Am. Chem. Soc. 1979, 101, 3060. doi:10.1021/ja00505a038 |
| 45. | Lamar, A. A.; Nicholas, K. M. J. Org. Chem. 2010, 75, 7644. doi:10.1021/jo1015213 |
| 46. | Kim, H. J.; Kim, J.; Cho, S. H.; Chang, S. J. Am. Chem. Soc. 2011, 133, 16382. doi:10.1021/ja207296y |
| 47. | Zhu, C.; Liang, Y.; Hong, X.; Sun, H.; Sun, W.-Y.; Houk, K. N.; Shi, Z. J. Am. Chem. Soc. 2015, 137, 7564. doi:10.1021/jacs.5b03488 |
| 51. | Dohi, T.; Takenaga, N.; Goto, A.; Fujioka, H.; Kita, Y. J. Org. Chem. 2008, 73, 7365. doi:10.1021/jo8012435 |
| 52. | Dohi, T.; Takenaga, N.; Goto, A.; Maruyama, A.; Kita, Y. Org. Lett. 2007, 9, 3129. doi:10.1021/ol071315n |
© 2018 Dohi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)