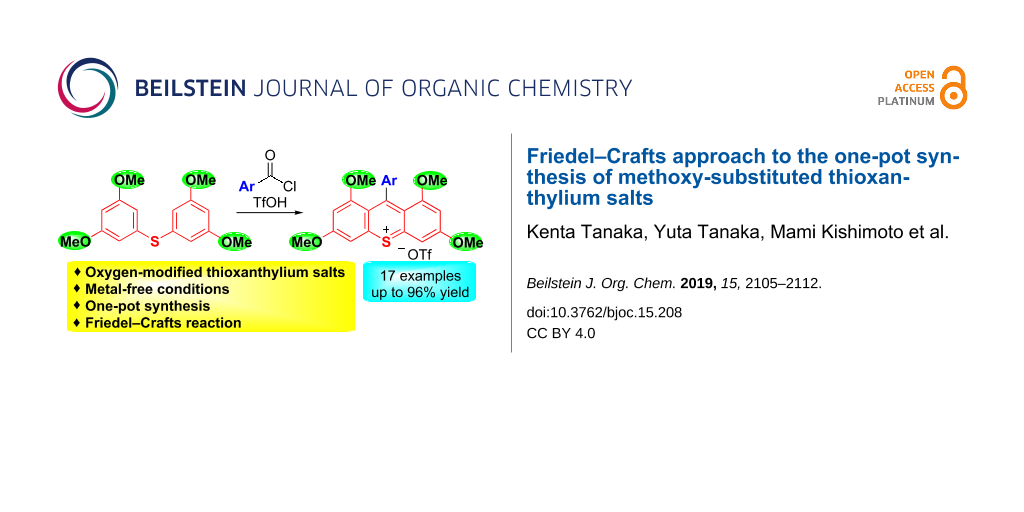
Abstract
An efficient synthesis of methoxy-substituted thioxanthylium salts has been developed. The reaction of diaryl sulfides with benzoyl chlorides in the presence of TfOH smoothly proceeded to give the desired thioxanthylium salts in good yields. In their UV–vis spectra, the maximum absorption wavelengths of methoxy-functionalized thioxanthylium salts were observed at around 460 nm, which show a drastic red shift compared to the parent thioxanthylium salts. The present reaction provides a versatile access to functionalized thioxanthylium salts, and therefore it constitutes a promising tool for the synthesis of biologically and photochemically active molecules.

Graphical Abstract
Introduction
Thioxanthylium salts are one of the important structural motifs found in biologically active compounds and photochemical materials [1-8]. Owing to these useful properties, several research groups have developed methodologies to synthesize them. The typical synthetic methods for thioxanthylium salts include the reaction of thioxanthone with aryl bromide in the presence of n-butyllithium or Grignard reagents followed by dehydration by acids such as hexafluorophosphoric acid (Scheme 1a and 1b) [3,4,9,10], oxidation of thioxanthene in the presence of PbO2 followed by dehydration by tetrafluoroboric acid [1], the reaction of 4,4’-bis(dimethylamino)diphenylmethane with sulfur in the presence of ZnCl2 [11], and the ring-closure reaction of diaryl sulfide in the presence of a Lewis acid such as SnCl4 and AlCl3 [12-14]. While these reactions were proven to be useful, they require the use of stoichiometric amounts of metals and/or toxic metal reagents. Moreover, there are only a few methods for the synthesis of thioxanthylium salts despite their useful active properties. Thus, developing efficient synthetic routes and more economic approaches is highly desirable. In addition, only amino groups were introduced to the thioxanthylium core except at the 9-position of the thioxanthylium salt (Scheme 1b), and the physical properties of these substituted compounds have not been examined.
![[1860-5397-15-208-i1]](/bjoc/content/inline/1860-5397-15-208-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The representative synthesis of thioxanthylium salts.
Scheme 1: The representative synthesis of thioxanthylium salts.
