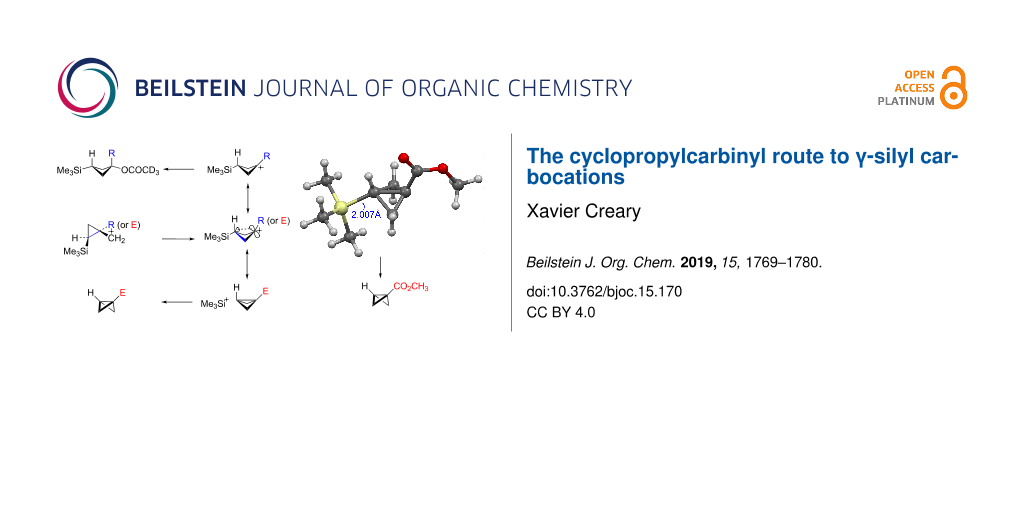
Abstract
The mesylate derivative of cis-1-hydroxymethyl-2-trimethylsilylcyclopropane has been prepared, along with a number of related mesylates and triflates with substituents on the 1-position. These substrates all solvolyze in CD3CO2D to give products derived from cyclopropylcarbinyl cations that undergo further rearrangement to give 3-trimethylsilylcyclobutyl cations. These 3-trimethylsilylcyclobutyl cations are stabilized by a long-range rear lobe interaction with the γ-trimethylsilyl group. When the substituent is electron-withdrawing (CF3, CN, or CO2CH3), significant amounts of bicyclobutane products are formed. The bicyclobutanes are a result of γ-trimethylsilyl elimination from the cationic intermediate that has an unusually long calculated Si–C bond. The solvolysis chemistry of mesylate and triflate derivatives of trans-1-hydroxymethyl-2-trimethylsilylcyclopropane and 1-substituted analogs can be quite different since these substrates do not generally lead to 3-trimethylsilylcyclobutyl cations.

Graphical Abstract
Introduction
Carbocations, positively charged trivalent carbon compounds and reactive intermediates, have continued to fascinate chemists since the early discoveries of tropylium [1,2] and trityl [3-7] salts. Many of the giants of organic chemistry during the last century contributed heavily to the development of carbocation chemistry. This article will deal with three types of carbocations that have been of intense and fundamental interest over the years, i.e., cyclopropylcarbinyl cations, electron-deficient cations, and silyl substituted carbocations. A brief overview of these types of carbocations is warranted.
Cyclopropylcarbinyl cations are an extensively studied system [8,9]. Initial interest was derived from the fact that both cyclopropylcarbinyl and cyclobutyl substrates 1 and 2, where X represents diazonium ion [10,11], chloride [10], or naphthalenesulfonate [12] leaving groups, reacted in aqueous solvents to give an identical mixture of products 3, 4, and 5 (Scheme 1). Additionally, solvolysis rates were far greater than expected for primary and strained secondary systems. To account for these facts, it has been suggested that there are common cationic intermediates in these solvolysis reactions of 1 and 2. Labelling [13-15], stable ion [16-19], and computational studies [19] implicate the involvement of three degenerate cyclopropylcarbinyl cations, 6a, 6b, and 6c, in equilibrium with cyclobutyl cation 7, as well as the homoallylic cation 8 (Scheme 2). Cations 6 are stabilized by the cyclopropyl ring and are therefore much more stable than simple primary carbocations. The cyclobutyl cation 7 is also quite stabilized relative to simple secondary carbocations. This cation has been called a “bicyclobutonium” cation, 7a, which is a nonclassical cation (a cation containing hypercoordinated carbon) that could be derived from protonation of bicyclobutane [20]. Another potential mode of stabilization is by an interaction of the cationic center with the adjacent strained cyclobutyl bonds as in 7b.
![[1860-5397-15-170-i1]](/bjoc/content/inline/1860-5397-15-170-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Solvolyses of cyclopropylcarbinyl and cyclobutyl substrates.
Scheme 1: Solvolyses of cyclopropylcarbinyl and cyclobutyl substrates.
![[1860-5397-15-170-i2]](/bjoc/content/inline/1860-5397-15-170-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The cyclopropylcarbinyl–cyclobutyl–homoallyl cation manifold.
Scheme 2: The cyclopropylcarbinyl–cyclobutyl–homoallyl cation manifold.
A second class of carbocations that this article will deal with is the so-called “electron-deficient” carbocation, i.e., carbocations 9 (Figure 1) substituted with electron-withdrawing groups E [21]. Many studies have shown that such cations can indeed be generated and that they can derive stabilization by a variety of mechanisms. Chief among these cations are the α-trifluoromethyl [22-24], α-cyano [22,25-29], α-carbonyl [30-33], and α-phosphoryl [34,35] analogs of 9. Carbocations of type 9 will be examined in conjunction with the cyclopropylcarbinyl–cyclobutyl manifold.
![[1860-5397-15-170-1]](/bjoc/content/figures/1860-5397-15-170-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Electron-deficient carbocations.
Figure 1: Electron-deficient carbocations.
The third type of carbocation that will be incorporated into this paper is the trimethylsilyl-substituted carbocation [36-44]. We have been interested in long-range interactions of silicon with both carbene [45-48] and carbocation centers [49,50]. Along these lines, γ-trimethylsilyl cations of general type 11 have been generated under stable-ion [51] as well as solvolytic conditions [52-54]. They are greatly stabilized by the “rear lobe” type of interaction shown involving the γ-trimethylsilyl group. A number of related cations are also stabilized by analogous γ-silyl interactions [55-59], which have also been termed ”percaudal” interactions [56]. Certain carbenes can also be stabilized in a similar fashion [60,61]. Thus substrates of type 10 solvolyze in protic solvents with large rate enhancements (anchimeric assistance) to generate carbocations 11 as reactive intermediates (Scheme 3). These cations 11 capture solvent molecules to give exclusively products 12 with net retention of configuration, a characteristic of carbocations that are stabilized by this type of rear lobe interaction.
![[1860-5397-15-170-i3]](/bjoc/content/inline/1860-5397-15-170-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Solvolyses of γ-trimethylsilylcyclobutyl substrates.
Scheme 3: Solvolyses of γ-trimethylsilylcyclobutyl substrates.
