Abstract
3-Methylbuta-1,2-dien-1-ylphosphonic acid derivatives (phosphorylallenes) [X2(O=)P–CR=C=CMe2, X = Cl, OMe, NR2, or SAr] undergo intramolecular cyclization into the corresponding 1,2-oxaphospholium ions in the Brønsted superacid TfOH. These cations have been thoroughly studied by means of NMR spectroscopy. The hydrolysis of superacidic solutions of these species afforded cyclic phosphonic acids and other phosphorus-containing compounds. Contrary to Brønsted acids, 3-methylbuta-1,2-dien-1-ylphosphonic dichloride [Cl2(O=)P–HC=C=CMe2] reacted with the Lewis acid AlCl3 in an intermolecular way forming noncyclic intermediates, which were investigated by NMR spectroscopy and DFT calculations. Hydrolysis of these species resulted in the formation of phosphoryl-substituted allyl alcohols and 1,3-butadienes. A strong coordination of the oxygen of the P=O group with AlCl3 prevented the formation of cyclic 1,2-oxaphospholium ions and played a crucial role in the different reactivity of such phosphorylallenes under the action of Brønsted or Lewis acids. Apart from that, the reaction of dichlorophosphorylallenes with arenes and AlCl3 led to products of hydroarylation of the allene system, phosphoryl-substituted alkenes and/or indanes. This is the first example of a Lewis acid-promoted intermolecular hydroarylation of allenes bearing electron-withdrawing substituents. Plausible reaction mechanisms have been proposed on the basis of the investigated reactions, and NMR analysis and DFT studies of the intermediate cationic species.

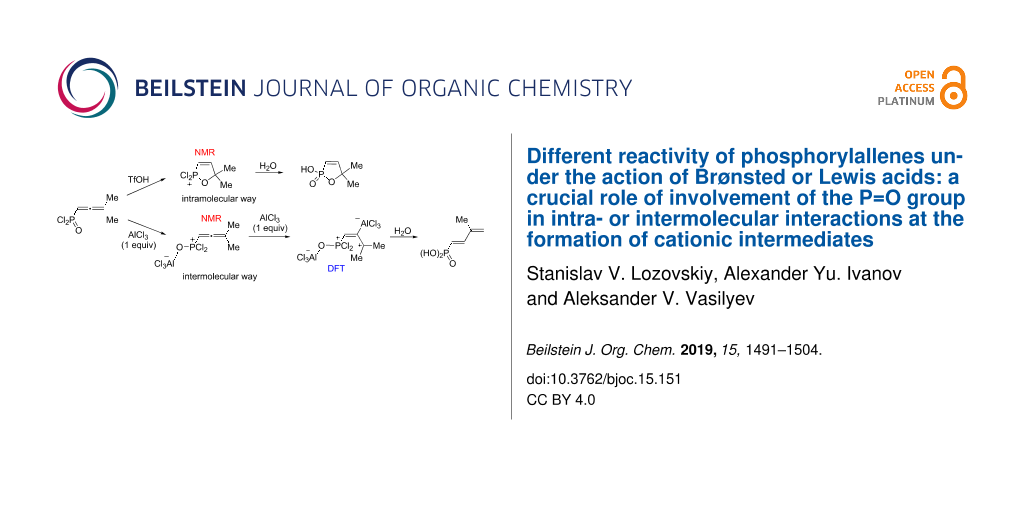
Graphical Abstract
Introduction
Electrophilic reactions of allenes have been intensely explored in organic synthesis [1-3]. In particular, reports on electrophilic activation of phosphorylallenes are numerous [4-10]. Miscellaneous electrophiles, such as sulfenyl, selenyl, and telluryl chlorides, were used in reactions with these allenes. However, only a few studies have been focused on reactions of phosphorylallenes with Brønsted acids [11,12]. These reactions proceed through an intermediate formation of the corresponding 2,5-dihydro-1,2-oxaphosphol-2-ium ions. The progenitor of the oxaphospholium ion family, 2,2-dichloro-5,5-dimethyl-1,2-oxaphosphol-2-ium, was postulated for the first time in 1978 [12].
We have recently reported on the generation, NMR characterization and reactions of oxaphospholium ions bearing phenyl or phenoxy substituents at the phosphorus atom of phosphorylallenes [13-16]. These cations were intermediates in Brønsted and Lewis acid-promoted intramolecular reactions of phosphorus-containing allenes with aromatic π-nucleophiles giving rise to various (bi)cyclic phosphorus-containing compounds [13-16].
It should be especially emphasized that intermolecular reactions of phosphorylallenes with arenes have not been yet achieved. In general, intermolecular hydroarylation of allenes has been developed for reactions catalyzed by complexes of various metals [17], such as Pd [18-20], Pt [21], Au [22-25], Ir [26], Rh [27,28], and Co [29]. However, only electron-rich allenes, bearing electron-donating substituents, take part in the metal-catalyzed reactions. There are just a few examples of Brønsted acid catalyzed intermolecular hydroarylations of allenes by electron-rich arenes, indoles [30] or phenols [31]. Other arenes (benzene and its substituted derivatives) have not been involved in these reactions. Concerning electron-deficient allenes, bearing electron-withdrawing groups, there is only one example of a trifluoroacetic acid-promoted hydroarylation with indoles [30]. To the best of our knowledge, up to the moment, there are no examples for an intermolecular hydroarylation of electron-deficient allenes by benzene derivatives under the action of strong Brønsted or Lewis acids.
The main goals of this work were to study transformations of various phosphorylallenes under electrophilic activation with Brønsted or Lewis (super)acids, including reactions with arenes as π-nucleophiles, and investigation of intermediate cationic species by means of NMR and DFT calculations.
