Abstract
The system of the hypervalent iodine(III) reagent FPID and (4-MeOC6H4)3P was successfully applied to solid-phase peptide synthesis and cyclic peptide synthesis. Four peptides with biological activities were synthesized through SPPS and the bioactive cyclic heptapeptide pseudostellarin D was obtained via solution-phase peptide synthesis. It is worth noting that FPID can be readily regenerated after the peptide coupling reaction.

Graphical Abstract
Introduction
The amide bond is one of the most fundamental functional groups in organic chemistry, and it plays a crucial role in the elaboration and composition of biological systems. Amide bonds are widely present not only in peptides and proteins but also in pharmaceuticals and many natural products. Among the methods for amide bond formation, the direct condensation of carboxylic acids and amines in the presence of a coupling reagent is the most convenient and simplest way [1-6]. The most commonly used coupling reagents such as carbodiimide [7], phosphonium [8], and uronium salts [9] are efficient and commercially available. In spite of these merits of traditional coupling reagents, it is still far from ideal because large amounts of chemical wastes are produced during the amide bond formation reaction using these reagents and the coupling reagents cannot be regenerated [10]. Thus, methods for the peptide synthesis which are efficient and atom-economic are still needed.
Hypervalent iodine reagents have drawn researchers’ considerable attentions due to their versatile reactivity, low toxicity, ready availability, environmental friendliness, and regenerability [11-27]. Our group has dedicated to the peptide synthesis mediated by hypervalent iodine(III) reagents in recent years. In 2012, for the first time, we reported that the hypervalent iodine(III) reagent iodosodilactone (Figure 1) can serve as a condensing reagent to promote esterification, macrolactonization, amidation and peptide coupling reactions in the presence of PPh3 [28]. In addition, the peptide coupling reaction proceeds without racemization in the absence of a racemization suppressant and iodosodilactone can be readily regenerated after the reaction. In order to further enhance the reactivity of iodosodilactone, we designed and synthesized a new derivative of iodosodilactone 6-(3,5-bis(trifluoromethyl)phenyl)-1H,4H-2aλ3-ioda-2,3-dioxacyclopenta[hi]indene-1,4-dione (abbreviated as FPID, Figure 1) [29].
![[1860-5397-14-97-1]](/bjoc/content/figures/1860-5397-14-97-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In combination with tris(4-methoxyphenyl)phosphine [(4-MeOC6H4)3P], FPID can efficiently mediate peptide coupling reactions within 30 minutes to obtain various dipeptides from standard amino acids as well as sterically hindered amino acids. Moreover, a pentapeptide Leu-enkephalin is successfully synthesized in its protected form using this coupling system. Similar to iodosodilactone, FPID can be easily regenerated after the reaction. The mechanism for this FPID-mediated amide bond formation reaction was proposed with the acyloxyphosphonium intermediate B being the key intermediate (Scheme 1). Herein, as part of our continuing exploration of the application of FPID in peptide synthesis, we disclose its successful application in solid-phase peptide synthesis and cyclic peptide synthesis.
![[1860-5397-14-97-i1]](/bjoc/content/inline/1860-5397-14-97-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Proposed mechanism for FPID-mediated amide bond formation.
Scheme 1: Proposed mechanism for FPID-mediated amide bond formation.
Results and Discussion
At the beginning of our study, we tried to utilize the system of FPID/(4-MeOC6H4)3P in the solid-phase peptide synthesis (SPPS). SPPS has been widely employed in peptide synthesis since its first report by Merrifield in 1963 [30,31]. Compared with classical solution-phase peptide synthesis, the fast development of SPPS is mainly due to its short reaction time, high efficiency, low racemization, simple work-up and automation. In recent decades, various strategies, for example, native chemical ligation (NCL) [32] and serine/threonine ligation (STL) [33], have been reported to solve the problems occurred during the development of SPPS. Peptide synthesis in solution mediated by FPID/(4-MeOC6H4)3P is rapid (within 30 min) and efficient, at the same time the reactions proceed without racemization. Thus, it is possible and significant to test whether the FPID/(4-MeOC6H4)3P system can be used in SPPS.