A series of cyclopropylcarbinyl substrates 13 and 14 (Figure 2), where X is a leaving group and R is an electron-donating group and E is an electron-withdrawing group, have now been examined. The goal was to evaluate the cyclopropylcarbinyl to cyclobutyl cation rearrangement. Can these substrates lead to γ-trimethylsilyl-substituted cyclobutyl cations 11 and what are the fates of such carbocations? Answers to these questions were sought.
![[1860-5397-15-170-2]](/bjoc/content/figures/1860-5397-15-170-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Phenyl-substituted systems
The first compounds to be examined were the mesylates 19 and 20. These substrates were prepared as shown in Scheme 4. Irradiation of ethyl 2-diazo-2-phenylacetate (15) in vinyltrimethylsilane as solvent gave an isomeric mixture of esters 16. Subsequent reduction with lithium aluminum hydride gave a mixture of alcohols 17 and 18, which could be readily separated by silica gel chromatography. The assignment of stereochemistry of these isomers was based on shielding effects in both 1H and 13C NMR spectra. For example, the trimethylsilyl singlet in 18 appears at δ −0.30 (shielded by the cis-phenyl group), while the trimethylsilyl singlet in 17 appears at δ 0.14 (deshielded by the trans-phenyl group). Such effects are in complete agreement with calculated shifts based on B3LYP/6-31G* calculated structures of 17 and 18. Additionally, nOe studies on 17 confirm the stereochemical assignment. Conversion to mesylates 19 and 20 using mesyl chloride and triethylamine was straightforward.
![[1860-5397-15-170-i4]](/bjoc/content/inline/1860-5397-15-170-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of mesylates 19 and 20.
Scheme 4: Synthesis of mesylates 19 and 20.
Mesylate 19 reacts readily in CD3CO2D at 20 °C (Table 1) to give the substituted cyclobutyl acetate 21 (92%) as the major product along with 8% of the alkene 22. It is proposed (Scheme 5) that these products arise from stepwise formation of the cyclopropylcarbinyl cation 23. This cation can rearrange via migration of bond a to give the cyclobutyl cation 24. The cis-nature of the phenyl group and the hydrogen in cation 23 necessarily results in the formation of the γ-silyl-stabilized cation 24. This cation is the source of the acetate 21. Alternatively, cation 23 can rearrange by migration of the b bond of the cyclopropane. This leads to the β-silylcyclobutyl cation 25, which can subsequently desilylate to give the minor product, the alkene 22. Interestingly, formation of the γ-silyl cation 24 is preferred over the β-silyl cation 25.
![[1860-5397-15-170-i5]](/bjoc/content/inline/1860-5397-15-170-i5.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of mesylate 19 in CD3CO2D.
Scheme 5: Reaction of mesylate 19 in CD3CO2D.
Reaction of the isomeric mesylate 20 in CD3CO2D gives the same rearranged products 21 and 22. These products are accounted for mechanistically in Scheme 6. The initially formed cyclopropylcarbinyl cation 26 rearranges by migration of the a bond of the cyclopropane to give the cyclobutyl cation 27. This cation 27 is different from the γ-silyl-stabilized cation 24 in that the cis-nature of the phenyl and TMS groups in 26 requires that these groups are closer to each other in 27. Shown in Figure 3 are M062X/6-311+G** calculated structures and energies of cations 27 and 24, which are distinct energy minima, along with the transition state 28 which connects these two cations. Cation 27 derives most of its stabilization from the phenyl group, while the TMS group in the 3-position provides no cross-ring stabilization. The calculated barrier for ring inversion of 27 to give the lower energy rear lobe stabilized γ-trimethylsilyl cation 24 is only 2.4 kcal/mol. Calculations at the B3LYP/6-31G*, B3LYP/6-311+G**, MP2/6-31G*, and the MP2/6-311+G** levels lead to the same conclusions, i.e., cations 24 and 27 are distinct energy minima with a very low barrier for conversion of 27 to 24. Therefore, formation of 27 under solvolytic conditions should readily yield 24, and subsequently the substitution product 21. The small amount (4%) of elimination product 22 is a result of rearrangement of 26 to the β-trimethylsilyl cation 25 as described in Scheme 5.
![[1860-5397-15-170-i6]](/bjoc/content/inline/1860-5397-15-170-i6.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of mesylate 20 in CD3CO2D.
Scheme 6: Reaction of mesylate 20 in CD3CO2D.
![[1860-5397-15-170-3]](/bjoc/content/figures/1860-5397-15-170-3.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 3: M062X/6-311+G** calculated structures and relative energies of cations 24, 27, and transition state 28.
Figure 3: M062X/6-311+G** calculated structures and relative energies of cations 24, 27, and transition state ...
Unsubstituted and methyl-substituted systems
Attention was next turned to potential γ-trimethylsilylcyclobutyl cation systems lacking phenyl stabilization. Thus pure Z- and E-alcohols 29 and 30 were each cyclopropanated under Simmons–Smith conditions, and the resultant stereochemically pure alcohols were converted to mesylates 31 and 32, respectively (Scheme 7). For rate comparisons, cyclopropylcarbinyl mesylate 33 [62,63] was also prepared.
![[1860-5397-15-170-i7]](/bjoc/content/inline/1860-5397-15-170-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of mesylates 31 and 32.
Scheme 7: Synthesis of mesylates 31 and 32.
Mesylate 31 reacted readily in CD3CO2D to give the cis-cyclobutyl acetate 34 as the major product (Scheme 8), along with a small amount of cyclobutene (35). The rate of 31 (Table 1) is not substantially enhanced relative to the unsubstituted cyclopropylcarbinyl mesylate (33). The small rate enhancement factor of 3.56 is consistent with a small inductive stabilization of the initially formed cationic intermediate. This behavior is completely analogous to that of the phenyl analog 19 and a similar mechanistic pathway is proposed. The initially formed cyclopropylcarbinyl cation 36 rearranges to the γ-silylcyclobutyl cation 37, the source of the major product 34. The desilylated product 35 arises from the alternative β-trimethylsilylcyclobutyl cation.
![[1860-5397-15-170-i8]](/bjoc/content/inline/1860-5397-15-170-i8.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reaction of mesylate 31 in CD3CO2D.
Scheme 8: Reaction of mesylate 31 in CD3CO2D.
The behavior of mesylate 32 is in contrast to that of 31 and the phenyl analog 20. Five products, 35, 38, 39, 40, and 41, are obtained and these products are formed in essentially the identical ratio as seen in our previous study of the trans-mesylate 42 [52]. The similarity of products formed from acetolysis of 32 and 42 implies that the same cation rearrangement manifold is involved. Scheme 9 gives a mechanistic rationale for these products. Capture of an unrearranged discrete cyclopropylcarbinyl cation 43 gives the major product 38, while migration of bond c to the cationic center gives rearranged cation 44, the source of the rearranged acetate 39. Ring expansion via migration of bond b in 43 gives the β-trimethylsilyl-stabilized cyclobutyl cation 45, and subsequent desilylation provides cyclobutene (35). Alternatively, cyclobutyl to homoallylic cation rearrangement leads to the homoallylic products 40 and 41 via internal mesylate return or solvent capture. Of interest is the fact that no product 34 (derived from γ-trimethylsilyl-stabilized cation 37) is formed. Our previous computational study [52] provided insight into the lack of involvement of cation 37. This study at the B3LYP/6-31G* level suggested that migration of bond a in 43 is not viable since the resultant cation 47 is not an energy minimum at this level, but a transition state. However, a current study at the M062X/6-311+G** level finds that both conformations 47a and 47b are energy minima. While 47a lies 10.8 kcal/mol above 37, the barrier for inversion of 47a to 37 is quite large (24.9 kcal/mol). Hence there is no viable route to 37.