Allenes used in this study are presented in Figure 1. We explored allenes having different substituents at the phosphoryl group: chloro (1a–d), amino (1e–g), arylsulfanyl (1h,i), and methoxy (1j).
![[1860-5397-15-151-1]](/bjoc/content/figures/1860-5397-15-151-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Allenes 1a–j used in this study.
Figure 1: Allenes 1a–j used in this study.
Results and Discussion
Reactions of allenes with Brønsted acids
Allenes 1a,b,e–j upon dissolving in TfOH in an NMR tube at room temperature formed intensively colored solutions of the corresponding 1,2-oxaphospholium ions A–H (Table 1). These species are formed by protonation of the central carbon atom of the allene system that gives the corresponding allyl cations, which undergo cyclization onto the oxygen of the P=O group. These ions have similar NMR data: the signal of the new proton H4 is located in the range 6.30–8.07 ppm, the signal of vinyl carbon C4 at 166.8–171.9 ppm, and the signal of quaternary carbon C5 at 96.0–116.3 ppm. It is worth noting that 2,2-dichloro (A, B) and 2,2-diarylsulfanyl (F, G)-substituted cations exhibit down field shifted signals in the 31P NMR (δ 87.82–115.37 ppm) in comparison with 2,2-diamino (C, D, E1) and 2,2-dimethoxy (H)-substituted species (δ 31P 52.87–70.79 ppm). This reveals that, for amino and methoxy substituents, positive charge is delocalized onto these groups to a greater extent than in the case of chloro or arylsulfanyl ones.
Table 1: Selected 1H, 13C and 31P NMR data for cations A–H derived from the protonation of the corresponding allenes 1a,b,e–j in TfOH at room temperature.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-151-i11.svg?max-width=637&scale=1.0)
|
||||||||||
| Allene | Cation | R | X | 1H NMR, δ, ppm (J, Hz) | 13C NMR, δ, ppm (J, Hz) | 31P NMR, δ, ppm | ||||
| H3 | H4 | C3 | C4 | C5 | ||||||
| 1a | A | H | Cl | 8.11 dd (68.3, 8.3) | 7.00 dd (49.4, 8.3) | 116.4 d (111.3) | 169.9 d (14.3) | 110.8 d (10.8) | 97.04 | |
| 1b | B | Br | Cl | – | 8.07 dd (55.1) | 104.9 d (137.0) | 169.8 d (33.2) | 113.6 d (5.4) | 87.82 | |
| 1e | C | H | O(CH2CH2)2N | 7.89 dd (49.2, 8.3) | 6.45 dd (36.9, 8.3) | 109.3 d (131.9) | 170.3 d (14.3) | 98.9 d (10.0) | 64.33 | |
| 1f | D | H | Et2N | 7.49 dd (49.3, 8.2) | 6.56 dd (38.8, 8.2) | 109.1 d (126.6) | 170.0 | 102.0 | 70.79 | |
| 1g | E1 | H | PhNH | m (overlapping with other signals) | 6.42 dd (37.1, 7.8) | 111.6 d (137.5) | 168.3 d (12.8) | 96.0 | 52.87 | |
| 1h | F | H | 4-MeC6H4S | 7.26 dd (54.0, 7.9) | 6.33 dd (45.5, 7.9) | 113.1 d (82.7) | 167.1 d (10.0) | 116.3 d (7.6) | 115.37 | |
| 1i | Ga | H | 4-ClC6H4S | 7.41 dd (54.4, 7.9) | 6.40 dd (45.9, 7.9) | 111.1 d (81.0) | 166.8 d (10.3) | 101.8 | 114.56 | |
| 1j | H | H | MeO | 7.93 dd (54.6, 8.5) | 6.30 dd (35.8, 8.4) | 107.3 d (159.4) | 171.9 d (14.4) | 97.8 d (13.2) | 57.82 | |
aContent of cation G in reaction solution was ≈50% based on 31P NMR data.
Cations A–D, and F–H are stable in TfOH at room temperature for a long time, they are not transformed into other species under the superacidic conditions. Unlike the others, allene 1g undergoes consequent transformations in TfOH at room temperature (see Scheme 1 and Figure 2). First, when dissolved in the acid, allene 1g forms oxaphospholium ion E1 (Table 1) through an intermediate formation of allyl cation E (Scheme 1). Ion E1 is transformed very fast into another species; after one minute new signals appear in the NMR spectra (see 31P NMR monitoring of this process in Figure 2), and after 12 hours it is completely converted to this new cation. It is most likely that this species is 1,2-azaphosphol-2-ium ion E2, which is formed through allyl cation E.
![[1860-5397-15-151-i1]](/bjoc/content/inline/1860-5397-15-151-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Transformations of allene 1g in TfOH leading to the formation of cations E1, E2 and E4 including selected spectral data for cation E4.
Scheme 1: Transformations of allene 1g in TfOH leading to the formation of cations E1, E2 and E4 including se...
![[1860-5397-15-151-2]](/bjoc/content/figures/1860-5397-15-151-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 31P NMR monitoring of the progress of transformation of E1 into E2 and E4 in TfOH at room temperature.
Figure 2: 31P NMR monitoring of the progress of transformation of E1 into E2 and E4 in TfOH at room temperatu...
However, cation E2, in turn, is further transformed into one more species during several days. The set of spectral data (see below) for this final species indicates that, most likely it should be seven-membered heterocyclic cation E4, which is formed through the P–N bond cleavage in E2 and formation of intermediate cation E3 (Scheme 1).
In the 31P NMR spectra, the signal of E4 is the most up field shifted (δ 24 ppm, see Figure 2 and Scheme 1) in comparison with signals of the species E1 (δ 53 ppm) and E2 (δ 43 ppm). This difference may reveal that phosphorus in cation E4 is bound to a carbon atom, rather than to a heteroatom O or N, like in E1 and E2. Structurally close six-membered ring cations, having the C–P bond, resonate at 30.5–31.9 in 31P NMR [16], that is close to the spectrum for species E4.