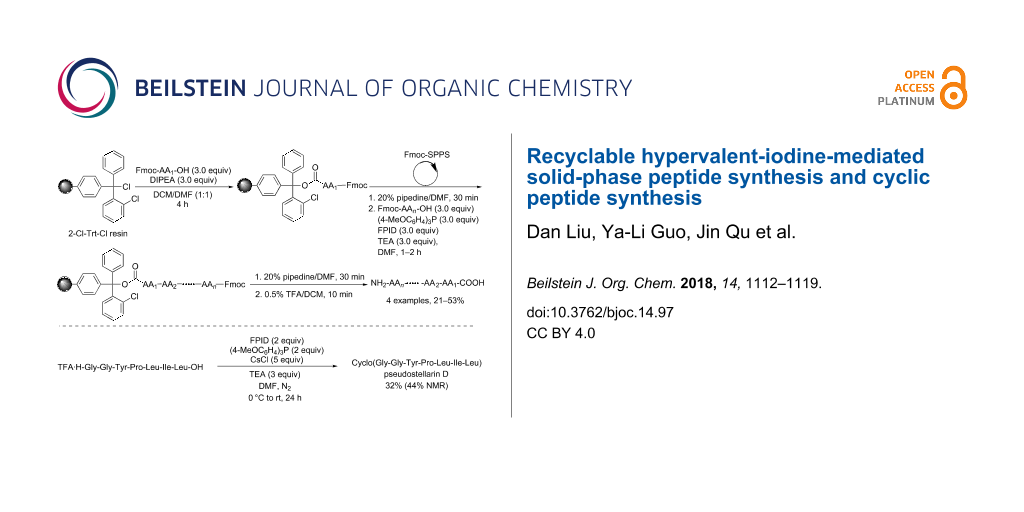
We selected the commercially available 2-chlorotrityl chloride resin (2-Cl-Trt-Cl resin) as the solid support and [(9-fluorenylmethyl)oxy]carbonyl (Fmoc) as the α-amino protecting group. The peptides were synthesized following the route as shown in Scheme 2. The C-terminal amino acid was immobilized onto the 2-Cl-Trt-Cl resin in the presence of 3.0 equiv of DIPEA in DCM/DMF (v:v 1:1). Subsequent peptide chain elongation was completed via Fmoc-SPPS protocol, which includes deprotection with 20% piperidine/DMF and peptide coupling with 3.0 equiv of Fmoc-protected amino acids, 3.0 equiv of FPID, 3.0 equiv of (4-MeOC6H4)3P and 3.0 equiv of TEA in DMF. After chain elongation and deprotection of Fmoc, the resulting resins were treated with 0.5% TFA/DCM to give the N,C-unprotected peptides as final products. The peptides were purified by reversed-phase HPLC (RP-HPLC).
![[1860-5397-14-97-i2]](/bjoc/content/inline/1860-5397-14-97-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Solid-phase peptide synthesis mediated by FPID/(4-MeOC6H4)3P. Conditions: The resin loading for 2-Cl-Trt-Cl resin is 0.98 mmol/g. For each peptide synthesized through SPPS, 200 mg 2-Cl-Trt-Cl resin was used. Fmoc-Ser-OH, Fmoc-Thr-OH, Fmoc-Tyr-OH, Fmoc-Trp-OH were directly used without any protecting group on OH or NH. During the synthesis of 3, Fmoc-Lys(Boc)-OH was used.
Scheme 2: Solid-phase peptide synthesis mediated by FPID/(4-MeOC6H4)3P. Conditions: The resin loading for 2-C...
For the target peptides, we aimed at peptides with specific biological activities. Leu-enkephalin, which is isolated from pig brains, acts as an endogenous mediator at central morphine receptor sites and thus possesses potent opiate agonist activity [34-36]. Leu-enkephalin 1 (Table 1, entry 1) could be successfully synthesized following the route mentioned above (Scheme 2). Besides, the precursor 2 of a cyclic heptapeptide pseudostellarin D [37-39] was also obtained via SPPS in good yield (Table 1, entry 2), the cyclization of 2 to give pseudostellarin D using FPID/(4-MeOC6H4)3P will be described in the following part. Moreover, angiotensin I converting enzyme (ACE) inhibitory peptides, widely exist in plants and animals, can serve as potential antihypertensive pharmaceuticals [40-42]. The synthesis of two ACE inhibitory peptides proceeded smoothly in moderate yield (Table 1, entries 3 and 4). Notably, it is unnecessary to protect the hydroxy group of serine, threonine, or tyrosine in advance in the synthesis of these four peptides. The presence of an unprotected hydroxy group does not affect the coupling efficiency, which is consistent with peptide coupling in solution phase [29]. The HRMS spectra of these peptides are consistent with their molecular formula.
Similar to the solution-phase peptide synthesis, FPID can be easily regenerated after SPPS (Scheme 3). After completion of peptide elongation, the washing solution of peptide coupling in each cycle was collected and evaporated. Then the mixture was acidified with 3 N HCl and extracted with EtOAc, dried and concentrated in vacuo. The synthetic precursor of FPID 6 could be purified by flash chromatography in order to remove excess Fmoc-protected amino acids during the peptide coupling. Compound 6 was subsequently oxidized with NaOCl/HCl to obtain FPID in 90% yield.
