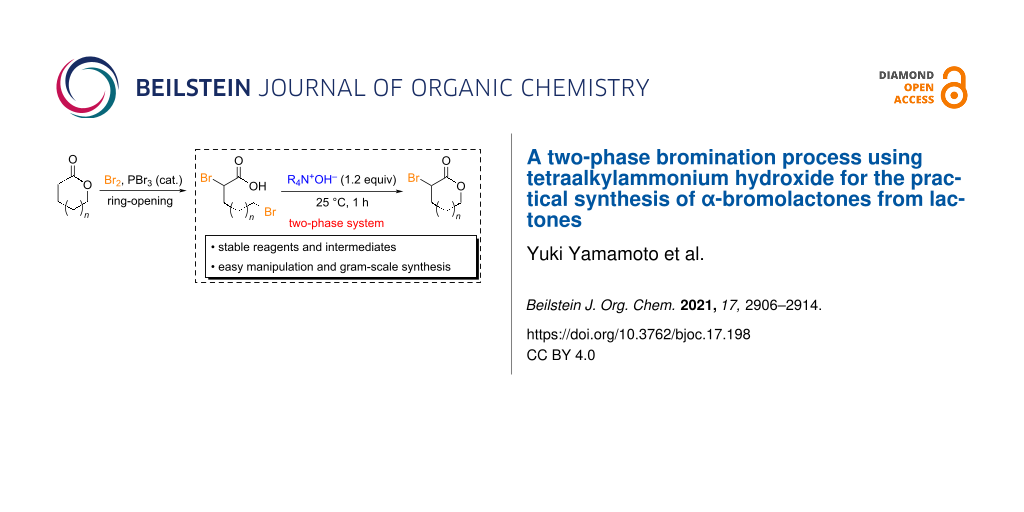
Abstract
A simple and efficient method for α-brominating lactones that affords α-bromolactones under mild conditions using tetraalkylammonium hydroxide (R4N+OH−) as a base was developed. Lactones are ring-opened with Br2 and a substoichiometric amount of PBr3, leading to good yields of the corresponding α-bromocarboxylic acids. Subsequent intramolecular cyclization over 1 h using a two-phase system (H2O/CHCl3) containing R4N+OH− afforded α-bromo lactones in good yields. This method can be applied at the 10 mmol scale using simple operations. α-Bromo-δ-valerolactone, which is extremely reactive and difficult to isolate, could be isolated and stored in a freezer for about one week using the developed method. Optimizing the solvent for environmentally friendly large-scale syntheses revealed that methyl ethyl ketone (MEK) was as effective. In addition, in situ-generated α-bromo-δ-valerolactone was directly converted into a sulfur-substituted functional lactone without difficulty by reacting it with a sulfur nucleophile in one pot without isolation. This new bromination system is expected to facilitate the industrial use of α-bromolactones as important intermediates.

Graphical Abstract
Introduction
Lactones are important heterocycles in the organic chemistry, materials science, and medicinal chemistry fields, and bromolactones are important synthetic intermediates for selectively, effectively, and practically introducing lactone units into organic molecules [1-18]. Among brominated lactones, the α-bromolactone, in which the bromine atom is located at the α-position relative to the carbonyl group, is the most versatile synthetic intermediate [19-28]. α-Bromolactones are widely used as synthetic intermediates for functional materials and pharmaceuticals, as well as initiators in atom-transfer living radical polymerization (ATRP) reactions and functional polymer synthesis [29-34].
Although α-bromo-γ-butyrolactone, which is a five-membered lactone, is easily accessible from the five-membered lactone by some bromination methods [35,36], the bromination method for the six-membered lactone, δ-valerolactone (1a), has been limited. The main process for the bromination of δ-valerolactone (1a) was treating the lactone with lithium diisopropylamide (LDA) at −78 °C to first generate the corresponding enolate, trapping it with trimethylsilyl chloride (TMSCl) to form the enol silyl ether, followed by reaction with bromine (Scheme 1a) [37,38]. While the industrial demand for α-bromolactones has grown in recent years, the above-mentioned laboratory-level synthetic methods are not suitable for scale-up because LDA, TMSCl, and enol silyl ethers are sensitive to moisture and air, as well as being expensive for large-scale syntheses. Therefore, the development of an innovative, cost-effective method for the production of α-bromolactones in large quantities is highly desirable.