We have developed the synthesis of multisubstituted condensed heterocyclic compounds in the presence of an acid catalyst [15-23]. More recently, we have reported the design and synthesis of thioxanthylium organophotoredox catalysts, which can work under green light irradiation [24,25]. In the course of this study, we found that these thioxanthylium photocatalysts efficiently oxidized styrene derivatives such as trans-anethole, and promoted radical cation Diels–Alder reactions. Based on the background mentioned above, in order to expand the utility of the synthesis of thioxanthylium salts and investigate their physical properties, we report the Friedel–Crafts approach as an efficient synthetic method of methoxy-substituted thioxanthylium salts (Scheme 1c).
Results and Discussion
Initially, we screened the reaction of bis(3,5-dimethoxyphenyl) sulfide (1a) with benzoyl chloride (2a) in the presence of Brønsted acids in chlorobenzene at several temperatures (Table 1). When we used a strong Brønsted acid such as trifluoromethanesulfonic acid (TfOH) at room temperature, the desired thioxanthylium salt 3a was obtained with 21% yield while other typical Brønsted acids did not work efficiently (Table 1, entries 1–8) [2,9,10]. At 60 °C, the yield effectively improved to 60% (Table, entry 9). Moreover, when the reaction temperature was increased to 90 °C, 120 °C, and reflux, higher yields were observed, especially under reflux conditions providing the product 3a in 82% yield (Table 1, entries 10–12). Decreasing the amount of TfOH did not improve the yield (Table 1, entry 13). It is suggested that the cyclization and dehydration were efficiently promoted at high temperature. Fortunately, when the reaction was carried out with benzoic acid, which is a more easily available substrate in comparison with benzyl chloride, the desired product was obtained in good yield. It was found that the reaction can be applied to not only benzoyl chloride but also to benzoic acid.
Table 1: Optimization of the reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-208-i2.svg?max-width=637&scale=1.0)
|
|||
| Entry | Brønsted acidb | Temperature (°C) | Yield (%) |
| 1 | H3PO4 | rt | 0 |
| 2 | HCl | rt | 0 |
| 3 | TsOH | rt | 0 |
| 4 | MsOH | rt | 0 |
| 5 | HBF4 | rt | 0 |
| 6 | HPF6 | rt | 0 |
| 7 | HClO4 | rt | traces |
| 8 | TfOH | rt | 21 |
| 9 | TfOH | 60 | 60 |
| 10 | TfOH | 90 | 72 |
| 11 | TfOH | 120 | 75 |
| 12 | TfOH | reflux | 82 |
| 13c | TfOH | reflux | 78 |
| 14d | TfOH | reflux | 72 |
aAll reactions were carried out with 1a (0.25 mmol), 2a (0.75 mmol), acid (3.0 equiv) in chlorobenzene (5.0 mL) for 1 h under N2. bH3PO4 (85% aq), HCl (0.5 M in MeOH), HBF4 (42% aq), HPF6 (65% aq), HClO4 (70% aq). cTfOH (2.0 equiv) was used. dbenzoic acid (0.75 mmol) was used instead of benzoyl chloride (2a).
With the optimized conditions in hand, we investigated the generality of diaryl sulfide 1 and benzoyl chloride 2 (Figure 1). o-Toluoyl chloride smoothly afforded the desired product 3b in excellent yield. Moreover, the reaction was performed on the 2 mmol scale to furnish the desired product in 73% yield, suggesting that the reaction can be applied to large scale conditions. In addition, 2-methoxy and 2-trifluoromethyl-functionalized benzoyl chloride can be applied to the reaction (3c,d). The 4-methoxy group was also tolerated in the reaction (3e). Substrates with strong electron-withdrawing groups such as 4-trifluoromethyl, 4-nitro and 4-cyano groups reacted with moderate to excellent yields (3f–h). The benzoyl chlorides bearing a variety of halogens were suitable for this reaction (3i–n). Although naphthalene is a sterically large group, the reaction proceeded smoothly (3o). The diaryl sulfide with ethoxy substituents furnished the corresponding product in moderate yield (3p). Interestingly, when bis(3,4-dimethoxyphenyl) sulfide was used as a substrate, the reaction proceeded to afford the desired 2,3,6,7-tetramethoxy-substituted thioxanthylium salt (3q). It was found that the present reaction can be applied to various benzoyl chlorides bearing either electron-donating or electron-withdrawing groups.