![[1860-5397-15-170-i9]](/bjoc/content/inline/1860-5397-15-170-i9.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reaction of mesylate 32 in CD3CO2D.
Scheme 9: Reaction of mesylate 32 in CD3CO2D.
In order to complete the study of substrates 13 with electron-donating groups, the methyl analog 48 was prepared from the corresponding cyclopropylcarbinyl alcohol, which was available from methyl 2-diazopropanoate by a process completely analogous to the synthesis of the phenyl analog 17. The mesylate derivative was too reactive for rates to be measured and hence the trifluoroacetate derivative 48 was studied. Acetolysis gave the acetate 50 along with a smaller amount of methylcyclobutene (51, Scheme 10). This reactivity is completely analogous to that seen in the phenyl and hydrogen analogs 19 and 31, i.e., a mechanistic scheme involving the γ-trimethylsilyl-stabilized cation 52 is likely.
![[1860-5397-15-170-i10]](/bjoc/content/inline/1860-5397-15-170-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Reaction of trifluoroacetate 48 in CD3CO2D.
Scheme 10: Reaction of trifluoroacetate 48 in CD3CO2D.
The isomeric trifluoroacetate 49 (shown in Table 1) gives methylcyclobutene (51) (68%) as the major acetolysis product, along with minor products that are identical to those previously reported [52] in solvolysis of the trifluoroacetate derivative of (1r,3r)-1-methyl-3-(trimethylsilyl)cyclobutanol. As in the case of mesylate 32, the γ-trimethylsilyl-stabilized cation 52 is apparently not formed from trifluoroacetate 49 due to stereochemical constraints.
Table 1: Solvolysis rates for substrates in CD3CO2D at 20.0 °C.
| Compound | k (s−1) |
krel
(for ROMs) |
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-170-i17.svg?max-width=637&scale=1.0)
33 |
1.71 × 10−4 | 1.0 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-170-i18.svg?max-width=637&scale=1.0)
19 |
6.50 × 10−4 | 3.8 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-170-i19.svg?max-width=637&scale=1.0)
20 |
1.26 × 10−3 | 7.4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-170-i20.svg?max-width=637&scale=1.0)
31 |
6.09 × 10−4 | 3.6 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-170-i21.svg?max-width=637&scale=1.0)
32 |
6.89 × 10−4 | 4.0 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-170-i22.svg?max-width=637&scale=1.0)
48 |
1.31 × 10−7a | 76b,c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-170-i23.svg?max-width=637&scale=1.0)
49 |
8.91 × 10−8a | 52b,c |
aExtrapolated from data at higher temperatures. k for 48 at 60.0 °C = 2.58 × 10−5 s−1; k for 48 at 80.0 °C = 2.33 × 10−4 s−1; k for 49 at 60.0 °C = 1.62 × 10−5 s−1; k for 49 at 80.0 °C = 1.42 × 10−4 s−1. bMesylate is too reactive for rate to be measured. cAssuming mesylate reacts 105 faster than trifluoroacetate.
Systems with electron-withdrawing groups
Attention was next turned to cyclopropylcarbinyl systems substituted with electron-withdrawing groups. Previously Tilley and co-workers [55] have examined the triflate 53 and found that this system solvolyzes with rear lobe TMS participation (Scheme 11). The unusual feature in solvolysis of 53 is the formation of the highly strained bicyclobutane 55 as the sole product. It was therefore of interest to see if the cyclopropylcarbinyl to cyclobutyl rearrangement could be used to access the carbocation 54, and subsequently, bicyclobutane 55. It was also of interest to see if other bicyclobutanes could be formed if the CF3 group were replaced by other electron-withdrawing groups that we have previously examined in carbocation forming reactions.
![[1860-5397-15-170-i11]](/bjoc/content/inline/1860-5397-15-170-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Bicyclobutane formation from a γ-trimethylsilyl cation.
Scheme 11: Bicyclobutane formation from a γ-trimethylsilyl cation.
The requisite trifluoromethyl-substituted cyclopropylcarbinyl systems were prepared by addition of the carbene derived from the diazoester 56 to vinyltrimethylsilane as shown in Scheme 12. Reduction of the ester mixture 57 with lithium aluminum hydride gave a chromatographically separable mixture of alcohols 58 and 59. Stereochemistry of the alcohol 58 was established by long-range 19F coupling to the cis-trimethylsilyl group hydrogens (JH-F = 0.9 Hz). Long-range 19F coupling to the TMS methyl groups of 58 was also observed in the 13C NMR spectrum (JC-F = 2.1 Hz) [64,65]. This long-range 19F coupling is not observed when the CF3 group is trans to the TMS group in the isomer 59.
![[1860-5397-15-170-i12]](/bjoc/content/inline/1860-5397-15-170-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Formation of triflates 60 and 61.
Scheme 12: Formation of triflates 60 and 61.
Additional cyclopropylcarbinyl systems containing the electron-withdrawing cyano and carbomethoxy groups were prepared in an analogous fashion as shown in Scheme 13. Carbomethoxycyano carbene addition to vinyltrimethylsilane followed by lithium borohydride reduction of the ester functionality of 63 gave a separable mixture of alcohols 64 and 65. The stereochemistry of the product 65 was established using nOe studies. Cyano to carbomethoxy conversion in 65 to give alcohol 66 was straightforward. Triflate derivatives 67 and 68 were prepared since analogous mesylate derivatives were relatively unreactive. Triflate 69 was a highly reactive substrate that could only be prepared in about 80% purity. The less reactive mesylate derivative 75 was therefore prepared and used for kinetic studies.
![[1860-5397-15-170-i13]](/bjoc/content/inline/1860-5397-15-170-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Formation of triflates 67, 68, and 69.
Scheme 13: Formation of triflates 67, 68, and 69.
The triflates 61, 68, and 69 (with electron-withdrawing groups trans to trimethylsilyl) were all solvolyzed in CD3CO2D and results are shown in Scheme 14. Since the triflate 69 was highly reactive and could not be isolated in pure form, the mesylate derivative 75 was used in kinetic studies that were carried out in the 40–60 °C range. Rates of reaction of mesylate derivatives (Table 2) were all substantially slower than the parent mesylate 33 or the phenyl, methyl, or H analogs. This is attributed to a significant inductive destabilizing β-effect of the group E on the initially formed cation 73. The triflates all produced significant amounts of bicyclobutane products 55 and 72 along with some unrearranged substitution products 70. In the cases of 68 and 69, some rearranged substitution products 71 were also formed. The mesylate 75 gave the same initial products as the triflate 69. However, the bicyclobutane 72c formed from mesylate 75 was not completely stable at 40–60 °C, but degraded slowly to a mixture of other products. The bicyclobutanes 55, 72b, and 72c were quite stable in CD3CO2D at 20 °C, where triflate studies were carried out.