In this study, we aimed to establish a new method for the synthesis of α-bromolactones and successfully developed an innovative synthetic method that relies on a two-phase reaction system. Specifically, lactones are ring-opened and converted into dibromocarboxylic acids when treated with Br2 and a substoichiometric amount of PBr3. Subsequent treatment of these carboxylic acids with a base leads to the corresponding α-bromolactones through ring-closing reactions that involve the elimination of HBr; notably, ring-closure is successfully promoted in a two-phase system (Scheme 1b). Furthermore, we also report the simple one-pot transformations of lactone derivatives using α-bromolactones as key intermediates.
![[1860-5397-17-198-i1]](/bjoc/content/inline/1860-5397-17-198-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General procedure for α-bromination of δ-valerolactone (1a) and the method described in this work.
Scheme 1: General procedure for α-bromination of δ-valerolactone (1a) and the method described in this work.
Results and Discussion
We begin by first discussing the properties and stabilities of industrially important five- and six-membered lactones. γ-Butyrolactone and its α-brominated derivative are both stable at room temperature; α-bromo-γ-butyrolactone is readily synthesized by brominating the five-membered lactone under basic conditions. In sharp contrast, the corresponding six-membered α-bromo-δ-valerolactone has a more-distorted ring and is extremely unstable, even at room temperature [39-41]. In fact, it must be stored in a freezer because ring-opening polymerization and ring-contraction reactions occur readily at room temperature (see Supporting Information File 1). Therefore, in this study, we chose unstable δ-valerolactone (1a) as a model compound during the development of a new and innovative method for the synthesis of α-bromolactones, and investigated the reaction conditions in detail.
We first examined the Hell–Volhard–Zelinsky-type ring-opening reaction of 1a (Table 1). In this reaction, the corresponding acid bromide is formed in situ by heating with Br2 and a substoichiometric amount of PBr3; the acid bromide is then converted into 2,5-dibromopentanoic acid (2a) via hydrolysis during the workup under open-air. Lactone 1a (5 mmol) was allowed to react with Br2 (2.0 equiv) and PBr3 (5 mol %) at 80 °C for 24 h, with subsequent hydrolysis successfully affording in 2a in 90% yield (Table 1, entry 1). In the absence of a substoichiometric amount of PBr3, the transformation of 1a to 2a hardly proceeded (Table 1, entry 2). 2a was obtained in 89% yield when this protocol was used on a 31 mmol scale (Table 1, entry 3). γ-Butyrolactone (1b) and ε-caprolactone (1c) were also converted into the corresponding dibromocarboxylic acids 2b and 2c in yields of 76% and 70%, respectively (Table 1, entries 4 and 5).
Table 1: Ring-opening reactions of lactones with Br2 in the presence of a substoichiometric amount of PBr3a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-198-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | n | 1 (mmol) | Br2 (equiv) | PBr3 (mol %) | Temp. (°C) | Time (h) | Yield 2 (%) |
| 1 | 1a: n = 1 | 5 | 2.0 | 5 | 80 | 24 | 2a: 90 |
| 2 | 1a: n = 1 | 5 | 2.0 | – | 80 | 24 | 2a: 1 |
| 3 | 1a: n = 1 | 31 | 2.0 | 10 | 80 | 24 | 2a: 89 |
| 4 | 1b: n = 0 | 31 | 2.0 | 10 | 90 | 24 | 2b: 76 |
| 5 | 1c: n = 2 | 31 | 2.0 | 10 | 90 | 24 | 2c: 70 |
aYields were determined by 1H NMR spectroscopy using 1,3,5-trioxane as an internal standard.
Since we successfully synthesized carboxylic acid 2a from lactone 1a in good yield, we next investigated the ring-closing reaction of 2a. Various acids and bases (PTSA, hydrochloric acid, NaOH, KOH, and NaHCO3) were used to promote the intramolecular cyclization of 2a; however, no reaction was observed using any of these acids/bases. Interestingly, 2a was converted into 3a in very low yield when 1.2 equiv of n-Bu4N+F− was used, despite n-Bu4N+F− itself being less basic than the other bases (Scheme 2a) [42,43]. These results suggest that the properties of the counter cation may be important for the intramolecular cyclization of 2a. Based on this observation, we next examined n-Bu4N+OH−, a more-basic R4N+X− system, for the ring-closure of 2a. Surprisingly, the reaction proceeded smoothly to give α-bromo-δ-valerolactone (3a) in 52% yield in 43 h (Scheme 2b). However, further extending the reaction time to 72 h resulted in a dramatically lower yield of 3a (8%), most likely because 3a is unstable to base at room temperature and may decompose or polymerize (see Supporting Information File 1).