![[1860-5397-15-208-1]](/bjoc/content/figures/1860-5397-15-208-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The generality of diaryl sulfide 1 and benzoyl chloride 2. aThe reaction was carried out with 1a (2.0 mmol), 2a (6.0 mmol), TfOH (3.0 equiv) in chlorobenzene (40.0 mL) at reflux for 1 h under N2. b2 (2.0 equiv) and TfOH (2.0 equiv) were used at 120 °C. c2 (2.0 equiv) and TfOH (2.0 equiv) were used. d20 h. e2 h.
Figure 1: The generality of diaryl sulfide 1 and benzoyl chloride 2. aThe reaction was carried out with 1a (2...
Subsequently, we measured the UV–vis spectra of thioxanthylium salts. As shown in Figure 2, almost all measured UV–vis spectra are nearly identical in spite of different substituents on the benzene ring at the 9-position of the thioxanthylium core. Moreover, when the solvent effects were examined using MeCN, CH3NO2, DMSO and MeOH, no substantial shifts of the main peak at around 460 nm in the UV–vis absorption spectra were observed [24], indicating that the main absorption of these catalysts would be due to π–π* transition, which is supported by DFT calculations (TD-DFT B3LYP method) (Figure 3a,b). Based on these calculations, it was found that tetramethoxy substituents at the thioxanthylium core lead to an up-shift of both HOMO/LUMO energy levels compared to thioxanthylium salts without methoxy groups (Figure 3c,d). The maximum absorption wavelength of thioxanthylium salt 3b (λmax = 464 nm) showed a large red shift compared to thioxanthylium salt 4b (λmax = 383 nm), which has no methoxy groups (Figure 4 and Figure 5).
![[1860-5397-15-208-2]](/bjoc/content/figures/1860-5397-15-208-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The UV–vis spectra of thioxanthylium salt (0.1 mM) in CH3CN.
Figure 2: The UV–vis spectra of thioxanthylium salt (0.1 mM) in CH3CN.
![[1860-5397-15-208-3]](/bjoc/content/figures/1860-5397-15-208-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Frontier orbitals of thioxanthylium salts, calculated by DFT at the B3LYP/6-31G(d,p) level of Orca. (a) LUMO localization of 3a (energy level: −5.745 eV), (b) HOMO localization of 3a (energy level: −8.842 eV), (c) LUMO localization of 4a (energy level: −6.812 eV), (d) HOMO localization of 4a (energy level: −9.788 eV).
Figure 3: Frontier orbitals of thioxanthylium salts, calculated by DFT at the B3LYP/6-31G(d,p) level of Orca....
![[1860-5397-15-208-4]](/bjoc/content/figures/1860-5397-15-208-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis spectra of thioxanthylium salts 3b and 4b (0.1 mM) in CH3CN.
Figure 4: UV–vis spectra of thioxanthylium salts 3b and 4b (0.1 mM) in CH3CN.
![[1860-5397-15-208-5]](/bjoc/content/figures/1860-5397-15-208-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Structure of thioxanthylium salt 4.
Figure 5: Structure of thioxanthylium salt 4.
Finally, we measured the cyclic voltammograms (CV) of thioxanthylium salts 3b and 4b (Figure 6). The CV data analysis implies that the reduction potential of 3b (E°’ = −0.79 V vs Fc/Fc+) afforded a negative shift compared to 4b (E°’ = −0.56 V vs Fc/Fc+). It is obviously indicated that the methoxy groups lower the reduction potential by their strong electron-donating effect.