Apart from that, in 13C NMR spectra, the signals of quaternary carbon bearing two methyl groups in E2 and E4 are very close (δ 70.3–70.9 ppm, see Scheme 1). Contrary to that, the signal of this carbon for E2 is very much down field shifted (δ 96.1 ppm). This indicates that in species E2 and E4 this carbon is connected to a protonated amino group, and in E1 it is bound to oxygen. The same range of absorbance around 100 ppm for this carbon was observed previously for other oxaphospholium ions [14,16].
Then, we carried out hydrolysis of cations A–H (Scheme 2). Results of hydrolysis strongly depend on the substituent X on the phosphorus atom. Ions containing a labile P–X bond (X = Cl, O, S), namely A, B, and F–H, gave unstable adducts 2 (registered by GC–MS), which are further transformed into acids 3. The structure of compound 3a was confirmed by X-ray analysis (see Supporting Information File 1). On the other hand, hydrolysis of cations C,D, bearing a stable P–N bond, resulted in the formation of allyl alcohols 4. Aqueous work-up of a superacidic solution of cation E4 led to azaphosphepine-5-oxide 5. This substance is insoluble in organic solvents, however, we were able to measure its 1H NMR spectrum in D2O at elevated temperature (80 °C, see Supporting Information File 1).
![[1860-5397-15-151-i2]](/bjoc/content/inline/1860-5397-15-151-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Results of the hydrolysis of cations A–H.
Scheme 2: Results of the hydrolysis of cations A–H.
Taking into account the stability of the P–N bond against hydrolysis, we conducted reactions of the cations A, B, and F–H with morpholine (Scheme 3). Amides 6a,b were isolated as products of these reactions in excellent yields. The plausible reaction mechanism includes at the first stage nucleophilic attack of morpholine onto the phosphorus cationic center that gives cation I, which is transformed into species J. Hydrolysis of the latter leads to cation K and then finally to amides 6a,b.
![[1860-5397-15-151-i3]](/bjoc/content/inline/1860-5397-15-151-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of amides 6a,b from cations A, B, and F–H.
Scheme 3: Preparation of amides 6a,b from cations A, B, and F–H.
We carried out a large-scale one-pot solvent-free synthesis of amides 6a,b starting from propargyl alcohols 7a,b at room temperature (Scheme 4). At the first step, alcohols 7a,b in the reaction with PCl3 were transformed into the corresponding allenes 1a,h. Then, the addition of Brønsted acid (TfOH or H2SO4) gave cations A and B, respectively. The interaction of these species with morpholine followed by hydrolysis furnished the target amides 6a,b in total yields of 60–90% (see procedures in Supporting Information File 1).
![[1860-5397-15-151-i4]](/bjoc/content/inline/1860-5397-15-151-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Large-scale one-pot solvent-free synthesis of amides 6a,b from the corresponding propargylic alcohols.
Scheme 4: Large-scale one-pot solvent-free synthesis of amides 6a,b from the corresponding propargylic alcoho...
It should be noted that allene 1d bearing no alkyl groups and monoalkylated allene 1c formed complex mixtures of oligomeric products under the action of various Brønsted acids (H2SO4, FSO3H, TfOH). In this case, the intermediate oxaphospholium ions are unstable and undergo consequent transformations. Apart from that, attempts to quench cations A–H with external aromatic π-nucleophiles failed. No products of intermolecular electrophilic aromatic substitution were obtained.
Reactions of allenes with Lewis acid AlCl3
Then, we checked reactions of allenes 1a–j with and without benzene under the action of the strong Lewis acid AlCl3, using benzene or dichloromethane as a solvent, followed by hydrolysis of the reaction mixtures. Allenes 1c–j gave complex mixtures of oligomeric products under these conditions. However, allenes 1a,b afforded the desired product of hydroarylation with benzene (vide infra).
AlCl3-promoted reactions of allene 1a were studied under various conditions (Table 2). This compound in reaction with AlCl3 without benzene afforded a mixture of allyl alcohol Z-9 and diene E-10a after aqueous work-up (Table 2, entries 1 and 2). The amount of 2.1 equivalents of AlCl3 is sufficient for activation of this transformation (compared to the amount of AlCl3 in entries 1 and 2, Table 2). On the other hand, 1 equivalent of AlCl3 is not enough to activate allene 1a; thus, under these conditions, only acid 2 was obtained as a product of the hydrolysis of starting compound 1a (Table 2, entry 3). Methanolysis of the reaction mixture gave diene E-10b (Table 2, entry 7). The reaction of allene 1a with benzene resulted in the formation of alkene Z-11a, as a product of intermolecular hydroarylation of the carbon–carbon double bond (Table 2, entries 4–6). This reaction required 2.1 equivalents of AlCl3, 1.05 equivalents of benzene and five minutes at room temperature (Table 2, entry 4). It is worth noting, that the use of other Lewis acids, NiCl2, EuCl3, FeCl3, CuOTf, AgNO3, did not activate allene 1a; in these reactions only the product of the hydrolysis 8 was finally isolated.
Table 2: AlCl3-promoted reactions of allene 1a at room temperature with/without benzene at various conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-151-i12.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Equiv of AlCl3 | Equiv of benzene | Time, min | Yield of 8, % | Yield of 9 + 10a,b, % | Yield of 11a, % |
| 1 | 5 | no benzene | 15 | – | 33 (9) + 32 (10a) | – |
| 2 | 2.1 | no benzene | 15 | – | 35 (9) + 36 (10a) | – |
| 3 | 1 | 1.05 | 15 | 98 | – | – |
| 4 | 2.1 | 1.05 | 5 | – | – | 82 |
| 5 | 2.1 | 1.05 | 15 | – | – | 78 |
| 6 | 2.1 | 1.05 | 60 | – | – | 81 |
| 7a | 2.1 | no benzene | 15 | – | 45 (10b) | – |
aReaction mixture was quenched with methanol.