![[1860-5397-17-198-i2]](/bjoc/content/inline/1860-5397-17-198-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Tetraalkylammonium salt-mediated intramolecular cyclization of 2a.
Scheme 2: Tetraalkylammonium salt-mediated intramolecular cyclization of 2a.
To avoid decomposition or polymerization, 3a produced in situ by the intramolecular cyclization of 2a should be separated immediately from the reaction mixture containing n-Bu4N+OH−. Tetraalkylammonium salts are used as phase-transfer catalysts as they are soluble in both organic solvents and water. With these properties in mind, we next investigated the ring-closure of 2a using a two-phase CHCl3/H2O system (Table 2). Intramolecular cyclization of the salt forms 3a, which is extracted into the organic layer due to its low solubility in water. Because n-Bu4N+OH− is less-soluble in organic solvents than water, 3a is phase-separable from the base. To our delight, 2a was smoothly converted into 3a in 74% yield in this two-phase system, with the reaction time successfully reduced to 1 h (Table 2, entries 1–5). The use of a co-solvent to increase the solubility of 2a was investigated in detail; DMSO was found to be the most effective solvent, with 3a produced in 82% yield (Table 2, entries 5–12).
Table 2: Optimizing the intramolecular cyclization of 2a in a two-phase systema.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-198-i8.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Time (h) | Yield 3a (%) |
| 1 | CH3CN | 24 | 29 |
| 2 | CH3CN | 18 | 38 |
| 3 | CH3CN | 9 | 58 |
| 4 | CH3CN | 3 | 65 |
| 5 | CH3CN | 1 | 74 |
| 6 | MeOH | 1 | 29 |
| 7 | EtOH | 1 | 51 |
| 8 | iPrOH | 1 | 68 |
| 9 | THF | 1 | 61 |
| 10 | DMSO | 1 | 82 |
| 11 | DMF | 1 | 64 |
| 12 | none | 1 | 65 |
aYields were determined by 1H NMR spectroscopy using 1,3,5-trioxane as an internal standard.
We next optimized the base used to cyclize 2a in the two-phase system under the optimized conditions (entry 10, Table 2), the results of which are summarized in Table 3. The use of tetraalkylammonium hydroxides with longer alkyl chains tended to increase the yield of 3a (Table 3, entries 1–5), while the use of diisopropylethylamine, triethylamine, DBU, or Cs2CO3 was less effective (Table 3, entries 6–9); furthermore, the reaction did not proceed in the absence of a base (Table 3, entry 10). This investigation revealed that medium-chain tetraalkylammonium hydroxides, namely n-Bu4N+OH− and n-Pr4N+OH−, effectively transform 2a into 3a through intramolecular cyclization.
Table 3: Optimizing the base for the intramolecular cyclization of 2a to 3aa.
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-198-i9.svg?max-width=637&scale=1.0)
|
||
| Entry | Base | Yield 3a (%) |
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-198-i10.svg?max-width=637&scale=1.0)
n-Bu4N+OH– (40% in H2O) |
82 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-198-i11.svg?max-width=637&scale=1.0)
n-Pr4N+OH– (40% in H2O) |
67 |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-198-i12.svg?max-width=637&scale=1.0)
Et4N+OH– (40% in H2O) |
36 |
| 4 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-198-i13.svg?max-width=637&scale=1.0)
(10% in H2O) |
37 |
| 5 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-198-i14.svg?max-width=637&scale=1.0)
Me4N+OH– (40% in H2O) |
21 |
| 6 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-198-i15.svg?max-width=637&scale=1.0)
|
34 |
| 7 | Et3N | 34 |
| 8 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-198-i16.svg?max-width=637&scale=1.0)
|
39 |
| 9 | Cs2CO3 | 15 |
| 10 | none | – |
aYields were determined by 1H NMR spectroscopy using 1,3,5-trioxane as an internal standard.