![[1860-5397-15-208-6]](/bjoc/content/figures/1860-5397-15-208-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Cyclic voltammograms of thioxanthylium salts 3b and 4b.
Figure 6: Cyclic voltammograms of thioxanthylium salts 3b and 4b.
Conclusion
We have developed the Friedel–Crafts approach as an efficient method to synthesize oxygen-modified thioxanthylium salts. When the reaction of diaryl sulfide with benzoyl chloride in the presence of TfOH was carried out under reflux conditions in chlorobenzene, the desired thioxanthylium salt was obtained in good yield. A variety of benzoyl chlorides bearing both electron-donating and electron-withdrawing groups can be applied to the reaction. It was found that the main absorption of thioxanthylium salts around 460 nm in UV–vis spectra would be due to π–π* transitions, which was supported by DFT calculations. The present reaction provides a versatile access to functionalized thioxanthylium salts, and therefore constitutes a promising tool for the synthesis of biologically and photochemically active molecules.
Experimental
General
Infrared (IR) spectra were recorded on a JASCO FT/IR-4100 spectrophotometer. 1H NMR spectra were recorded on a Bruker DRX-300 (300 MHz) spectrometer, a Bruker DRX-500 (500 MHz) spectrometer or a JEOL JNM ECA-500 (500 MHz) spectrometer with tetramethylsilane (TMS) as internal standard. Chemical shifts are reported in ppm from TMS. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants, integration. 13C NMR spectra were recorded on a Bruker DRX-500 (126 MHz) or a JEOL JNM ECA-500 (126 MHz) spectrometer with complete proton decoupling. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3: δ 77.0). 19F NMR spectra were recorded on a JEOL JNM AL-400 (376 MHz) or a JEOL JNM ECA-500 (471 MHz) spectrometer with hexafluorobenzene (C6F6: δ −164.9 ppm) as internal standard. High-resolution mass spectra (HRMS) were obtained with a Hitachi Nanofrontier LD Spectrometer (ESI/TOF). Elemental analyses of carbon, hydrogen, nitrogen, and sulfur were performed with a CHNOS Elemental Analyzer Vario ELIII Elemental (Elementar Co.). Column chromatography was carried out with Cicareagent silica gel 60 N (spherical, particle size 63–210 mm). Thin-layer chromatography (TLC) was carried out with Merck TLC plates with silica gel 60 F254. Unless otherwise noted, reagents were commercially available and were used without further purification. The UV absorption spectra were measured with a JASCO V-630 spectrometer. Cyclic voltammetry measurements were carried out with a computer-controlled potentiostat Model 660C (ALS Co., Ltd.).
General procedure for the synthesis of thioxanthylium salt 3
Analogously as described in [24] thioxanthylium salts 3a–q were prepared according to the following procedure.
A solution of diaryl sulfide 1 (0.25 mmol) and benzoyl chloride 2 (0.75 mmol) in chlorobenzene (5.0 mL) was placed in a 50 mL recovery flask under N2. Trifluoromethanesulfonic acid (0.75 mmol) was slowly added to the solution, which was heated to reflux for 1 h. The solution was cooled to room temperature and excess Et2O was added to precipitate a solid. After stirred for 1 h, the mixture was filtered. The solid was washed with Et2O and dried in vacuo, affording the desired thioxanthylium salt 3.
9-Phenyl-1,3,6,8-tetramethoxythioxanthylium trifluoromethanesulfonate (3a)
Red solid (0.1109 g, 82% yield). 1H NMR (500 MHz, CDCl3) δ 7.51 (d, J = 2.2 Hz, 2H), 7.44–7.38 (m, 3H), 7.14–7.10 (m, 2H), 6.51 (d, J = 2.5 Hz, 2H), 4.15 (s, 6H), 3.37 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 168.3. 165.5. 165.2. 147.8. 142.2. 127.2. 127.0. 125.6. 116.9. 101.8. 101.4. 57.7. 56.7; 19F NMR (376 MHz, CDCl3) δ −81.3; IR (ATR): 1585, 1219, 1143, 1026, 634 cm−1; HRMS (ESI+) m/z: [M]+ calcd for C23H21O4S, 393.1155; found, 393.1171; anal. calcd for C24H21F3O7S2: C, 53.13; H, 3.90. found: C, 52.80; H, 4.001.