![[1860-5397-15-170-i14]](/bjoc/content/inline/1860-5397-15-170-i14.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Reactions of substrates with electron-withdrawing groups in CD3CO2D.
Scheme 14: Reactions of substrates with electron-withdrawing groups in CD3CO2D.
Table 2: Solvolysis rates for substrates in CD3CO2D at 20.0 °C.
| Compound | k (s−1) |
krel
(for ROMs) |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-170-i24.svg?max-width=637&scale=1.0)
33 |
1.71 × 10−4 | 1.00 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-170-i25.svg?max-width=637&scale=1.0)
75 |
1.26 × 10−7a | 7.3 × 10−4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-170-i26.svg?max-width=637&scale=1.0)
61 |
2.25 × 10−4 | 1.3 × 10−5b |
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-170-i27.svg?max-width=637&scale=1.0)
60 |
1.25 × 10−3 | 7.3 × 10−5b |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-170-i28.svg?max-width=637&scale=1.0)
68 |
2.14 × 10−4 | 1.3 × 10−5b |
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-170-i29.svg?max-width=637&scale=1.0)
67 |
1.61 × 10−3 | 9.4 × 10−5b |
aExtrapolated from data at higher temperatures. k at 40.0 °C = 2.24 × 10−6 s−1; k at 50.0 °C = 8.40 × 10−6 s−1; k at 60.0 °C = 2.85 × 10−5 s−1. bAssuming triflate reacts 105 faster than mesylate.
The bicyclobutane products 55 and 72 are a result of desilylation of the γ-silyl cations 54 and 74. Why are bicylobutanes formed from cations 54 and 74 and not from cations 24, 37, and 52, which do not have electron-withdrawing groups? Previous studies have shown that “electron-deficient” cations 9, where E = COR [66], CN [25], CF3 [67], and PO(OEt)2 [34], readily eliminate β-hydrogens to form alkenes as major products. They do not readily capture solvent at the cationic center. It is therefore expected that nucleophilic attack at the cationic centers of 54 and 74 will be slowed. Table 3 shows results of calculations on the γ-trimethylsilylcyclobutyl cations shown in Figure 4 at different levels of theory. The presence of the electron-withdrawing group results in an increase in the Si–C3 bond length relative to the cations 24 and 52. Also, the cross-ring C1–C3 distance is decreased. In the language of resonance theory, these features are in line with increased contributions of form 74a to the overall structure of the cation. These features suggest more facile nucleophilic attack should occur at silicon, favoring bicyclobutane formation. Also included in Table 3 are calculated bond lengths in the phosphoryl-substituted cation 74d, which also shows a very long Si–C bond. Preferred trimethylsilyl elimination from this intermediate is in line with the behavior of mesylate 76, which gives exclusively the bicyclobutane 77 on solvolysis in CH3CO2H (Scheme 15).
![[1860-5397-15-170-4]](/bjoc/content/figures/1860-5397-15-170-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Table 3: Calculated bond lengths (Å) of γ-trimethylsilyl cations.
| Cation | Bond |
B3LYP/
6-31G* |
B3LYP/
6-311+G** |
MP2/
6-31G* |
MP2/
6-311+G** |
M062X/
6-311+G** |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-170-i30.svg?max-width=637&scale=1.0)
24 |
Si–C3
C1–C3 |
1.962
1.916 |
1.959
1.914 |
1.975
1.760 |
1.970
1.759 |
1.970
1.736 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-170-i31.svg?max-width=637&scale=1.0)
52 |
Si–C3
C1–C3 |
1.999
1.717 |
1.994
1.719 |
1.990
1.665 |
1.983
1.675 |
1.984
1.652 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-170-i32.svg?max-width=637&scale=1.0)
37 |
Si–C3
C1–C3 |
2.016
1.662 |
2.013
1.659 |
2.004
1.636 |
1.998
1.645 |
2.000
1.616 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-170-i33.svg?max-width=637&scale=1.0)
74c |
Si–C3
C1–C3 |
2.018
1.658 |
2.018
1.655 |
2.009
1.625 |
2.002
1.632 |
2.007
1.601 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-15-170-i34.svg?max-width=637&scale=1.0)
74d |
Si–C3
C1–C3 |
2.013
1.663 |
2.012
1.659 |
2.008
1.624 |
2.003
1.630 |
2.004
1.602 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-15-170-i35.svg?max-width=637&scale=1.0)
74b |
Si–C3
C1–C3 |
2.046
1.694 |
2.045
1.688 |
2.037
1.652 |
2.028
1.663 |
2.031
1.623 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-15-170-i36.svg?max-width=637&scale=1.0)
54 |
Si–C3
C1–C3 |
2.034
1.646 |
2.037
1.642 |
2.024
1.616 |
2.019
1.623 |
2.024
1.595 |
![[1860-5397-15-170-i15]](/bjoc/content/inline/1860-5397-15-170-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Bicyclobutane formation from mesylate 76 in CH3CO2H.
Scheme 15: Bicyclobutane formation from mesylate 76 in CH3CO2H.
The final item to be addressed is the behavior of triflates 60 and 67 with electron-withdrawing CF3 and CN groups cis to the trimethylsilyl group. These substrates gave exclusively unrearranged substitution products 78 and 79 when reacted in CD3CO2D (Scheme 16). The lack of rearrangement products suggests that these potent electron-withdrawing groups make further rearrangement of cations 80 untenable. Indeed, M062X/6-311+G** calculations show that the potential rearranged cation 81 (E = CN) is not even an energy minimum, but a transition state.
![[1860-5397-15-170-i16]](/bjoc/content/inline/1860-5397-15-170-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Reactions of triflates 60 and 67 in CD3CO2D.
Scheme 16: Reactions of triflates 60 and 67 in CD3CO2D.
Conclusion
1-Substituted-cis-2-trimethylsilylyclopropylcarbinyl mesylates and triflates 13 solvolyze in CD3CO2D to give products derived from 3-trimethylsilylcyclobutyl cations. These cationic intermediates are stabilized by a long-range rear lobe interaction with the γ-trimethylsilyl group. When the substituent is electron-withdrawing (CF3, CN, or CO2CH3), significant amounts of bicyclobutane products are formed. The bicyclobutanes are a result of γ-trimethylsilyl elimination from the cationic intermediate. Computational studies support a carbocation intermediate with an unusually long Si–C bond, indicative of increased demand for Si–C hyperconjugation due to the electron-withdrawing group. With the exception of the phenyl substitution, the chemistry of trans-derivatives 14 is quite different since these substrates are geometrically precluded from forming γ-trimethylsilyl-stabilized cyclobutyl cations.
Supporting Information
Full experimental details, 1H and 13C NMR spectra of new compounds, and M062X/6-311+G** computational studies are presented as Supporting Information.
| Supporting Information File 1: Experimental details and 1H and 13C NMR spectra of new compounds. | ||
| Format: PDF | Size: 6.6 MB | Download |
| Supporting Information File 2: M062X/6-611+G** calculated structures, energies, and Cartesian coordinates for carbocations and transition states. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Merling, G. Ber. Dtsch. Chem. Ges. 1891, 24, 3108–3126. doi:10.1002/cber.189102402151
Return to citation in text: [1] -
von E. Doering, W.; Knox, L. H. J. Am. Chem. Soc. 1954, 76, 3203–3206. doi:10.1021/ja01641a027
Return to citation in text: [1] -
Norris, J. F.; Sanders, W. W. Am. Chem. J. 1901, 25, 54–62.