The configuration of the carbon–carbon double bond in compounds Z-9, E-10b and Z-11a was determined on the basis of the observed values of the spin–spin interaction constants for vinyl protons (13–14 Hz for cis-isomers and 17–18 Hz for trans-isomers), and using H,H-NOESY correlations for Z-11a (see Supporting Information File 1).
Having these conditions for hydroarylation of allene 1a in hand (Table 2, entry 4), we conducted reactions with the series of arenes (Table 3). An excess of methanol was used for quenching of reaction mixtures instead of water. This treatment produced dimethoxyphosphoryl groups [(MeO)2P=O] in the reaction products, rather than the acidic group [(HO)2P=O] in compounds 8–11a (Table 2). The presence of the (MeO)2P=O group in the structures of reaction products makes them more soluble in organic solvents and easy to isolate in preparative reactions.
Table 3: AlCl3-promoted reactions of allene 1a with arenes leading to alkenes 11 and indanes 12 at room temperature for 5 min.
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-151-i13.svg?max-width=637&scale=1.0)
|
||
| Entry | Starting arene, ArH | Reaction products 11 and 12, yield, % |
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-151-i14.svg?max-width=637&scale=1.0)
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-151-i15.svg?max-width=637&scale=1.0)
|
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-151-i16.svg?max-width=637&scale=1.0)
|
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-151-i17.svg?max-width=637&scale=1.0)
|
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-151-i18.svg?max-width=637&scale=1.0)
|
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-151-i19.svg?max-width=637&scale=1.0)
|
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-151-i20.svg?max-width=637&scale=1.0)
|
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-151-i21.svg?max-width=637&scale=1.0)
|
| 5 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-151-i22.svg?max-width=637&scale=1.0)
|
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-151-i23.svg?max-width=637&scale=1.0)
|
| 6a |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-151-i24.svg?max-width=637&scale=1.0)
|
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-151-i25.svg?max-width=637&scale=1.0)
|
| 7 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-151-i26.svg?max-width=637&scale=1.0)
|
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-151-i27.svg?max-width=637&scale=1.0)
|
| 8 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-15-151-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 19]](/bjoc/content/inline/1860-5397-15-151-i29.svg?max-width=637&scale=1.0)
|
| 9 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-15-151-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-15-151-i31.svg?max-width=637&scale=1.0)
|
aReaction was run with 3.1 equiv of AlCl3.
Depending on the structure of the starting arene, allene 1a gave two kinds of reaction products, E-/Z-alkenes 11 and/or indanes 12 (Table 3). Thus, in reactions with benzene, only cis-alkene Z-11b was obtained in 88% yield (Table 3, entry 1). The sole formation of alkenes E-11g, Z-11h and Z-11i, and E/Z-11l was also observed in reactions with 1,2-dimethoxybenzene (veratrole) (Table 3, entry 6), fluorobenzene (Table 3, entry 7) and bromobenzene (Table 3, entry 9), respectively. On the other hand, reactions with methylbenzenes (toluene, o- and m-xylenes, o-fluorotoluene) led to mixtures of alkenes 5 and indanes 6 (Table 3, entries 2–4, and 8). However, p-xylene gave the only reaction product, indane 6d, in nearly quantitative yield of 95% (Table 3, entry 5).
It should be emphasized that compounds 11 and 12 were obtained as inseparable mixtures after TLC separation due to their close chromatographic retention parameters. However, E- and Z-isomers of alkenes 11 can be separated by preparative thin-layer chromatography, for instance, compounds E-11m and Z-11m (Table 3, entry 9 and Supporting Information File 1).
The E/Z-stereochemistry of compounds 11 was determined on the basis of the values of spin–spin interaction constants of vinyl protons, which were 13–14 Hz for Z-isomers and 17–18 Hz for E-isomers (see Supporting Information File 1).
Reactions of allene 1a with strongly donating arenes, 1,3,5-trimethylbenzene (mesitylene), 1,2,4-trimethylbenzene (pseudocumene), phenol, thiophenol, 1,3-dimethoxybenzene, 1,4-dimethoxybenzene, and other arenes, such as 1,2-dichlorobenzene, 1,4-dibromobenzene, gave rise to complex mixtures of oligomeric compounds.
In the same reaction with benzene, allene 1b afforded alkene Z-11n in high yield (Scheme 5).
![[1860-5397-15-151-i5]](/bjoc/content/inline/1860-5397-15-151-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: AlCl3-promoted hydroarylation of allene 1b by benzene leading to alkene Z-11n.
Scheme 5: AlCl3-promoted hydroarylation of allene 1b by benzene leading to alkene Z-11n.
The use of morpholine for quenching of the superacidic reaction mixture gave amide Z-11o in the reaction of 1a with benzene (Scheme 6).
![[1860-5397-15-151-i6]](/bjoc/content/inline/1860-5397-15-151-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of allene 1a with benzene under the action of AlCl3 followed by quenching of the reaction mixture with morpholine leading to amide Z-11o.
Scheme 6: Reaction of allene 1a with benzene under the action of AlCl3 followed by quenching of the reaction ...
We also conducted a large-scale one-pot synthesis of indane 12d starting from 2-methylbut-3-yn-2-ol (Scheme 7, see procedure in Supporting Information File 1). The first stage of this procedure gave allene 1a, which was dissolved in CH2Cl2 and subjected to reaction with p-xylene under the action of AlCl3. Finally, methanolysis of the reaction mixture resulted in the formation of indane 12d in a total yield of 78%.