![[1860-5397-14-97-i3]](/bjoc/content/inline/1860-5397-14-97-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The regeneration of FPID after SPPS.
Scheme 3: The regeneration of FPID after SPPS.
Cyclic peptides, an important kind of peptides, possess several favorable properties such as target selectivity, good binding affinity and low toxicity, which make them attractive candidates in the development of therapeutics [43,44]. Due to the particular significance of cyclic peptides, chemists pay considerable attention to the efficient synthesis of cyclic peptides [45]. In the second part of the investigation of the synthetic utility of the FPID/(4-MeOC6H4)3P system, we tested this system in the cyclic peptide synthesis.
The roots of Pseudostellaria heterophylla are well-known traditional Chinese medicine, which are often used as a lung and spleen tonic. There are more than 10 cyclic peptides isolated from it, for example, pseudostellarin A–H and heterophyllin A–D [37,46-49]. Cyclo(Gly-Gly-Tyr-Pro-Leu-Ile-Leu), a cyclic heptapeptide named pseudostellarin D (Figure 2), is one of these cyclic peptides. Pseudostellarin D was first isolated and identified in 1994 by Itokawa and co-workers [37]. In addition, the authors reported that pseudostellarin D showed potent tyrosinase inhibitory activities. In 1999, Belagali and co-workers further evaluated the antimicrobial, anti-inflammatory and anthelmintic activities of pseudostellarin D [38]. As for the synthesis of pseudostellarin D, the existing methods utilized the active ester method to complete the cyclization of the linear peptide precursor with the same amino acid sequence of Gly-Gly-Tyr-Pro-Leu-Ile-Leu as a result of the amide bond between “Gly-Leu” being the cyclization position. The first one was reported by Belagali in 1999, pseudostellarin D was obtained by the cyclization of the linear heptapeptide peptide-PNP ester, which is known as the p-nitrophenyl ester method (Scheme 4, method A). The other one was described by Agrigento and co-workers, the cyclization was completed via the p-chlorophenyl thioester method with peptide-thioester being the precursor (Scheme 4, method B) [39]. Herein, we realized the synthesis of pseudostellarin D following the same cyclization strategy mentioned above by direct coupling of the precursor 2 without any protecting group utilizing the system of FPID/(4-MeOC6H4)3P (Scheme 4, method C).
![[1860-5397-14-97-2]](/bjoc/content/figures/1860-5397-14-97-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of pseudostellarin D.
Figure 2: Structure of pseudostellarin D.
![[1860-5397-14-97-i4]](/bjoc/content/inline/1860-5397-14-97-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthetic strategies of pseudostellarin D.
Scheme 4: Synthetic strategies of pseudostellarin D.
We have obtained the precursor 2 through SPPS mediated by FPID/(4-MeOC6H4)3P system (Table 1, entry 2). Firstly, the C-terminal amino acid Leu was immobilized onto 2-Cl-Trt-Cl resin. Then the chain elongation was completed by the deprotection of Fmoc and coupling with Fmoc-protected Ile, Leu, Pro, Tyr, Gly, Gly in turn. The successive deprotection of Fmoc, cleavage from the resin and purification via RP-HPLC yielded the precursor 2 (see Supporting Information File 1). Alternatively, the synthesis of the precursor 2 can be achieved through a convergent [3 + 4] segment condensation strategy in solution phase (Scheme 5). Notably, during the synthesis of tetrapeptide segment 10, the stepwise manner was adopted in order to avoid racemization. In the synthesis of tripeptide segment 13, our condensing system, FPID/(4-MeOC6H4)3P was utilized. Besides, the coupling between 10 and 13 to yield heptapeptide segment 14 was carried out using 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT) as coupling reagent, which was developed by Ye’s group in order to reduce racemization and side reactions [50]. The precursor 2 was obtained via successive deprotection of the C-terminal and the N-terminal protecting group of 14. The overall yield of this route is 28%.
![[1860-5397-14-97-i5]](/bjoc/content/inline/1860-5397-14-97-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparation of the precursor of pseudostellarin D.
Scheme 5: Preparation of the precursor of pseudostellarin D.