Return to citation in text: [1] -
Kehrmann, F.; Wentzel, F. Ber. Dtsch. Chem. Ges. 1901, 34, 3815–3819. doi:10.1002/cber.19010340393
Return to citation in text: [1] -
Gomberg, M. Ber. Dtsch. Chem. Ges. 1907, 40, 1847–1888. doi:10.1002/cber.19070400289
Return to citation in text: [1] -
Freedman, H. H. In Carbonium Ions; Olah, G. A.; von Ragué Schleyer, P., Eds.; Wiley Interscience: New York, U.S.A., 1973; Vol. IV, pp 1501–1578.
See for a review and leading references.
Return to citation in text: [1] -
Horn, M.; Mayr, H. J. Phys. Org. Chem. 2012, 25, 979–988. doi:10.1002/poc.2979
See for a review and leading references.
Return to citation in text: [1] -
Richey, H. G., Jr. In Carbonium Ions; Olah, G. A.; von Ragué Schleyer, P., Eds.; Wiley Interscience: New York, U.S.A., 1972; Vol. III, pp 1201–1294.
See for a leading review.
Return to citation in text: [1] -
Wiberg, K. B.; Hess, B. A., Jr.; Ashe, A. J., III. In Carbonium Ions; Olah, G. A.; von Ragué Schleyer, P., Eds.; Wiley Interscience: New York, U.S.A., 1972; Vol. III, pp 1295–1345.
See for a leading review.
Return to citation in text: [1] -
Demjanow, N. J. Ber. Dtsch. Chem. Ges. 1907, 40, 4961–4963. doi:10.1002/cber.190704004168
Return to citation in text: [1] [2] -
Roberts, J. D.; Mazur, R. H. J. Am. Chem. Soc. 1951, 73, 2509–2520. doi:10.1021/ja01150a029
Return to citation in text: [1] -
Sneen, R. A.; Lewandowski, K. M.; Taha, I. A. I.; Smith, B. R. J. Am. Chem. Soc. 1961, 83, 4843–4848. doi:10.1021/ja01484a035
Return to citation in text: [1] -
Roberts, J. D.; Mazur, R. H. J. Am. Chem. Soc. 1951, 73, 3542–3543. doi:10.1021/ja01151a550
Return to citation in text: [1] -
Caserio, M. C.; Graham, W. H.; Roberts, J. D. Tetrahedron 1960, 11, 171–182. doi:10.1016/0040-4020(60)80068-3
Return to citation in text: [1] -
von Ragué Schleyer, P.; Majerski, Z. J. Am. Chem. Soc. 1971, 93, 665–671. doi:10.1021/ja00732a019
Return to citation in text: [1] -
Olah, G. A.; Reddy, V. P.; Prakash, G. K. S. Chem. Rev. 1992, 92, 69–95. doi:10.1021/cr00009a003
Return to citation in text: [1] -
Saunders, M.; Siehl, H. U. J. Am. Chem. Soc. 1980, 102, 6868–6869. doi:10.1021/ja00542a045
Return to citation in text: [1] -
Staral, J. S.; Yavari, I.; Roberts, J. D.; Prakash, G. K. S.; Donovan, D. J.; Olah, G. A. J. Am. Chem. Soc. 1978, 100, 8016–8018. doi:10.1021/ja00493a045
Return to citation in text: [1] -
Koch, W.; Liu, B.; DeFrees, D. J. J. Am. Chem. Soc. 1988, 110, 7325–7328. doi:10.1021/ja00230a008
Return to citation in text: [1] [2] -
Siehl, H.-U. Adv. Phys. Org. Chem. 2018, 52, 1–47. doi:10.1016/bs.apoc.2018.10.001
Return to citation in text: [1] -
Creary, X. Chem. Rev. 1991, 91, 1625–1678. doi:10.1021/cr00008a001
Return to citation in text: [1] -
Gassman, P. G.; Tidwell, T. T. Acc. Chem. Res. 1983, 16, 279–285. doi:10.1021/ar00092a003
Return to citation in text: [1] [2] -
Tidwell, T. T. Angew. Chem., Int. Ed. Engl. 1984, 23, 20–32. doi:10.1002/anie.198400201
Return to citation in text: [1] -
Allen, A. D.; Tidwell, T. T. In Advances in Carbocation Chemistry; Creary, X., Ed.; Jai Press Inc.: Greenwich, CT, 1989; Vol. 1, pp 1–44.
Return to citation in text: [1] -
Gassman, P. G.; Talley, J. J. J. Am. Chem. Soc. 1980, 102, 1214–1216. doi:10.1021/ja00523a076
Return to citation in text: [1] [2] -
Gassman, P. G.; Talley, J. J. J. Am. Chem. Soc. 1980, 102, 4138–4143. doi:10.1021/ja00532a026
Return to citation in text: [1] -
Dixon, D. A.; Charlier, P. A.; Gassman, P. G. J. Am. Chem. Soc. 1980, 102, 3957–3959. doi:10.1021/ja00531a051
Return to citation in text: [1] -
Gassman, P. G.; Saito, K. Tetrahedron Lett. 1981, 22, 1311–1314. doi:10.1016/s0040-4039(01)90304-1
Return to citation in text: [1] -
Gassman, P. G.; Guggenheim, T. L. J. Org. Chem. 1982, 47, 3023–3026. doi:10.1021/jo00136a048
Return to citation in text: [1] -
Begue, J. P.; Charpentier-Morize, M. Acc. Chem. Res. 1980, 13, 207–212. doi:10.1021/ar50151a003
Return to citation in text: [1] -
Creary, X. Acc. Chem. Res. 1985, 18, 3–8. doi:10.1021/ar00109a002
Return to citation in text: [1] -
Creary, X. In Advances in Carbocation Chemistry; Creary, X., Ed.; Jai Press Inc.: Greenwich, CT, 1989; Vol. 1, pp 45–92.
Return to citation in text: [1] -
Charpentier-Morize, M.; Begue, J.-P. In Advances in Carbocation Chemistry; Creary, X., Ed.; Jai Press Inc.: Greenwich, CT, 1989; Vol. 1, pp 219–253.
Return to citation in text: [1] -
Creary, X.; Geiger, C. C.; Hilton, K. J. Am. Chem. Soc. 1983, 105, 2851–2858. doi:10.1021/ja00347a054
Return to citation in text: [1] [2] -
Creary, X.; Underiner, T. L. J. Org. Chem. 1985, 50, 2165–2170. doi:10.1021/jo00212a033
Return to citation in text: [1] -
Siehl, H.-U. In Recent Developments in Carbocation and Onium Ion Chemistry; Laali, K. K., Ed.; ACS Symposium Series No. 965; American Chemical Society: Washington, DC, 2007; pp 1–31.
See for a review and leading references.