![[1860-5397-15-151-i7]](/bjoc/content/inline/1860-5397-15-151-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Multigram-scale one-pot synthesis of indane 12d from 2-methylbut-3-yn-2-ol.
Scheme 7: Multigram-scale one-pot synthesis of indane 12d from 2-methylbut-3-yn-2-ol.
To elucidate the reaction mechanism additional experiments were conducted. First of all, alkenes 11 were subjected to the action of five-fold excess of AlCl3 at room temperature or elevated temperature. However, no formation of indanes 12 was detected. Then we carried out an NMR study to catch the reaction intermediates. Upon mixing of allene 1a with 1 equivalent of AlCl3 in CD2Cl2 in an NMR tube at room temperature, a yellow solution was formed, which was most likely a complex of 1a with AlCl3, which is coordinated onto oxygen of the P=O group. The comparison of 1H, 13C, and 31P NMR spectra of starting 1a and its complex with AlCl3 13 is presented in Figure 3 (see full spectral data in Supporting Information File 1). It is clear that the complex formation led to significant broadening of NMR spectral lines and, mainly, a downfield shift of the corresponding signals, due to large positive charge on the phosphorus atom. This solution was stable for a long time (several days) and complex 13 was not converted into other compounds. It must be reminded here, that allene 1a did not react with benzene under the action of 1 equivalent of AlCl3 (see Table 2, entry 3).
![[1860-5397-15-151-3]](/bjoc/content/figures/1860-5397-15-151-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: NMR spectra of starting allene 1a (black) and its complex with 1 equivalent of AlCl3 13 (red) in CD2Cl2 at room temperature: (a) 1H NMR, (b) 13C NMR (selected signals, doublets due to 13C–31P spin–spin interaction), (c) 31P NMR.
Figure 3: NMR spectra of starting allene 1a (black) and its complex with 1 equivalent of AlCl3 13 (red) in CD2...
Addition of more than 1 equivalent of AlCl3 (2–5 equivalents) to a solution of 1a in CD2Cl2 in an NMR tube resulted in an immediate formation of diene E-14 as a part of a complex mixture (Scheme 8, see Supporting Information File 1 for NMR). Compare with the same transformations of 1a followed by hydrolysis of the reaction mixture affording a mixture of alcohol Z-9 and diene E-10a (Table 2, entries 1 and 2). The formation of compound 14 in an NMR monitoring experiment may also indicate that alcohol Z-9 is formed upon hydrolysis of allene 8 (Table 2, entries 1 and 2). Reaction of allene 1a with deuterobenzene C6D6 (1 equivalent) under the action of AlCl3 (2 equivalents) in CD2Cl2 in an NMR tube gave alkene 15 (Scheme 8) analogously to the formation of alkenes 11 (Table 3).
![[1860-5397-15-151-i8]](/bjoc/content/inline/1860-5397-15-151-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: 1H, 13C, and 31P NMR monitoring of AlCl3-promoted reactions of allene 1a leading to compounds E-14 or 15 at room temperature.
Scheme 8: 1H, 13C, and 31P NMR monitoring of AlCl3-promoted reactions of allene 1a leading to compounds E-14 ...
Thus, the different reactivity of these particular dichlorophosphorylallenes under the action of Brønsted or Lewis acids can be explained by involvement of the P=O group in intra- or intermolecular interactions at the formation of cationic intermediates. Strong coordination of Lewis acid AlCl3 with the P=O group completely deactivates it for further intramolecular reactions (Figure 3, Table 2). Despite solvation in the Brønsted superacid TfOH, the P=O group takes part in intramolecular cyclization into oxaphospholium ions (Table 1). These two different types of reaction intermediates, generated from such allenes in Brønsted and Lewis acids, lead to various reaction products.
Based on the data obtained, one may propose a plausible mechanism A for the transformation of allene 1a in the presence of AlCl3 (Scheme 9). When the first equivalent of AlCl3 is added to allene 1a, adduct 13 is formed as a result of electrophilic attack of AlCl3 on the oxygen atom. The second equivalent of AlCl3 is coordinated to the central atom of the allene system of the complex 13 and gives intermediate 16. The latter, in the absence of nucleophiles (arene molecules), undergoes deprotonation from the methyl group affording butadiene 14. Hydrolysis of the latter resulted in compounds Z-9 and E-10a. Whereas, in the presence of an arene, cation 16 reacts with it leading to species 17. The latter can be protonated with the formation of cation 18. This species may react in two different ways. The first option is it could lead to alkene 19 and finally to compounds 11 upon methanolysis of the reaction mixture. An alternative pathway for species 18 is cyclization into indane structure 20, which is further transformed into 21 and 12. At the same time, an alternative mechanism B, involving the formation of the protic superacid HCl–AlCl3 and its participation in the observed reaction should be considered (Scheme 10). The required catalytic amount of such superacid may be formed due to the presence of traces of HCl (byproduct in acetylene–allene rearrangement step) in the reaction mixture. Next, the protonaton of complex 13 occurs, leading to allylic cation 22. As analogue of cation 16 (Scheme 9), the latter can interact with arenes giving hydroarylated complex 23. Consequently, it eliminates AlCl3 and is transformed into P(O)Cl2 alkene 19. The latter can further undergo a protonaton–cyclization sequence (alkene 19→cation 24→P(O)Cl2 indane 21). Target P(O)OMe2 alkenes 11 and indanes 12 are formed during methanolysis of 19 and 21 consequently.
![[1860-5397-15-151-i9]](/bjoc/content/inline/1860-5397-15-151-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Plausible reaction mechanism A for the formation of compounds 9, 10, 11, 12 from aillene 1a involving AlCl3 attack on the central atom of allenic complex 13.
Scheme 9: Plausible reaction mechanism A for the formation of compounds 9, 10, 11, 12 from aillene 1a involvi...