With the precursor 2 in hand, we then investigated the reaction conditions of its cyclization. Considering the solubility of 2 and FPID, we chose DMF as the solvent. At the beginning of our investigation, the reaction was carried out under air within 10 h, we tested the influence of the adding sequence of TEA, FPID and (4-MeOC6H4)3P, and the results indicated that adding TEA first and FPID/(4-MeOC6H4)3P 5 minutes later performed better than the inverse sequence (Table 2, entries 1 and 2). When prolonging the reaction time to 24 h, the product was obtained in a yield of 23% and 25% according to different adding sequence of TEA and FPID/(4-MeOC6H4)3P, and the effect of the adding sequence was the same with that mentioned above (Table 2, entries 3 and 4). Then the reaction was run under N2 atmosphere instead of air, the cyclization yield increased to 27% (Table 2, entry 5). Increasing the equivalents of FPID and (4-MeOC6H4)3P to 2.0 equiv resulted in 33% NMR yield of the product (Table 2, entry 7). Ye’s group disclosed that adding univalent metal ions such as Na+, K+ and Cs+ to the reaction system for the synthesis of some cyclic pentapeptides and heptapeptide would not only enhance the cyclization yields but also the cyclization rates [51]. Inspired by this finding, we tried to add some metal chlorides into the reaction, such as LiCl, NaCl, KCl, CsCl (Table 2, entries 8–11). The results indicated that adding NaCl, KCl, CsCl could increase the cyclization yield, whereas LiCl decreased the yield. Among the efficient metal chlorides, adding 5 equiv of CsCl to the reaction yielded the product in 44% NMR yield. The isolated yield of pseudostellarin D under the optimized reaction conditions was 32%.
Table 2: Optimization of cyclization of linear heptapeptide 2.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-97-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | x equiv | additive | atmosphere | time | yield (%) |
| 1 | 1.2 | – | air | 10 h | <18 |
| 2b | 1.2 | – | air | 10 h | <15 |
| 3 | 1.2 | – | air | 24 h | <23 |
| 4b | 1.2 | – | air | 24 h | <20 |
| 5 | 1.2 | – | N2 | 24 h | <27 |
| 6c | 2.0 | – | N2 | 24 h | n.d. |
| 7 | 2.0 | – | N2 | 24 h | 33 (NMR) |
| 8d | 2.0 | CsCl (5 equiv) | N2 | 24 h | 44 (NMR) |
| 9d | 2.0 | NaCl (5 equiv) | N2 | 24 h | 42 (NMR) |
| 10d | 2.0 | LiCl (5 equiv) | N2 | 24 h | 28 (NMR) |
| 11d | 2.0 | KCl (5 equiv) | N2 | 24 h | 40 (NMR) |
aConditions: performed with 2 (0.05 mmol), TEA (0.15 mmol), DMF (50 mL). Unless otherwise mentioned, the adding sequence of TEA and FPID/(4-MeOC6H4)3P was adding TEA first and FPID/(4-MeOC6H4)3P 5 minutes later. “x equiv” meant the equivalents of FPID and (4-MeOC6H4)3P. The NMR yield was calculated by adding CH2ClBr as internal standard substance. bAdding FPID/(4-MeOC6H4)3P first and TEA 5 minutes later. c2 was dissolved in 2 mL of DMF and added portionwise to the reaction system within 2 h. dMetal chloride was dissolved in 0.33 mL of H2O and then added to the reaction.
Consequently, pseudostellarin D can be successfully synthesized in 32% isolated yield (44% NMR yield) under the optimized conditions: FPID (2 equiv), (4-MeOC6H4)3P (2 equiv), TEA (3 equiv), CsCl (5 equiv). The 1H NMR and HRMS spectra of pseudostellarin D are in agreement with data from the literature [37-39].
Conclusion
The system of the hypervalent iodine(III) reagent FPID and (4-MeOC6H4)3P can be applied to the solid-phase peptide synthesis because four bioactive peptides were smoothly obtained including the precursor 2 of cyclic peptide pseudostellarin D. Moreover, we have also successfully synthesized the bioactive cyclic heptapeptide pseudostellarin D using this system. Notably, FPID can be easily regenerated after peptide coupling reaction in SPPS. These results, along with the successful use of the FPID/(4-MeOC6H4)3P system in the solution-phase linear peptide synthesis [29], show its potential in the practical application in peptide synthesis.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of all products, copies of 1H, 13C, HPLC, HRMS spectra of some compounds. | ||
| Format: PDF | Size: 1.8 MB | Download |
Acknowledgements
This work was financially supported by National Key R&D Program of China (2017YFD020030202), The National Natural Science Foundation of China (21472094, 21172110, 21421062) and The Tianjin Natural Science Foundation (17JCYBJC20300). We are grateful to MS. Bo Sun for the preliminary investigation of this project.