Return to citation in text: [1] -
Siehl, H.-U.; Müller, T. In The Chemistry of Organosilicon Compounds; Rappoport, Z.; Apeloig, Y., Eds.; John Wiley and Sons: New York, U.S.A.; Vol. 2, pp 595–701.
See for a review and leading references.
Return to citation in text: [1] -
Lambert, J. B.; Zhao, Y.; Emblidge, R. W.; Salvador, L. A.; Liu, X.; So, J. H.; Chelius, E. C. Acc. Chem. Res. 1999, 32, 183–190. doi:10.1021/ar970296m
See for a review and leading references.
Return to citation in text: [1] -
Lambert, J. B.; Liu, X. J. Organomet. Chem. 1996, 521, 203–210. doi:10.1016/0022-328x(96)06228-6
See for a review and leading references.
Return to citation in text: [1] -
Lambert, J. B. Tetrahedron 1990, 46, 2677–2689. doi:10.1016/s0040-4020(01)88362-9
See for a review and leading references.
Return to citation in text: [1] -
Lambert, J. B.; Chelius, E. C. J. Am. Chem. Soc. 1990, 112, 8120–8126. doi:10.1021/ja00178a041
See for a review and leading references.
Return to citation in text: [1] -
Lambert, J. B.; Wang, G. T.; Finzel, R. B.; Teramura, D. H. J. Am. Chem. Soc. 1987, 109, 7838–7845. doi:10.1021/ja00259a036
See for a review and leading references.
Return to citation in text: [1] -
White, J. M. Aust. J. Chem. 1995, 48, 1227–1251. doi:10.1071/ch9951227
See for a review and leading references.
Return to citation in text: [1] -
Sommer, L. H.; Bailey, D. L.; Whitmore, F. C. J. Am. Chem. Soc. 1948, 70, 2869–2872. doi:10.1021/ja01189a009
See for a review and leading references.
Return to citation in text: [1] -
Creary, X.; Butchko, M. A. J. Am. Chem. Soc. 2001, 123, 1569–1578. doi:10.1021/ja002407+
Return to citation in text: [1] -
Creary, X.; Butchko, M. A. J. Org. Chem. 2001, 66, 1115–1121. doi:10.1021/jo001112b
Return to citation in text: [1] -
Creary, X.; Jiang, Z.; Butchko, M.; McLean, K. Tetrahedron Lett. 1996, 37, 579–582. doi:10.1016/0040-4039(95)02266-x
Return to citation in text: [1] -
Creary, X.; Wang, Y.-X. Tetrahedron Lett. 1989, 30, 2493–2496. doi:10.1016/s0040-4039(01)80433-0
Return to citation in text: [1] -
Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 2134–2144. doi:10.1021/jo802722z
Return to citation in text: [1] -
Creary, X.; O'Donnel, B. D.; Vervaeke, M. J. Org. Chem. 2007, 72, 3360–3368. doi:10.1021/jo062668n
Return to citation in text: [1] -
Siehl, H.-U.; Fulj, M. Pure Appl. Chem. 1998, 70, 2015–2022. doi:10.1351/pac199870102015
Return to citation in text: [1] -
Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 9044–9053. doi:10.1021/jo901821f
Return to citation in text: [1] [2] [3] [4] -
Creary, X.; Heffron, A. J. Org. Chem. 2014, 79, 2547–2555. doi:10.1021/jo500007p
Return to citation in text: [1] -
Creary, X.; Heffron, A.; Going, G.; Prado, M. J. Org. Chem. 2015, 80, 1781–1788. doi:10.1021/jo502691t
Return to citation in text: [1] -
Mercadante, M. A.; Kelly, C. B.; Hamlin, T. A.; Delle Chiaie, K. R.; Drago, M. D.; Duffy, K. K.; Dumas, M. T.; Fager, D. C.; Glod, B. L. C.; Hansen, K. E.; Hill, C. R.; Leising, R. M.; Lynes, C. L.; MacInnis, A. E.; McGohey, M. R.; Murray, S. A.; Piquette, M. C.; Roy, S. L.; Smith, R. M.; Sullivan, K. R.; Truong, B. H.; Vailonis, K. M.; Gorbatyuk, V.; Leadbeater, N. E.; Tilley, L. J. Chem. Sci. 2014, 5, 3983–3994. doi:10.1039/c4sc01732c
Return to citation in text: [1] [2] -
Shiner, V. J., Jr.; Ensinger, M. W.; Kriz, G. S. J. Am. Chem. Soc. 1986, 108, 842–844. doi:10.1021/ja00264a050
Return to citation in text: [1] [2] -
Shiner, V. J., Jr.; Ensinger, M. W.; Huffman, J. C. J. Am. Chem. Soc. 1989, 111, 7199–7205. doi:10.1021/ja00200a045
Return to citation in text: [1] -
Grob, C. A.; Gründel, M.; Sawlewicz, P. Helv. Chim. Acta 1988, 71, 1502–1507. doi:10.1002/hlca.19880710615
Return to citation in text: [1] -
Adcock, W.; Clark, C. I.; Schiesser, C. H. J. Am. Chem. Soc. 1996, 118, 11541–11547. doi:10.1021/ja961870c
Return to citation in text: [1] -
Creary, X. J. Am. Chem. Soc. 2013, 135, 6570–6578. doi:10.1021/ja400747u
Return to citation in text: [1] -
Creary, X. J. Org. Chem. 2015, 80, 11378–11387. doi:10.1021/acs.joc.5b01955
Return to citation in text: [1] -
Mascitti, V.; Corey, E. J. J. Am. Chem. Soc. 2006, 128, 3118–3119. doi:10.1021/ja058370g
Return to citation in text: [1] -
Ohta, H.; Ishizaka, T.; Tatsuzuki, M.; Yoshinaga, M.; Iida, I.; Yamaguchi, T.; Tomishima, Y.; Futaki, N.; Toda, Y.; Saito, S. Bioorg. Med. Chem. 2008, 16, 1111–1124. doi:10.1016/j.bmc.2007.10.087
Return to citation in text: [1] -
Hsee, L. C.; Sardella, D. J. Magn. Reson. Chem. 1990, 28, 688–692. doi:10.1002/mrc.1260280806
Return to citation in text: [1] -
Chen, J.; Reibenspies, J.; Derecskei-Kovacs, A.; Burgess, K. Chem. Commun. 1999, 2501–2502. doi:10.1039/a907559c
Return to citation in text: [1] -
Creary, X. J. Am. Chem. Soc. 1984, 106, 5568–5577. doi:10.1021/ja00331a029
Return to citation in text: [1] -
Jansen, M. P.; Koshy, K. M.; Mangru, N. N.; Tidwell, T. T. J. Am. Chem. Soc. 1981, 103, 3863–3867. doi:10.1021/ja00403a040
Return to citation in text: [1]
| 55. | Mercadante, M. A.; Kelly, C. B.; Hamlin, T. A.; Delle Chiaie, K. R.; Drago, M. D.; Duffy, K. K.; Dumas, M. T.; Fager, D. C.; Glod, B. L. C.; Hansen, K. E.; Hill, C. R.; Leising, R. M.; Lynes, C. L.; MacInnis, A. E.; McGohey, M. R.; Murray, S. A.; Piquette, M. C.; Roy, S. L.; Smith, R. M.; Sullivan, K. R.; Truong, B. H.; Vailonis, K. M.; Gorbatyuk, V.; Leadbeater, N. E.; Tilley, L. J. Chem. Sci. 2014, 5, 3983–3994. doi:10.1039/c4sc01732c |
| 64. | Hsee, L. C.; Sardella, D. J. Magn. Reson. Chem. 1990, 28, 688–692. doi:10.1002/mrc.1260280806 |
| 65. | Chen, J.; Reibenspies, J.; Derecskei-Kovacs, A.; Burgess, K. Chem. Commun. 1999, 2501–2502. doi:10.1039/a907559c |
| 1. | Merling, G. Ber. Dtsch. Chem. Ges. 1891, 24, 3108–3126. doi:10.1002/cber.189102402151 |
| 2. | von E. Doering, W.; Knox, L. H. J. Am. Chem. Soc. 1954, 76, 3203–3206. doi:10.1021/ja01641a027 |
| 10. | Demjanow, N. J. Ber. Dtsch. Chem. Ges. 1907, 40, 4961–4963. doi:10.1002/cber.190704004168 |
| 34. | Creary, X.; Geiger, C. C.; Hilton, K. J. Am. Chem. Soc. 1983, 105, 2851–2858. doi:10.1021/ja00347a054 |
| 35. | Creary, X.; Underiner, T. L. J. Org. Chem. 1985, 50, 2165–2170. doi:10.1021/jo00212a033 |
| 10. | Demjanow, N. J. Ber. Dtsch. Chem. Ges. 1907, 40, 4961–4963. doi:10.1002/cber.190704004168 |
| 11. | Roberts, J. D.; Mazur, R. H. J. Am. Chem. Soc. 1951, 73, 2509–2520. doi:10.1021/ja01150a029 |
| 36. |
Siehl, H.-U. In Recent Developments in Carbocation and Onium Ion Chemistry; Laali, K. K., Ed.; ACS Symposium Series No. 965; American Chemical Society: Washington, DC, 2007; pp 1–31.