![[1860-5397-15-151-i10]](/bjoc/content/inline/1860-5397-15-151-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Plausible reaction mechanism B of formation of compounds 11, 12 from allene 1a involving HCl–AlCl3 attack on the central atom of allenic complex 13.
Scheme 10: Plausible reaction mechanism B of formation of compounds 11, 12 from allene 1a involving HCl–AlCl3 ...
In accordance with both mechanisms A and B, yields of indanes 12 should be increased for substrates having electron-donating groups, Ar. Indeed, the highest yields of indanes 12 were achieved for the reactions of allene 1a with the electron-rich arenes toluene and xylenes (Table 3, entries 2–5).
We carried out a DFT study [at the B3LYP/6-311+G(2d,2p) level of theory] for the observed AlCl3-involved reactions (Scheme 9, Scheme 10, Table 4 and Supporting Information File 1 for details of DFT calculations). First, the thermochemistry (ΔG of reaction) for selected transformations (1a→13→16→17 for mechanism A, 1a→13→22→23 for mechanism B) was explored. Formation of complex 13 from allene 1a and AlCl3 is exergonic (−26.7 kcal/mol) and thermodynamically favorable. The arylation stage for mechanism A (16→17) is significantly less endergonic (22.6 kcal/mol) than that in mechanism B (22→23, 122.0 kcal/mol, Scheme 9). At the same time, formation of allylic cation 22 (mechanism B) is accompanied by positive changes in ΔG (76.9 kcal/mol), whereas its analogue species 16 formed with slightly negative ΔG (−1.6 kcal/mol, Scheme 10).
Table 4: Comparison of selected electronic characteristics of species 16 (mechanism A) and species 22 (mechanism B) derived from allene 1a.
![[Graphic 22]](/bjoc/content/inline/1860-5397-15-151-i32.svg?max-width=637&scale=1.0)
|
|||||||
| Species | ω,a eV | q (P),b e | q (C1),b e | q(C3),b e | kLUMO(P),c % | kLUMO(C1),c % | kLUMO(C3),c % |
| 16 | 3.42 | 1.78 | -0.64 | 0.11 | 16.6 | 3.2 | 32.1 |
| 22 | 17.1 | 1.73 | -0.52 | 0.44 | 0.73 | 7.4 | 56.6 |
aGlobal electrophilicity index ω = (EHOMO + ELUMO)2/8 (ELUMO − EHOMO); bnatural charges; ccontribution of atomic orbital into the molecular orbital.
Next, we compared electronic characteristics (global electrophilicity indexes ω, natural charges (NBO) and atomic orbital contributions into LUMO) of species 16 and 22 as key intermediates from mechanisms A and B. The calculations reveal that both charge and orbital factors coincide in electrophilic reactivity of carbon C3 in species 16, 22 (Scheme 9 and Scheme 10). At the same time, the carbon C3 in C-protonated intermediate 22 bears a more positive charge (0.44 e) and gives a rather big contribution into LUMO (56.6%) compared to that of 16 (0.11 e, 32.1%). Also, species 22 is five times more electrophilic than 16 according to values of ω. Visualizations of the LUMO for 16 and 22 are shown on Figure 4.
![[1860-5397-15-151-4]](/bjoc/content/figures/1860-5397-15-151-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Visualization of LUMO, only positive values are shown, isosurface value 0.043: (a) species 16, (b) species 22.
Figure 4: Visualization of LUMO, only positive values are shown, isosurface value 0.043: (a) species 16, (b) ...
Conclusion
Transformations of various phosphorylallenes under the action of strong Brønsted or Lewis acids were studied. These allenes showed different reactivity depending on the type of the acid. In the Brønsted superacid TfOH, the allenes were transformed into oxophospholium cations. Hydrolysis (or morpholinolysis) of these species afforded a series of phosphorous-containing compounds, cyclic phosphoric acids and their derivatives, and other substances. Contrarily, reactions of dichlorophosphorylallenes with the Lewis acid AlCl3 proceeded through the formation of non-cyclic intermediates. Hydrolysis of the latter afforded phosphorylallyl alcohols and butadienes. For the first time, the intermolecular hydroarylation of the allene system of dichlorophosphorylallenes by arenes under the action of AlCl3 was achieved. This reaction gave rise to phosphoryl-substituted alkenes and indanes. The intermediates of these reactions were investigated by means of NMR and DFT calculations, that shed light on the reaction mechanisms.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 7.5 MB | Download |
Acknowledgement
This work was supported by the Russian Scientific Foundation (grant no. 18-13-00008). Spectral studies were performed at the Center for Magnetic Resonance, Center for Chemical Analysis and Materials Research, and Research Center for X-ray Diffraction Studies of Saint Petersburg State University, Saint Petersburg, Russia.
References
-
Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004.