We next investigated the effect of the size of the lactone ring on the α-bromination reaction in this two-phase system. Table 4 shows that 2a and 2b were transformed to 3a and 3b in good yields, but 2c did not react under these conditions due to the entropic cost associated with forming a seven-membered ring. Lactones 3a and 3b were obtained in good yields even when n-Pr4N+OH− was used as the base [44].
Table 4: Reaction scope for the the intramolecular cyclization of 2 in a two-phase systema.
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-198-i17.svg?max-width=637&scale=1.0)
|
|||
| Entry | n | Base | Yield 3 (%) |
| 1 | 2a: n = 1 | n-Bu4N+OH− | 3a: 74 |
| n-Pr4N+OH− | 3a: 75b | ||
| 2 | 2b: n = 0 | n-Bu4N+OH− | 3b: 69 |
| n-Pr4N+OH− | 3b: 73 (61) | ||
| 3 | 2c: n = 2 | n-Bu4N+OH− | 3c: trace |
| n-Pr4N+OH− | 3c: trace | ||
aYields were determined by 1H NMR spectroscopy using 1,3,5-trioxane as an internal standard (isolated yield). bSee reference [44].
While the developed two-phase CHCl3/H2O system performed well for the syntheses of α-bromolactones, the use of CHCl3 as the reaction solvent should ideally be avoided because it is toxic and an environmental pollutant. Therefore, we further optimized the solvent combination to construct an eco-friendlier reaction system (Table 5). Various solvents were used as the organic layer instead of CHCl3, with 3a produced in good yield using methyl ethyl ketone (MEK) as the solvent (Table 5, entries 1–5). Although the use of other ketones as solvents also afforded 3a in moderate yields, MEK proved to be the most suitable replacement for CHCl3 (Table 5, entries 5–8).
Table 5: Optimizing the organic solvent in the two-phase systema.
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-198-i18.svg?max-width=637&scale=1.0)
|
||
| Entry | Solvent | Yield 3a (%) |
| 1 | CHCl3 | 74 |
| 2 | FC-72 | 56 |
| 3 | BTF | 43 |
| 4 | 2-bromopropane | 33 |
| 5 | MEK | 75 |
| 6 | acetylacetone | 64 |
| 7 | 3-methyl-2-butanone | 55 |
| 8 | pinacolone | 49 |
aYields were determined by 1H NMR spectroscopy using 1,3,5-trioxane as an internal standard.
This environmentally friendly procedure was used to synthesize other lactones. For instance, this method was used to prepare 3b from 2b in 84% yield on a 10 mmol scale (Scheme 3a). Furthermore, this system also provided 2,2-diphenyl-γ-butyrolactone (3d), which bears two phenyl groups at the α-position, in 78% yield (Scheme 3b).
![[1860-5397-17-198-i3]](/bjoc/content/inline/1860-5397-17-198-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of α-functionalized lactones using the two-phase system.
Scheme 3: Synthesis of α-functionalized lactones using the two-phase system.
α-Bromolactones were obtained without any handling difficulties using our developed system, with synthesis scale-up tolerated under mild conditions. To facilitate the construction of various functional scaffolds using this system, lactones were subsequently α-functionalized via the corresponding α-bromolactones using this two-phase system. Interestingly, 3b synthesized using this method was smoothly substituted at the α-position with benzenethiol (4) in the presence of K2CO3 to afford the unsymmetrically functionalized sulfide 5 in 86% yield; 5 is a precursor to some pharmaceutical cores (Scheme 4) [45-48].
![[1860-5397-17-198-i4]](/bjoc/content/inline/1860-5397-17-198-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of unsymmetrically functionalized sulfide 5 via the two-phase system-promoted intramolecular cyclization of 2b.
Scheme 4: Synthesis of unsymmetrically functionalized sulfide 5 via the two-phase system-promoted intramolecu...