References
-
Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606–631. doi:10.1039/b701677h
Return to citation in text: [1] -
El-Faham, A.; Albericio, F. Chem. Rev. 2011, 111, 6557–6602. doi:10.1021/cr100048w
Return to citation in text: [1] -
Al-Warhi, T. I.; Al-Hazimi, H. M. A.; El-Faham, A. J. Saudi Chem. Soc. 2012, 16, 97–116. doi:10.1016/j.jscs.2010.12.006
Return to citation in text: [1] -
Prasad, K. V. S. R. G.; Bharathi, K.; Haseena Banu, B. Int. J. Pharm. Sci. Rev. Res. 2011, 8, 108–119.
Return to citation in text: [1] -
Dunetz, J. R.; Magano, J.; Weisenburger, G. A. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s
Return to citation in text: [1] -
Yang, J.; Zhao, J. Sci. China: Chem. 2018, 61, 97–112. doi:10.1007/s11426-017-9056-5
Return to citation in text: [1] -
Sheehan, J. C.; Hess, G. P. J. Am. Chem. Soc. 1955, 77, 1067–1068. doi:10.1021/ja01609a099
Return to citation in text: [1] -
Gawne, G.; Kenner, G. W.; Sheppard, R. C. J. Am. Chem. Soc. 1969, 91, 5669–5671. doi:10.1021/ja01048a057
Return to citation in text: [1] -
Carpino, L. A.; Henklein, P.; Foxman, B. M.; Abdelmoty, I.; Costisella, B.; Wray, V.; Domke, T.; El-Faham, A.; Mügge, C. J. Org. Chem. 2001, 66, 5245–5247. doi:10.1021/jo001616+
Return to citation in text: [1] -
Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c
Return to citation in text: [1] -
Zhdankin, V. V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: New York, 2014.
Return to citation in text: [1] -
Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: London, 1997.
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. doi:10.1021/cr010003+
Return to citation in text: [1] -
Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950–971. doi:10.1002/asia.201301582
Return to citation in text: [1] -
Brown, M.; Farid, U.; Wirth, T. Synlett 2013, 24, 424–431. doi:10.1055/s-0032-1318103
Return to citation in text: [1] -
Wirth, T. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. doi:10.1002/anie.200500115
Return to citation in text: [1] -
Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073
Return to citation in text: [1] -
Brand, J. P.; González, D. F.; Nicolai, S. F.; Nicolai, S.; Waser, J. Chem. Commun. 2011, 47, 102–115. doi:10.1039/c0cc02265a
Return to citation in text: [1] -
Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h
Return to citation in text: [1] -
Dohi, T.; Kita, Y. Chem. Commun. 2009, 2073–2085. doi:10.1039/b821747e
Return to citation in text: [1] -
Duan, Y.; Jiang, S.; Han, Y.; Sun, B.; Zhang, C. Chin. J. Org. Chem. 2016, 36, 1973–1984.
Return to citation in text: [1] -
Zhang, X.; Cong, Y.; Lin, G.; Guo, X.; Cao, Y.; Lei, K.; Du, Y. Chin. J. Org. Chem. 2016, 36, 2513–2529.
Return to citation in text: [1] -
Chen, J.; Qu, H.-M.; Peng, J.; Chen, C. Chin. J. Org. Chem. 2015, 35, 937–946.
Return to citation in text: [1] -
Duschek, A.; Kirsch, S. F. Angew. Chem., Int. Ed. 2011, 50, 1524–1552. doi:10.1002/anie.201000873
Return to citation in text: [1] -
Merritt, E. A.; Olofsson, B. Synthesis 2011, 517–538. doi:10.1055/s-0030-1258328
Return to citation in text: [1] -
Tian, J.; Gao, W.-C.; Zhou, D.-M.; Zhang, C. Org. Lett. 2012, 14, 3020–3023. doi:10.1021/ol301085v
Return to citation in text: [1] -
Zhang, C.; Liu, S.-S.; Sun, B.; Tian, J. Org. Lett. 2015, 17, 4106–4109. doi:10.1021/acs.orglett.5b02045