Return to citation in text: [1] -
Back, T. G.; Clary, K. N.; Gao, D. Chem. Rev. 2010, 110, 4498–4553. doi:10.1021/cr1000546
Return to citation in text: [1] -
Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. doi:10.1002/anie.201101460
Return to citation in text: [1] -
Essid, I.; Laborde, C.; Legros, F.; Sevrain, N.; Touil, S.; Rolland, M.; Ayad, T.; Volle, J.-N.; Pirat, J.-L.; Virieux, D. Org. Lett. 2017, 19, 1882–1885. doi:10.1021/acs.orglett.7b00648
Return to citation in text: [1] -
Berton, J. K. E. T.; Salemi, H.; Pirat, J.-L.; Virieux, D.; Stevens, C. V. J. Org. Chem. 2017, 82, 12439–12446. doi:10.1021/acs.joc.7b02227
Return to citation in text: [1] -
Ismailov, I. E.; Ivanov, I. K.; Christov, V. C. Molecules 2014, 19, 11056–11076. doi:10.3390/molecules190811056
Return to citation in text: [1] -
Angelov, C. M.; Enchev, D. D. Phosphorus Sulfur Relat. Elem. 1988, 37, 125–128. doi:10.1080/03086648808079026
Return to citation in text: [1] -
Enchev, D. D. Phosphorus, Sulfur Silicon Relat. Elem. 2000, 165, 273–284. doi:10.1080/10426500008076346
Return to citation in text: [1] -
Ivanov, I. K.; Christov, V. C. Synth. Commun. 2013, 43, 800–809. doi:10.1080/00397911.2011.609957
Return to citation in text: [1] -
Yuan, J.; Ruan, X.; Yang, Y.; Huang, X. Synlett 2007, 2871–2874. doi:10.1055/s-2007-991082
Return to citation in text: [1] -
Macomber, R. S. J. Org. Chem. 1977, 42, 3297–3298. doi:10.1021/jo00440a021
Return to citation in text: [1] -
Mikhailova, T. S.; Skvortsov, I. K.; Ignatiev, V. M.; Ionin, B. I.; Petrov, A. A. Dokl. Akad. Nauk. SSSR 1978, 241, 1095.
Return to citation in text: [1] [2] -
Lozovskiy, S. V.; Bogachenkov, A. S.; Dogadina, A. V.; Vasilyev, A. V. Tetrahedron Lett. 2016, 57, 3167–3170. doi:10.1016/j.tetlet.2016.06.026
Return to citation in text: [1] [2] -
Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637
Return to citation in text: [1] [2] [3] -
Bogachenkov, A. S.; Dogadina, A. V.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2015, 13, 1333–1338. doi:10.1039/c4ob02269f
Return to citation in text: [1] [2] -
Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j
Return to citation in text: [1] [2] [3] [4] -
Widenhoefer, R. A. Transition metal-catalyzed hydroarylation of allenes. In Catalytic Hydroarylation of Carbon-Carbon Multiple Bonds; Ackermann, L., Ed.; Wiley-VCH: Weinheim, Germany, 2017; pp 361–388. doi:10.1002/9783527697649.ch9
See for a review.
Return to citation in text: [1] -
Fang, Z.; Fu, C.; Ma, S. Chem. – Eur. J. 2010, 16, 3910–3913. doi:10.1002/chem.200903012
Return to citation in text: [1] -
Suresh, R. R.; Swamy, K. C. K. J. Org. Chem. 2012, 77, 6959–6969. doi:10.1021/jo301149s
Return to citation in text: [1] -
Miller, Z. D.; Montgomery, J. Org. Lett. 2014, 16, 5486–5489. doi:10.1021/ol502766q
Return to citation in text: [1] -
Muñoz, M. P.; de la Torre, M. C.; Sierra, M. A. Chem. – Eur. J. 2012, 18, 4499–4504. doi:10.1002/chem.201103337
Return to citation in text: [1] -
Skouta, R.; Li, C.-J. Can. J. Chem. 2008, 86, 616–620. doi:10.1139/v08-067
Return to citation in text: [1] -
Toups, K. L.; Liu, G. T.; Widenhoefer, R. A. J. Organomet. Chem. 2009, 694, 571–575. doi:10.1016/j.jorganchem.2008.11.058
Return to citation in text: [1] -
Tarselli, M. A.; Liu, A.; Gagné, M. R. Tetrahedron 2009, 65, 1785–1789. doi:10.1016/j.tet.2008.10.110
Return to citation in text: [1] -
Sutherland, D. R.; Kinsman, L.; Angiolini, S. M.; Rosair, G. M.; Lee, A.-L. Chem. – Eur. J. 2018, 24, 7002–7009. doi:10.1002/chem.201800209
Return to citation in text: [1] -
Zhang, Y. J.; Skucas, E.; Krische, M. J. Org. Lett. 2009, 11, 4248–4250. doi:10.1021/ol901759t
Return to citation in text: [1] -
Zeng, R.; Fu, C.; Ma, S. J. Am. Chem. Soc. 2012, 134, 9597–9600. doi:10.1021/ja303790s
Return to citation in text: [1] -
Ye, B.; Cramer, N. J. Am. Chem. Soc. 2013, 135, 636–639. doi:10.1021/ja311956k
Return to citation in text: [1] -
Nakanowatari, S.; Mei, R.; Feldt, M.; Ackermann, L. ACS Catal. 2017, 7, 2511–2515. doi:10.1021/acscatal.7b00207
Return to citation in text: [1] -
Fang, Z.; Fu, C.; Ma, S. Eur. J. Org. Chem. 2011, 1227–1231. doi:10.1002/ejoc.201001661
Return to citation in text: [1] [2] -
Jin, H. J.; Kim, J. H.; Kang, E. J. Synthesis 2017, 49, 3137–3144. doi:10.1055/s-0036-1589004
Return to citation in text: [1]
| 16. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 14. | Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637 |