However, α-bromo-δ-valerolactone (3a) was extremely unstable under ambient conditions, and its purity quickly deteriorated even when stored in a freezer with shading (see Supporting Information File 1), resulting in trace amounts of α-functionalized lactones using the above-mentioned two-step method. Hence, we focused on sequential nucleophilic substitution in a two-phase system based on this bromination protocol. After ring-closing 2a, the generated α-bromolactone 3a was extracted into the organic layer, whereas the formed n-Bu4N+Br− dissolved in both the aqueous and organic layers. N-Bu4N+Nu− is formed when Na+Nu− (Nu−: nucleophile) is added to the reaction mixture, which is then phase-transferred into the organic phase, with subsequent nucleophilic substitution at the α-position of 3a proceeding directly to produce a variety of α-substituted lactones, along with the regeneration of n-Bu4N+Br− (Scheme 5).
![[1860-5397-17-198-i5]](/bjoc/content/inline/1860-5397-17-198-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Sequential nucleophilic substitution in the two-phase system.
Scheme 5: Sequential nucleophilic substitution in the two-phase system.
To demonstrate the applicability of this protocol, we investigated the synthesis of 2-phenylthio-α-valerolactone (6). Carboxylic acid 2a (2.0 mmol) directly reacted with n-Bu4N+OH− (1.2 equiv) and PhS−Na+ in the two-phase system under the optimized conditions for the synthesis of α-bromolactones, and 6 was successfully obtained in 72% yield (Scheme 6). These results demonstrate that this novel system facilitates the easy syntheses of functional molecules via α-bromolactones as key synthetic intermediates.
![[1860-5397-17-198-i6]](/bjoc/content/inline/1860-5397-17-198-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: One-pot synthesis of 2-phenylthio-α-valerolactone 6.
Scheme 6: One-pot synthesis of 2-phenylthio-α-valerolactone 6.
Conclusion
In this study, we developed a facile and efficient method for α-brominating lactones using tetraalkylammonium hydroxide (R4N+OH−) as the base under mild conditions. Lactones were ring-opened with Br2 and a substoichiometric amount of PBr3, which led to the corresponding α-bromocarboxylic acids in good yields. These carboxylic acids subsequently underwent intramolecular cyclization in 1 h using a two-phase system (H2O/CHCl3) with R4N+OH− to afford α-bromolactones in excellent yields. The use of methyl ethyl ketone (MEK) in the two-phase system led to an eco-friendly system amenable to large-scale synthesis. Furthermore, the α-bromolactones generated in situ by this method were transformed into functional molecules, such as α-thiolated lactones, in good yields without any handling difficulties. We expect that this new bromination system will lead to the use of various α-bromolactones as synthetic intermediates in organic chemistry.
Experimental
General comments. Unless otherwise stated, all starting materials and catalysts were purchased from commercial sources and used without further purification. All solvents were used without distillation. 1H NMR spectra were recorded on a JEOL JNM-ECS400 (400 MHz) FT NMR system or a JEOL JNM-ECX400 (400 MHz) FT NMR system in CDCl3 with Me4Si as an internal standard. 13C{1H} NMR spectra were recorded on a JEOL JNM-ECX400 (100 MHz) FT NMR or JEOL JNM-ECS400 (100 MHz) FT NMR system in CDCl3.
Ring-opening reaction of δ-valerolactone (1a) with Br2 in the presence of a catalytic amount of PBr3 (entry 1, Table 1). To a 50 mL three-neck flask were added δ-valerolactone (1a, 5 mmol) and PBr3 (5 mol %), then Br2 (1.0 equiv) was added dropwise for 2 h at 0 °C. After adding Br2, another amount of Br2 (1.0 equiv) was added to the reaction mixture for 30 min at 70 °C. The resulting solution was then stirred for 24 h at 80 °C. After the reaction was completed, the mixture was dissolved in CH3CN (30 mL) and bubbling N2 gas to remove excess amount of Br2 and the formed HBr (under open-air) then filtered. The filtrate was concentrated under reduced pressure to produce 2,5-dibromopentanoic acid 2a in 90% yield with trace amount of 1a. The purity of 2a was determined by 1H and 13C NMR spectroscopy, and 2a was used for the subsequent intramolecular cyclization without any further purification.