Return to citation in text: [1] [2] [3] -
Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149–2154. doi:10.1021/ja00897a025
Return to citation in text: [1] -
Behrendt, R.; White, P.; Offer, J. J. Pept. Sci. 2016, 22, 4–27. doi:10.1002/psc.2836
Return to citation in text: [1] -
Dawson, P. E.; Muir, T. W.; Clark-Lewis, I.; Kent, S. B. Science 1994, 266, 776–779. doi:10.1126/science.7973629
Return to citation in text: [1] -
Li, X.; Lam, H. Y.; Zhang, Y.; Chan, C. K. Org. Lett. 2010, 12, 1724–1727. doi:10.1021/ol1003109
Return to citation in text: [1] -
Hughes, J.; Smith, T. W.; Kosterlitz, H. W.; Fothergill, L. A.; Morgan, B. A.; Morris, H. R. Nature 1975, 258, 577–579. doi:10.1038/258577a0
Return to citation in text: [1] -
Hughes, J. Brain Res. 1975, 88, 295–308. doi:10.1016/0006-8993(75)90391-1
Return to citation in text: [1] -
Naot, D.; Cornish, J. Bone 2008, 43, 813–818. doi:10.1016/j.bone.2008.07.003
Return to citation in text: [1] -
Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 9975–9982. doi:10.1016/S0040-4020(01)89612-5
Return to citation in text: [1] [2] [3] [4] -
Himaja, M.; Harish Kumar, K.; Ramana, M. V.; Belagali, S. L. Eur. J. Med. Chem. 1999, 34, 525–529. doi:10.1016/S0223-5234(99)80101-9
Return to citation in text: [1] [2] [3] -
Agrigento, P.; Albericio, F.; Chamoin, S.; Dacquignies, I.; Koc, H.; Eberle, M. Org. Lett. 2014, 16, 3922–3925. doi:10.1021/ol501669n
Return to citation in text: [1] [2] [3] -
Ariyoshi, Y. Trends Food Sci. Technol. 1993, 4, 139–144. doi:10.1016/0924-2244(93)90033-7
Return to citation in text: [1] -
Wanasundara, P. K. J. P. D.; Ross, A. R. S.; Amarowicz, R.; Ambrose, S. J.; Pegg, R. B.; Shand, P. J. J. Agric. Food Chem. 2002, 50, 6981–6988. doi:10.1021/jf025592e
Return to citation in text: [1] -
Guang, C.; Phillips, R. D. J. Agric. Food Chem. 2009, 57, 5113–5120. doi:10.1021/jf900494d
Return to citation in text: [1] -
Zorzi, A.; Deyle, K.; Heinis, C. Curr. Opin. Chem. Biol. 2017, 38, 24–29. doi:10.1016/j.cbpa.2017.02.006
Return to citation in text: [1] -
Sewald, N.; Jakube, H.-D. Peptides: Chemistry and Biology, 2nd ed.; Wiley-VCH: Weinheim, 2009.
Return to citation in text: [1] -
White, C. J.; Yudin, A. K. Nat. Chem. 2011, 3, 509–524. doi:10.1038/NCHEM.1062
Return to citation in text: [1] -
Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 6797–6804. doi:10.1016/S0040-4020(01)81333-8
Return to citation in text: [1] -
Morita, H.; Kobata, H.; Takeya, K.; Itokawa, H. Tetrahedron Lett. 1994, 35, 3563–3564. doi:10.1016/S0040-4039(00)73238-2
Return to citation in text: [1] -
Morita, H.; Kayashita, T.; Takeya, K.; Itokawa, H. J. Nat. Prod. 1995, 58, 943–947. doi:10.1021/np50120a021
Return to citation in text: [1] -
Tan, N.-H.; Zou, J.; Chen, C.-X.; Zhao, S.-X. Phytochemistry 1993, 32, 1327–1330. doi:10.1016/S0031-9422(00)95114-8
Return to citation in text: [1] -
Fan, C.-X.; Hao, X.-L.; Ye, Y.-H. Synth. Commun. 1996, 26, 1455–1460. doi:10.1080/00397919608003508
Return to citation in text: [1] -
Ye, Y.-h.; Liu, M.; Tang, Y.-c.; Jiang, X. Chem. Commun. 2002, 532–533. doi:10.1039/b108133k
Return to citation in text: [1]
| 1. | Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606–631. doi:10.1039/b701677h |
| 2. | El-Faham, A.; Albericio, F. Chem. Rev. 2011, 111, 6557–6602. doi:10.1021/cr100048w |