For the synthesis of 9-(naphthalene-1-yl)-1,3,6,8-tetramethoxythioxanthylium trifluoromethanesulfonate (3o)
To a solution of 1-naphthoic acid (0.1746 g, 1.0 mmol) in dry CH2Cl2 (2.5 mL) at 0 °C under N2 (COCl)2 (0.100 mL, 1.2 mmol) was dropwise added. After the addition of a catalytic amount of dry DMF (2 drops), the solution was allowed to warm to room temperature, and stirred at that temperature for 2 h. The reaction mixture was concentrated in vacuo to afford the corresponding crude acid chloride. After the residue was dissolved in chlorobenzene, bis(3,5-dimethoxyphenyl) sulfide 1a (0.1005 g, 0.33 mmol) and trifluoromethanesulfonic acid (0.087 mL, 0.99 mmol) were added. The reaction temperature was increased to reflux and the solution was stirred for 1 h. The solution was cooled to room temperature and excess Et2O was added. After stirring for 1 h, the solution was filtered, and the solid was washed with Et2O and dried in vacuo to afford the desired thioxanthylium 3o (0.1420 g, 73% yield). Red solid. 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 8.2 Hz, 1H), 7.88 (d, J = 8.2 Hz, 1H), 7.56 (d, J = 2.2 Hz, 2H), 7.53–7.45 (m, 2H), 7.36–7.32 (m, 1H), 7.28 (d, J = 8.0 Hz, 1H), 6.93 (dd, J = 7.1, 1.1 Hz, 1H), 6.43 (d, J = 2.2 Hz, 2H), 4.15 (s, 6H), 2.96 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 168.4, 165.2, 164.7, 147.6, 140.7, 132.1, 132.0, 128.2, 127.5, 126.4, 125.9, 125.0, 124.4, 121.3, 117.6, 102.0, 101.5, 57.7, 56.6; 19F NMR (376 MHz, CDCl3) δ −81.3; IR (ATR): 1593, 1245, 1148, 1026, 634 cm−1; HRMS (ESI+) m/z: [M]+ calcd for C27H23O4S, 443.1312; found, 443.1316.
Supporting Information
| Supporting Information File 1: Copies of 1H and 13C NMR spectra, procedures for the synthesis of diaryl sulfides and thioxanthylium 4, computational data, absorption spectra, and cyclic voltammetry data. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Wu, D.; Pisula, W.; Haberecht, M. C.; Feng, X.; Müllen, K. Org. Lett. 2009, 11, 5686–5689. doi:10.1021/ol902366y
Return to citation in text: [1] [2] -
Gannon, M. K., II; Holt, J. J.; Bennett, S. M.; Wetzel, B. R.; Loo, T. W.; Bartlett, M. C.; Clarke, D. M.; Sawada, G. A.; Higgins, J. W.; Tombline, G.; Raub, T. J.; Detty, M. R. J. Med. Chem. 2009, 52, 3328–3341. doi:10.1021/jm900253g
Return to citation in text: [1] [2] -
McKnight, R. E.; Onogul, B.; Polasani, S. R.; Gannon, M. K., II; Detty, M. R. Bioorg. Med. Chem. 2008, 16, 10221–10227. doi:10.1016/j.bmc.2008.10.051
Return to citation in text: [1] [2] -
Gibson, S. L.; Holt, J. J.; Ye, M.; Donnelly, D. J.; Ohulchanskyy, T. Y.; You, Y.; Detty, M. R. Bioorg. Med. Chem. 2005, 13, 6394–6403. doi:10.1016/j.bmc.2005.06.056