See for a review and leading references. |
| 37. |
Siehl, H.-U.; Müller, T. In The Chemistry of Organosilicon Compounds; Rappoport, Z.; Apeloig, Y., Eds.; John Wiley and Sons: New York, U.S.A.; Vol. 2, pp 595–701.
See for a review and leading references. |
| 38. |
Lambert, J. B.; Zhao, Y.; Emblidge, R. W.; Salvador, L. A.; Liu, X.; So, J. H.; Chelius, E. C. Acc. Chem. Res. 1999, 32, 183–190. doi:10.1021/ar970296m
See for a review and leading references. |
| 39. |
Lambert, J. B.; Liu, X. J. Organomet. Chem. 1996, 521, 203–210. doi:10.1016/0022-328x(96)06228-6
See for a review and leading references. |
| 40. |
Lambert, J. B. Tetrahedron 1990, 46, 2677–2689. doi:10.1016/s0040-4020(01)88362-9
See for a review and leading references. |
| 41. |
Lambert, J. B.; Chelius, E. C. J. Am. Chem. Soc. 1990, 112, 8120–8126. doi:10.1021/ja00178a041
See for a review and leading references. |
| 42. |
Lambert, J. B.; Wang, G. T.; Finzel, R. B.; Teramura, D. H. J. Am. Chem. Soc. 1987, 109, 7838–7845. doi:10.1021/ja00259a036
See for a review and leading references. |
| 43. |
White, J. M. Aust. J. Chem. 1995, 48, 1227–1251. doi:10.1071/ch9951227
See for a review and leading references. |
| 44. |
Sommer, L. H.; Bailey, D. L.; Whitmore, F. C. J. Am. Chem. Soc. 1948, 70, 2869–2872. doi:10.1021/ja01189a009
See for a review and leading references. |
| 8. |
Richey, H. G., Jr. In Carbonium Ions; Olah, G. A.; von Ragué Schleyer, P., Eds.; Wiley Interscience: New York, U.S.A., 1972; Vol. III, pp 1201–1294.
See for a leading review. |
| 9. |
Wiberg, K. B.; Hess, B. A., Jr.; Ashe, A. J., III. In Carbonium Ions; Olah, G. A.; von Ragué Schleyer, P., Eds.; Wiley Interscience: New York, U.S.A., 1972; Vol. III, pp 1295–1345.
See for a leading review. |
| 22. | Gassman, P. G.; Tidwell, T. T. Acc. Chem. Res. 1983, 16, 279–285. doi:10.1021/ar00092a003 |
| 25. | Gassman, P. G.; Talley, J. J. J. Am. Chem. Soc. 1980, 102, 1214–1216. doi:10.1021/ja00523a076 |
| 26. | Gassman, P. G.; Talley, J. J. J. Am. Chem. Soc. 1980, 102, 4138–4143. doi:10.1021/ja00532a026 |
| 27. | Dixon, D. A.; Charlier, P. A.; Gassman, P. G. J. Am. Chem. Soc. 1980, 102, 3957–3959. doi:10.1021/ja00531a051 |
| 28. | Gassman, P. G.; Saito, K. Tetrahedron Lett. 1981, 22, 1311–1314. doi:10.1016/s0040-4039(01)90304-1 |
| 29. | Gassman, P. G.; Guggenheim, T. L. J. Org. Chem. 1982, 47, 3023–3026. doi:10.1021/jo00136a048 |
| 3. | Norris, J. F.; Sanders, W. W. Am. Chem. J. 1901, 25, 54–62. |
| 4. | Kehrmann, F.; Wentzel, F. Ber. Dtsch. Chem. Ges. 1901, 34, 3815–3819. doi:10.1002/cber.19010340393 |
| 5. | Gomberg, M. Ber. Dtsch. Chem. Ges. 1907, 40, 1847–1888. doi:10.1002/cber.19070400289 |
| 6. |
Freedman, H. H. In Carbonium Ions; Olah, G. A.; von Ragué Schleyer, P., Eds.; Wiley Interscience: New York, U.S.A., 1973; Vol. IV, pp 1501–1578.