| 3. | Al-Warhi, T. I.; Al-Hazimi, H. M. A.; El-Faham, A. J. Saudi Chem. Soc. 2012, 16, 97–116. doi:10.1016/j.jscs.2010.12.006 |
| 4. | Prasad, K. V. S. R. G.; Bharathi, K.; Haseena Banu, B. Int. J. Pharm. Sci. Rev. Res. 2011, 8, 108–119. |
| 5. | Dunetz, J. R.; Magano, J.; Weisenburger, G. A. Org. Process Res. Dev. 2016, 20, 140–177. doi:10.1021/op500305s |
| 6. | Yang, J.; Zhao, J. Sci. China: Chem. 2018, 61, 97–112. doi:10.1007/s11426-017-9056-5 |
| 10. | Constable, D. J. C.; Dunn, P. J.; Hayler, J. D.; Humphrey, G. R.; Leazer, J. L., Jr.; Linderman, R. J.; Lorenz, K.; Manley, J.; Pearlman, B. A.; Wells, A.; Zaks, A.; Zhang, T. Y. Green Chem. 2007, 9, 411–420. doi:10.1039/b703488c |
| 29. | Zhang, C.; Liu, S.-S.; Sun, B.; Tian, J. Org. Lett. 2015, 17, 4106–4109. doi:10.1021/acs.orglett.5b02045 |
| 9. | Carpino, L. A.; Henklein, P.; Foxman, B. M.; Abdelmoty, I.; Costisella, B.; Wray, V.; Domke, T.; El-Faham, A.; Mügge, C. J. Org. Chem. 2001, 66, 5245–5247. doi:10.1021/jo001616+ |
| 43. | Zorzi, A.; Deyle, K.; Heinis, C. Curr. Opin. Chem. Biol. 2017, 38, 24–29. doi:10.1016/j.cbpa.2017.02.006 |
| 44. | Sewald, N.; Jakube, H.-D. Peptides: Chemistry and Biology, 2nd ed.; Wiley-VCH: Weinheim, 2009. |
| 8. | Gawne, G.; Kenner, G. W.; Sheppard, R. C. J. Am. Chem. Soc. 1969, 91, 5669–5671. doi:10.1021/ja01048a057 |
| 37. | Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 9975–9982. doi:10.1016/S0040-4020(01)89612-5 |
| 38. | Himaja, M.; Harish Kumar, K.; Ramana, M. V.; Belagali, S. L. Eur. J. Med. Chem. 1999, 34, 525–529. doi:10.1016/S0223-5234(99)80101-9 |
| 39. | Agrigento, P.; Albericio, F.; Chamoin, S.; Dacquignies, I.; Koc, H.; Eberle, M. Org. Lett. 2014, 16, 3922–3925. doi:10.1021/ol501669n |
| 7. | Sheehan, J. C.; Hess, G. P. J. Am. Chem. Soc. 1955, 77, 1067–1068. doi:10.1021/ja01609a099 |
| 40. | Ariyoshi, Y. Trends Food Sci. Technol. 1993, 4, 139–144. doi:10.1016/0924-2244(93)90033-7 |
| 41. | Wanasundara, P. K. J. P. D.; Ross, A. R. S.; Amarowicz, R.; Ambrose, S. J.; Pegg, R. B.; Shand, P. J. J. Agric. Food Chem. 2002, 50, 6981–6988. doi:10.1021/jf025592e |
| 42. | Guang, C.; Phillips, R. D. J. Agric. Food Chem. 2009, 57, 5113–5120. doi:10.1021/jf900494d |
| 30. | Merrifield, R. B. J. Am. Chem. Soc. 1963, 85, 2149–2154. doi:10.1021/ja00897a025 |
| 31. | Behrendt, R.; White, P.; Offer, J. J. Pept. Sci. 2016, 22, 4–27. doi:10.1002/psc.2836 |
| 33. | Li, X.; Lam, H. Y.; Zhang, Y.; Chan, C. K. Org. Lett. 2010, 12, 1724–1727. doi:10.1021/ol1003109 |
| 29. | Zhang, C.; Liu, S.-S.; Sun, B.; Tian, J. Org. Lett. 2015, 17, 4106–4109. doi:10.1021/acs.orglett.5b02045 |
| 34. | Hughes, J.; Smith, T. W.; Kosterlitz, H. W.; Fothergill, L. A.; Morgan, B. A.; Morris, H. R. Nature 1975, 258, 577–579. doi:10.1038/258577a0 |
| 35. | Hughes, J. Brain Res. 1975, 88, 295–308. doi:10.1016/0006-8993(75)90391-1 |
| 36. | Naot, D.; Cornish, J. Bone 2008, 43, 813–818. doi:10.1016/j.bone.2008.07.003 |
| 28. | Tian, J.; Gao, W.-C.; Zhou, D.-M.; Zhang, C. Org. Lett. 2012, 14, 3020–3023. doi:10.1021/ol301085v |
| 11. | Zhdankin, V. V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: New York, 2014. |
| 12. | Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: London, 1997. |
| 13. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 14. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299–5358. doi:10.1021/cr800332c |