Return to citation in text: [1] [2] -
Freund, T.; Müllen, K.; Scherf, U. Macromolecules 1995, 28, 547–551. doi:10.1021/ma00106a020
Return to citation in text: [1] -
Nishida, J.-i.; Miyagawa, T.; Yamashita, Y. Org. Lett. 2004, 6, 2523–2526. doi:10.1021/ol049216m
Return to citation in text: [1] -
Suzuki, T.; Nishida, J.-i.; Ohkita, M.; Tsuji, T. Angew. Chem., Int. Ed. 2000, 39, 1804–1806. doi:10.1002/(sici)1521-3773(20000515)39:10<1804::aid-anie1804>3.0.co;2-3
Return to citation in text: [1] -
Okada, K.; Imakura, T.; Oda, M.; Kajiwara, A.; Kamachi, M.; Yamaguchi, M. J. Am. Chem. Soc. 1997, 119, 5740–5741. doi:10.1021/ja9635084
Return to citation in text: [1] -
Hagel, M.; Liu, J.; Muth, O.; Estevez Rivera, H. J.; Schwake, E.; Sripanom, L.; Henkel, G.; Dyker, G. Eur. J. Org. Chem. 2007, 3573–3582. doi:10.1002/ejoc.200700177
Return to citation in text: [1] [2] -
Erabi, T.; Asahara, M.; Miyamoto, M.; Goto, K.; Wada, M. Bull. Chem. Soc. Jpn. 2002, 75, 1325–1332. doi:10.1246/bcsj.75.1325
Return to citation in text: [1] [2] -
Nealey, R. H.; Driscoll, J. S. J. Heterocycl. Chem. 1966, 3, 228–229. doi:10.1002/jhet.5570030227
Return to citation in text: [1] -
Ashby, J.; Ayad, M.; Meth-Cohn, O. J. Chem. Soc., Perkin Trans. 1 1973, 1104–1107. doi:10.1039/p19730001104
Return to citation in text: [1] -
Krollpfeiffer, F.; Wißner, A. Justus Liebigs Ann. Chem. 1951, 572, 195–211. doi:10.1002/jlac.19515720116
Return to citation in text: [1] -
Rudorf, W.-D. Sci. Synth. 2003, 14, 719–771.
Return to citation in text: [1] -
Tanaka, K.; Kishimoto, M.; Ohtsuka, N.; Iwama, Y.; Wada, H.; Hoshino, Y.; Honda, K. Synlett 2019, 30, 189–192. doi:10.1055/s-0037-1611361
Return to citation in text: [1] -
Tanaka, K.; Hoshino, Y.; Honda, K. J. Synth. Org. Chem., Jpn. 2018, 76, 1341–1351. doi:10.5059/yukigoseikyokaishi.76.1341
Return to citation in text: [1] -
Tanaka, K.; Sukekawa, M.; Hoshino, Y.; Honda, K. Chem. Lett. 2018, 47, 440–443. doi:10.1246/cl.171124
Return to citation in text: [1] -
Tanaka, K.; Sukekawa, M.; Kishimoto, M.; Hoshino, Y.; Honda, K. Heterocycles 2019, 99, 145–170. doi:10.3987/com-18-s(f)5
Return to citation in text: [1] -
Tanaka, K.; Kishimoto, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 1841–1845. doi:10.1016/j.tetlet.2018.03.090
Return to citation in text: [1] -
Tanaka, K.; Sukekawa, M.; Shigematsu, Y.; Hoshino, Y.; Honda, K. Tetrahedron 2017, 73, 6456–6464. doi:10.1016/j.tet.2017.09.045
Return to citation in text: [1] -
Tanaka, K.; Hoshino, Y.; Honda, K. Heterocycles 2017, 95, 474–486. doi:10.3987/com-16-s(s)38
Return to citation in text: [1] -