See for a review and leading references. |
| 7. |
Horn, M.; Mayr, H. J. Phys. Org. Chem. 2012, 25, 979–988. doi:10.1002/poc.2979
See for a review and leading references. |
| 30. | Begue, J. P.; Charpentier-Morize, M. Acc. Chem. Res. 1980, 13, 207–212. doi:10.1021/ar50151a003 |
| 31. | Creary, X. Acc. Chem. Res. 1985, 18, 3–8. doi:10.1021/ar00109a002 |
| 32. | Creary, X. In Advances in Carbocation Chemistry; Creary, X., Ed.; Jai Press Inc.: Greenwich, CT, 1989; Vol. 1, pp 45–92. |
| 33. | Charpentier-Morize, M.; Begue, J.-P. In Advances in Carbocation Chemistry; Creary, X., Ed.; Jai Press Inc.: Greenwich, CT, 1989; Vol. 1, pp 219–253. |
| 19. | Koch, W.; Liu, B.; DeFrees, D. J. J. Am. Chem. Soc. 1988, 110, 7325–7328. doi:10.1021/ja00230a008 |
| 34. | Creary, X.; Geiger, C. C.; Hilton, K. J. Am. Chem. Soc. 1983, 105, 2851–2858. doi:10.1021/ja00347a054 |
| 16. | Olah, G. A.; Reddy, V. P.; Prakash, G. K. S. Chem. Rev. 1992, 92, 69–95. doi:10.1021/cr00009a003 |
| 17. | Saunders, M.; Siehl, H. U. J. Am. Chem. Soc. 1980, 102, 6868–6869. doi:10.1021/ja00542a045 |
| 18. | Staral, J. S.; Yavari, I.; Roberts, J. D.; Prakash, G. K. S.; Donovan, D. J.; Olah, G. A. J. Am. Chem. Soc. 1978, 100, 8016–8018. doi:10.1021/ja00493a045 |
| 19. | Koch, W.; Liu, B.; DeFrees, D. J. J. Am. Chem. Soc. 1988, 110, 7325–7328. doi:10.1021/ja00230a008 |
| 22. | Gassman, P. G.; Tidwell, T. T. Acc. Chem. Res. 1983, 16, 279–285. doi:10.1021/ar00092a003 |
| 23. | Tidwell, T. T. Angew. Chem., Int. Ed. Engl. 1984, 23, 20–32. doi:10.1002/anie.198400201 |
| 24. | Allen, A. D.; Tidwell, T. T. In Advances in Carbocation Chemistry; Creary, X., Ed.; Jai Press Inc.: Greenwich, CT, 1989; Vol. 1, pp 1–44. |
| 13. | Roberts, J. D.; Mazur, R. H. J. Am. Chem. Soc. 1951, 73, 3542–3543. doi:10.1021/ja01151a550 |
| 14. | Caserio, M. C.; Graham, W. H.; Roberts, J. D. Tetrahedron 1960, 11, 171–182. doi:10.1016/0040-4020(60)80068-3 |
| 15. | von Ragué Schleyer, P.; Majerski, Z. J. Am. Chem. Soc. 1971, 93, 665–671. doi:10.1021/ja00732a019 |
| 25. | Gassman, P. G.; Talley, J. J. J. Am. Chem. Soc. 1980, 102, 1214–1216. doi:10.1021/ja00523a076 |
| 12. | Sneen, R. A.; Lewandowski, K. M.; Taha, I. A. I.; Smith, B. R. J. Am. Chem. Soc. 1961, 83, 4843–4848. doi:10.1021/ja01484a035 |
| 20. | Siehl, H.-U. Adv. Phys. Org. Chem. 2018, 52, 1–47. doi:10.1016/bs.apoc.2018.10.001 |
| 67. | Jansen, M. P.; Koshy, K. M.; Mangru, N. N.; Tidwell, T. T. J. Am. Chem. Soc. 1981, 103, 3863–3867. doi:10.1021/ja00403a040 |
| 51. | Siehl, H.-U.; Fulj, M. Pure Appl. Chem. 1998, 70, 2015–2022. doi:10.1351/pac199870102015 |
| 45. | Creary, X.; Butchko, M. A. J. Am. Chem. Soc. 2001, 123, 1569–1578. doi:10.1021/ja002407+ |
| 46. | Creary, X.; Butchko, M. A. J. Org. Chem. 2001, 66, 1115–1121. doi:10.1021/jo001112b |
| 47. | Creary, X.; Jiang, Z.; Butchko, M.; McLean, K. Tetrahedron Lett. 1996, 37, 579–582. doi:10.1016/0040-4039(95)02266-x |
| 48. | Creary, X.; Wang, Y.-X. Tetrahedron Lett. 1989, 30, 2493–2496. doi:10.1016/s0040-4039(01)80433-0 |
| 49. | Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 2134–2144. doi:10.1021/jo802722z |
| 50. | Creary, X.; O'Donnel, B. D.; Vervaeke, M. J. Org. Chem. 2007, 72, 3360–3368. doi:10.1021/jo062668n |
| 52. | Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 9044–9053. doi:10.1021/jo901821f |
| 52. | Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 9044–9053. doi:10.1021/jo901821f |
| 62. | Mascitti, V.; Corey, E. J. J. Am. Chem. Soc. 2006, 128, 3118–3119. doi:10.1021/ja058370g |
| 63. | Ohta, H.; Ishizaka, T.; Tatsuzuki, M.; Yoshinaga, M.; Iida, I.; Yamaguchi, T.; Tomishima, Y.; Futaki, N.; Toda, Y.; Saito, S. Bioorg. Med. Chem. 2008, 16, 1111–1124. doi:10.1016/j.bmc.2007.10.087 |
| 52. | Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 9044–9053. doi:10.1021/jo901821f |
| 56. | Shiner, V. J., Jr.; Ensinger, M. W.; Kriz, G. S. J. Am. Chem. Soc. 1986, 108, 842–844. doi:10.1021/ja00264a050 |
| 60. | Creary, X. J. Am. Chem. Soc. 2013, 135, 6570–6578. doi:10.1021/ja400747u |
| 61. | Creary, X. J. Org. Chem. 2015, 80, 11378–11387. doi:10.1021/acs.joc.5b01955 |
| 52. | Creary, X.; Kochly, E. D. J. Org. Chem. 2009, 74, 9044–9053. doi:10.1021/jo901821f |
| 53. | Creary, X.; Heffron, A. J. Org. Chem. 2014, 79, 2547–2555. doi:10.1021/jo500007p |
| 54. | Creary, X.; Heffron, A.; Going, G.; Prado, M. J. Org. Chem. 2015, 80, 1781–1788. doi:10.1021/jo502691t |
| 55. | Mercadante, M. A.; Kelly, C. B.; Hamlin, T. A.; Delle Chiaie, K. R.; Drago, M. D.; Duffy, K. K.; Dumas, M. T.; Fager, D. C.; Glod, B. L. C.; Hansen, K. E.; Hill, C. R.; Leising, R. M.; Lynes, C. L.; MacInnis, A. E.; McGohey, M. R.; Murray, S. A.; Piquette, M. C.; Roy, S. L.; Smith, R. M.; Sullivan, K. R.; Truong, B. H.; Vailonis, K. M.; Gorbatyuk, V.; Leadbeater, N. E.; Tilley, L. J. Chem. Sci. 2014, 5, 3983–3994. doi:10.1039/c4sc01732c |
| 56. | Shiner, V. J., Jr.; Ensinger, M. W.; Kriz, G. S. J. Am. Chem. Soc. 1986, 108, 842–844. doi:10.1021/ja00264a050 |
| 57. | Shiner, V. J., Jr.; Ensinger, M. W.; Huffman, J. C. J. Am. Chem. Soc. 1989, 111, 7199–7205. doi:10.1021/ja00200a045 |
| 58. | Grob, C. A.; Gründel, M.; Sawlewicz, P. Helv. Chim. Acta 1988, 71, 1502–1507. doi:10.1002/hlca.19880710615 |
| 59. | Adcock, W.; Clark, C. I.; Schiesser, C. H. J. Am. Chem. Soc. 1996, 118, 11541–11547. doi:10.1021/ja961870c |
© 2019 Creary; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)