| 16. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 1. | Krause, N.; Hashmi, A. S. K., Eds. Modern Allene Chemistry; Wiley-VCH: Weinheim, Germany, 2004. |
| 2. | Back, T. G.; Clary, K. N.; Gao, D. Chem. Rev. 2010, 110, 4498–4553. doi:10.1021/cr1000546 |
| 3. | Yu, S.; Ma, S. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. doi:10.1002/anie.201101460 |
| 13. | Lozovskiy, S. V.; Bogachenkov, A. S.; Dogadina, A. V.; Vasilyev, A. V. Tetrahedron Lett. 2016, 57, 3167–3170. doi:10.1016/j.tetlet.2016.06.026 |
| 14. | Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637 |
| 15. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2015, 13, 1333–1338. doi:10.1039/c4ob02269f |
| 16. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 31. | Jin, H. J.; Kim, J. H.; Kang, E. J. Synthesis 2017, 49, 3137–3144. doi:10.1055/s-0036-1589004 |
| 12. | Mikhailova, T. S.; Skvortsov, I. K.; Ignatiev, V. M.; Ionin, B. I.; Petrov, A. A. Dokl. Akad. Nauk. SSSR 1978, 241, 1095. |
| 30. | Fang, Z.; Fu, C.; Ma, S. Eur. J. Org. Chem. 2011, 1227–1231. doi:10.1002/ejoc.201001661 |
| 11. | Macomber, R. S. J. Org. Chem. 1977, 42, 3297–3298. doi:10.1021/jo00440a021 |
| 12. | Mikhailova, T. S.; Skvortsov, I. K.; Ignatiev, V. M.; Ionin, B. I.; Petrov, A. A. Dokl. Akad. Nauk. SSSR 1978, 241, 1095. |
| 29. | Nakanowatari, S.; Mei, R.; Feldt, M.; Ackermann, L. ACS Catal. 2017, 7, 2511–2515. doi:10.1021/acscatal.7b00207 |
| 4. | Essid, I.; Laborde, C.; Legros, F.; Sevrain, N.; Touil, S.; Rolland, M.; Ayad, T.; Volle, J.-N.; Pirat, J.-L.; Virieux, D. Org. Lett. 2017, 19, 1882–1885. doi:10.1021/acs.orglett.7b00648 |
| 5. | Berton, J. K. E. T.; Salemi, H.; Pirat, J.-L.; Virieux, D.; Stevens, C. V. J. Org. Chem. 2017, 82, 12439–12446. doi:10.1021/acs.joc.7b02227 |
| 6. | Ismailov, I. E.; Ivanov, I. K.; Christov, V. C. Molecules 2014, 19, 11056–11076. doi:10.3390/molecules190811056 |
| 7. | Angelov, C. M.; Enchev, D. D. Phosphorus Sulfur Relat. Elem. 1988, 37, 125–128. doi:10.1080/03086648808079026 |
| 8. | Enchev, D. D. Phosphorus, Sulfur Silicon Relat. Elem. 2000, 165, 273–284. doi:10.1080/10426500008076346 |
| 9. | Ivanov, I. K.; Christov, V. C. Synth. Commun. 2013, 43, 800–809. doi:10.1080/00397911.2011.609957 |
| 10. | Yuan, J.; Ruan, X.; Yang, Y.; Huang, X. Synlett 2007, 2871–2874. doi:10.1055/s-2007-991082 |
| 30. | Fang, Z.; Fu, C.; Ma, S. Eur. J. Org. Chem. 2011, 1227–1231. doi:10.1002/ejoc.201001661 |
| 21. | Muñoz, M. P.; de la Torre, M. C.; Sierra, M. A. Chem. – Eur. J. 2012, 18, 4499–4504. doi:10.1002/chem.201103337 |
| 26. | Zhang, Y. J.; Skucas, E.; Krische, M. J. Org. Lett. 2009, 11, 4248–4250. doi:10.1021/ol901759t |
| 18. | Fang, Z.; Fu, C.; Ma, S. Chem. – Eur. J. 2010, 16, 3910–3913. doi:10.1002/chem.200903012 |
| 19. | Suresh, R. R.; Swamy, K. C. K. J. Org. Chem. 2012, 77, 6959–6969. doi:10.1021/jo301149s |
| 20. | Miller, Z. D.; Montgomery, J. Org. Lett. 2014, 16, 5486–5489. doi:10.1021/ol502766q |
| 27. | Zeng, R.; Fu, C.; Ma, S. J. Am. Chem. Soc. 2012, 134, 9597–9600. doi:10.1021/ja303790s |
| 28. | Ye, B.; Cramer, N. J. Am. Chem. Soc. 2013, 135, 636–639. doi:10.1021/ja311956k |
| 17. |
Widenhoefer, R. A. Transition metal-catalyzed hydroarylation of allenes. In Catalytic Hydroarylation of Carbon-Carbon Multiple Bonds; Ackermann, L., Ed.; Wiley-VCH: Weinheim, Germany, 2017; pp 361–388. doi:10.1002/9783527697649.ch9
See for a review. |
| 13. | Lozovskiy, S. V.; Bogachenkov, A. S.; Dogadina, A. V.; Vasilyev, A. V. Tetrahedron Lett. 2016, 57, 3167–3170. doi:10.1016/j.tetlet.2016.06.026 |
| 14. | Lozovskiy, S. V.; Ivanov, A. Y.; Bogachenkov, A. S.; Vasilyev, A. V. ChemistrySelect 2017, 2, 4505–4510. doi:10.1002/slct.201700637 |
| 15. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2015, 13, 1333–1338. doi:10.1039/c4ob02269f |
| 16. | Bogachenkov, A. S.; Dogadina, A. V.; Boyarskaya, I. A.; Boyarskiy, V. P.; Vasilyev, A. V. Org. Biomol. Chem. 2016, 14, 1370–1381. doi:10.1039/c5ob02143j |
| 22. | Skouta, R.; Li, C.-J. Can. J. Chem. 2008, 86, 616–620. doi:10.1139/v08-067 |
| 23. | Toups, K. L.; Liu, G. T.; Widenhoefer, R. A. J. Organomet. Chem. 2009, 694, 571–575. doi:10.1016/j.jorganchem.2008.11.058 |
| 24. | Tarselli, M. A.; Liu, A.; Gagné, M. R. Tetrahedron 2009, 65, 1785–1789. doi:10.1016/j.tet.2008.10.110 |
| 25. | Sutherland, D. R.; Kinsman, L.; Angiolini, S. M.; Rosair, G. M.; Lee, A.-L. Chem. – Eur. J. 2018, 24, 7002–7009. doi:10.1002/chem.201800209 |
© 2019 Lozovskiy et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)