Tanaka, K.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2016, 57, 2448–2450. doi:10.1016/j.tetlet.2016.04.086
Return to citation in text: [1] -
Tanaka, K.; Shigematsu, Y.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2016, 57, 5914–5918. doi:10.1016/j.tetlet.2016.11.076
Return to citation in text: [1] -
Tanaka, K.; Kishimoto, M.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 3361–3364. doi:10.1016/j.tetlet.2018.07.058
Return to citation in text: [1] [2] [3] -
Tanaka, K.; Omata, D.; Asada, Y.; Hoshino, Y.; Honda, K. J. Org. Chem. 2019. doi:10.1021/acs.joc.9b01156
Return to citation in text: [1]
| 1. | Wu, D.; Pisula, W.; Haberecht, M. C.; Feng, X.; Müllen, K. Org. Lett. 2009, 11, 5686–5689. doi:10.1021/ol902366y |
| 2. | Gannon, M. K., II; Holt, J. J.; Bennett, S. M.; Wetzel, B. R.; Loo, T. W.; Bartlett, M. C.; Clarke, D. M.; Sawada, G. A.; Higgins, J. W.; Tombline, G.; Raub, T. J.; Detty, M. R. J. Med. Chem. 2009, 52, 3328–3341. doi:10.1021/jm900253g |
| 3. | McKnight, R. E.; Onogul, B.; Polasani, S. R.; Gannon, M. K., II; Detty, M. R. Bioorg. Med. Chem. 2008, 16, 10221–10227. doi:10.1016/j.bmc.2008.10.051 |
| 4. | Gibson, S. L.; Holt, J. J.; Ye, M.; Donnelly, D. J.; Ohulchanskyy, T. Y.; You, Y.; Detty, M. R. Bioorg. Med. Chem. 2005, 13, 6394–6403. doi:10.1016/j.bmc.2005.06.056 |
| 5. | Freund, T.; Müllen, K.; Scherf, U. Macromolecules 1995, 28, 547–551. doi:10.1021/ma00106a020 |
| 6. | Nishida, J.-i.; Miyagawa, T.; Yamashita, Y. Org. Lett. 2004, 6, 2523–2526. doi:10.1021/ol049216m |
| 7. | Suzuki, T.; Nishida, J.-i.; Ohkita, M.; Tsuji, T. Angew. Chem., Int. Ed. 2000, 39, 1804–1806. doi:10.1002/(sici)1521-3773(20000515)39:10<1804::aid-anie1804>3.0.co;2-3 |
| 8. | Okada, K.; Imakura, T.; Oda, M.; Kajiwara, A.; Kamachi, M.; Yamaguchi, M. J. Am. Chem. Soc. 1997, 119, 5740–5741. doi:10.1021/ja9635084 |
| 12. | Ashby, J.; Ayad, M.; Meth-Cohn, O. J. Chem. Soc., Perkin Trans. 1 1973, 1104–1107. doi:10.1039/p19730001104 |
| 13. | Krollpfeiffer, F.; Wißner, A. Justus Liebigs Ann. Chem. 1951, 572, 195–211. doi:10.1002/jlac.19515720116 |
| 14. | Rudorf, W.-D. Sci. Synth. 2003, 14, 719–771. |
| 11. | Nealey, R. H.; Driscoll, J. S. J. Heterocycl. Chem. 1966, 3, 228–229. doi:10.1002/jhet.5570030227 |
| 1. | Wu, D.; Pisula, W.; Haberecht, M. C.; Feng, X.; Müllen, K. Org. Lett. 2009, 11, 5686–5689. doi:10.1021/ol902366y |
| 3. | McKnight, R. E.; Onogul, B.; Polasani, S. R.; Gannon, M. K., II; Detty, M. R. Bioorg. Med. Chem. 2008, 16, 10221–10227. doi:10.1016/j.bmc.2008.10.051 |
| 4. | Gibson, S. L.; Holt, J. J.; Ye, M.; Donnelly, D. J.; Ohulchanskyy, T. Y.; You, Y.; Detty, M. R. Bioorg. Med. Chem. 2005, 13, 6394–6403. doi:10.1016/j.bmc.2005.06.056 |