| 15. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. doi:10.1021/cr010003+ |
| 16. | Singh, F. V.; Wirth, T. Chem. – Asian J. 2014, 9, 950–971. doi:10.1002/asia.201301582 |
| 17. | Brown, M.; Farid, U.; Wirth, T. Synlett 2013, 24, 424–431. doi:10.1055/s-0032-1318103 |
| 18. | Wirth, T. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. doi:10.1002/anie.200500115 |
| 19. | Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073 |
| 20. | Brand, J. P.; González, D. F.; Nicolai, S. F.; Nicolai, S.; Waser, J. Chem. Commun. 2011, 47, 102–115. doi:10.1039/c0cc02265a |
| 21. | Charpentier, J.; Früh, N.; Togni, A. Chem. Rev. 2015, 115, 650–682. doi:10.1021/cr500223h |
| 22. | Dohi, T.; Kita, Y. Chem. Commun. 2009, 2073–2085. doi:10.1039/b821747e |
| 23. | Duan, Y.; Jiang, S.; Han, Y.; Sun, B.; Zhang, C. Chin. J. Org. Chem. 2016, 36, 1973–1984. |
| 24. | Zhang, X.; Cong, Y.; Lin, G.; Guo, X.; Cao, Y.; Lei, K.; Du, Y. Chin. J. Org. Chem. 2016, 36, 2513–2529. |
| 25. | Chen, J.; Qu, H.-M.; Peng, J.; Chen, C. Chin. J. Org. Chem. 2015, 35, 937–946. |
| 26. | Duschek, A.; Kirsch, S. F. Angew. Chem., Int. Ed. 2011, 50, 1524–1552. doi:10.1002/anie.201000873 |
| 27. | Merritt, E. A.; Olofsson, B. Synthesis 2011, 517–538. doi:10.1055/s-0030-1258328 |
| 32. | Dawson, P. E.; Muir, T. W.; Clark-Lewis, I.; Kent, S. B. Science 1994, 266, 776–779. doi:10.1126/science.7973629 |
| 37. | Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 9975–9982. doi:10.1016/S0040-4020(01)89612-5 |
| 45. | White, C. J.; Yudin, A. K. Nat. Chem. 2011, 3, 509–524. doi:10.1038/NCHEM.1062 |
| 37. | Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 9975–9982. doi:10.1016/S0040-4020(01)89612-5 |
| 46. | Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 6797–6804. doi:10.1016/S0040-4020(01)81333-8 |
| 47. | Morita, H.; Kobata, H.; Takeya, K.; Itokawa, H. Tetrahedron Lett. 1994, 35, 3563–3564. doi:10.1016/S0040-4039(00)73238-2 |
| 48. | Morita, H.; Kayashita, T.; Takeya, K.; Itokawa, H. J. Nat. Prod. 1995, 58, 943–947. doi:10.1021/np50120a021 |
| 49. | Tan, N.-H.; Zou, J.; Chen, C.-X.; Zhao, S.-X. Phytochemistry 1993, 32, 1327–1330. doi:10.1016/S0031-9422(00)95114-8 |
| 37. | Morita, H.; Kayashita, T.; Kobata, H.; Gonda, A.; Takeya, K.; Itokawa, H. Tetrahedron 1994, 50, 9975–9982. doi:10.1016/S0040-4020(01)89612-5 |
| 38. | Himaja, M.; Harish Kumar, K.; Ramana, M. V.; Belagali, S. L. Eur. J. Med. Chem. 1999, 34, 525–529. doi:10.1016/S0223-5234(99)80101-9 |
| 39. | Agrigento, P.; Albericio, F.; Chamoin, S.; Dacquignies, I.; Koc, H.; Eberle, M. Org. Lett. 2014, 16, 3922–3925. doi:10.1021/ol501669n |
| 29. | Zhang, C.; Liu, S.-S.; Sun, B.; Tian, J. Org. Lett. 2015, 17, 4106–4109. doi:10.1021/acs.orglett.5b02045 |
| 50. | Fan, C.-X.; Hao, X.-L.; Ye, Y.-H. Synth. Commun. 1996, 26, 1455–1460. doi:10.1080/00397919608003508 |
| 51. | Ye, Y.-h.; Liu, M.; Tang, Y.-c.; Jiang, X. Chem. Commun. 2002, 532–533. doi:10.1039/b108133k |
| 38. | Himaja, M.; Harish Kumar, K.; Ramana, M. V.; Belagali, S. L. Eur. J. Med. Chem. 1999, 34, 525–529. doi:10.1016/S0223-5234(99)80101-9 |
| 39. | Agrigento, P.; Albericio, F.; Chamoin, S.; Dacquignies, I.; Koc, H.; Eberle, M. Org. Lett. 2014, 16, 3922–3925. doi:10.1021/ol501669n |
© 2018 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)