| 9. | Hagel, M.; Liu, J.; Muth, O.; Estevez Rivera, H. J.; Schwake, E.; Sripanom, L.; Henkel, G.; Dyker, G. Eur. J. Org. Chem. 2007, 3573–3582. doi:10.1002/ejoc.200700177 |
| 10. | Erabi, T.; Asahara, M.; Miyamoto, M.; Goto, K.; Wada, M. Bull. Chem. Soc. Jpn. 2002, 75, 1325–1332. doi:10.1246/bcsj.75.1325 |
| 24. | Tanaka, K.; Kishimoto, M.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 3361–3364. doi:10.1016/j.tetlet.2018.07.058 |
| 2. | Gannon, M. K., II; Holt, J. J.; Bennett, S. M.; Wetzel, B. R.; Loo, T. W.; Bartlett, M. C.; Clarke, D. M.; Sawada, G. A.; Higgins, J. W.; Tombline, G.; Raub, T. J.; Detty, M. R. J. Med. Chem. 2009, 52, 3328–3341. doi:10.1021/jm900253g |
| 9. | Hagel, M.; Liu, J.; Muth, O.; Estevez Rivera, H. J.; Schwake, E.; Sripanom, L.; Henkel, G.; Dyker, G. Eur. J. Org. Chem. 2007, 3573–3582. doi:10.1002/ejoc.200700177 |
| 10. | Erabi, T.; Asahara, M.; Miyamoto, M.; Goto, K.; Wada, M. Bull. Chem. Soc. Jpn. 2002, 75, 1325–1332. doi:10.1246/bcsj.75.1325 |
| 24. | Tanaka, K.; Kishimoto, M.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 3361–3364. doi:10.1016/j.tetlet.2018.07.058 |
| 25. | Tanaka, K.; Omata, D.; Asada, Y.; Hoshino, Y.; Honda, K. J. Org. Chem. 2019. doi:10.1021/acs.joc.9b01156 |
| 15. | Tanaka, K.; Kishimoto, M.; Ohtsuka, N.; Iwama, Y.; Wada, H.; Hoshino, Y.; Honda, K. Synlett 2019, 30, 189–192. doi:10.1055/s-0037-1611361 |
| 16. | Tanaka, K.; Hoshino, Y.; Honda, K. J. Synth. Org. Chem., Jpn. 2018, 76, 1341–1351. doi:10.5059/yukigoseikyokaishi.76.1341 |
| 17. | Tanaka, K.; Sukekawa, M.; Hoshino, Y.; Honda, K. Chem. Lett. 2018, 47, 440–443. doi:10.1246/cl.171124 |
| 18. | Tanaka, K.; Sukekawa, M.; Kishimoto, M.; Hoshino, Y.; Honda, K. Heterocycles 2019, 99, 145–170. doi:10.3987/com-18-s(f)5 |
| 19. | Tanaka, K.; Kishimoto, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 1841–1845. doi:10.1016/j.tetlet.2018.03.090 |
| 20. | Tanaka, K.; Sukekawa, M.; Shigematsu, Y.; Hoshino, Y.; Honda, K. Tetrahedron 2017, 73, 6456–6464. doi:10.1016/j.tet.2017.09.045 |
| 21. | Tanaka, K.; Hoshino, Y.; Honda, K. Heterocycles 2017, 95, 474–486. doi:10.3987/com-16-s(s)38 |
| 22. | Tanaka, K.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2016, 57, 2448–2450. doi:10.1016/j.tetlet.2016.04.086 |
| 23. | Tanaka, K.; Shigematsu, Y.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2016, 57, 5914–5918. doi:10.1016/j.tetlet.2016.11.076 |
| 24. | Tanaka, K.; Kishimoto, M.; Sukekawa, M.; Hoshino, Y.; Honda, K. Tetrahedron Lett. 2018, 59, 3361–3364. doi:10.1016/j.tetlet.2018.07.058 |
© 2019 Tanaka et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)