General procedure for the synthesis of α-substituted lactones 3 via intramolecular cyclization of 2 with R4N+OH− in two-phase system (Table 4, Table 5 and Scheme 3b). To a 30 mL flask were added 2 (1.0 mmol, 2a–c: synthesized and used without further purification; 2d: purchased from commercial sources), CH3CN (1.0 mL), H2O (4 mL), CHCl3 (5.0 mL) or MEK (5.0 mL), and R4N+OH− (1.2 equiv in aqueous solution). The mixture was stirred vigorously at 25 °C for 1 h. After the reaction was completed, the mixture was extracted with CHCl3 (15 mL × 3). The organic layer was washed with H2O (10 mL × 2), dried by anhydrous Na2SO4, then filtered. The filtrate was concentrated under reduced pressure. Finally, the residue was purified by gel permeation chromatography (eluent: CH2Cl2) or distillation to give pure product 3.
Gram-scale synthesis of α-bromolactone 3b in two-phase system with n-Pr4N+OH− (Scheme 3a). To a 300 mL flask were added 2b (10 mmol, synthesized by the procedure above mentioned and used without further purification), CH3CN (10 mL), H2O (40 mL), MEK (50 mL), and n-Pr4N+OH− (1.2 equiv in aqueous solution). The mixture was stirred vigorously at 25 °C for 3 h. After the reaction was completed, the solvent was removed under reduced pressure. The residue was extracted with CHCl3 (20 mL × 3). The organic layer was washed with H2O (10 mL × 2), dried by anhydrous Na2SO4, then filtered. The filtrate was concentrated under reduced pressure. Finally, the residue was purified by distillation to give pure product 3b in 64% yield (1.05 g).
Cascade synthesis of 5 via two-phase ring closing of 3b and following substitution with benzenethiol 4 in the presence of K2CO3 (Scheme 4). To a 100 mL flask were added 2b (4.0 mmol), CH3CN (4 mL), H2O (16 mL), MEK (20 mL), and n-Pr4N+OH− (1.2 equiv in aqueous solution). The mixture was stirred vigorously at 25 °C for 1 h. After the reaction was completed, the mixture was extracted with CHCl3 (15 mL × 3). The organic layer was washed with H2O (10 mL × 2), dried by anhydrous Na2SO4, then filtered. The filtrate was concentrated under reduced pressure to give crude 3b. To a 50 mL flask were added 3b (used without isolation), benzenethiol 4 (2.4 mmol), DMF (5 mL), and K2CO3 (0.45 mmol), and the mixture was stirred at 25 °C overnight. The resulting mixture was extracted with CH2Cl2 (15 mL × 3). The organic layer was washed with H2O (10 mL × 2), dried by anhydrous Na2SO4, then filtered. The filtrate was concentrated under reduced pressure. Finally, the residue was purified by silica-gel column chromatography (AcOMe/isohexane) to give pure product 5.
One-pot synthesis of a functional lactone 6 using PhS−Na+ as the nucleophiles in the two-phase system (Scheme 6). To a three-necked flask were added 2a (2.0 mmol), DMSO (2 mL), H2O (4 mL), CHCl3 (10 mL), and n-Bu4N+OH− (1.2 equiv in aqueous solution), and stirred vigorously for 10 min at 25 °C. Then, a solution of PhS−Na+ (1.2 mmol) in H2O (4 mL) was slowly added over 30 min. After the addition, the solution was further stirred for 80 min. The resulting mixture was extracted with Et2O (15 mL × 3), and the organic layer was washed with 1% HCl aq (10 mL), H2O (10 mL × 2), and dried by anhydrous MgSO4. The filtration was carried out, and the filtrate was concentrated under reduced pressure. Finally, the residue was purified by silica-gel column chromatography (Et2O/isohexane) to give pure product 6.
Supporting Information
| Supporting Information File 1: Evaluation of the stability of α-bromo-δ-valerolactone (3a, Table S1), characterization data of compounds (3a, 3b, 3d, 5, and 6), and copies of 1H NMR and 13C{1H} NMR spectra. | ||
| Format: PDF | Size: 1.2 MB | Download |
Acknowledgements
The authors would like to express their sincere gratitude to Nippoh Chemicals Co., Ltd. for the collaboration on this project. The authors also acknowledge Mr. Yuta Yabe (Nippoh Chemicals Co., Ltd.) and Ms. Qiqi Chen (Osaka Prefecture University) for the helpful suggestion and experimental support.
References
-
Hamlin, T. A.; Swart, M.; Bickelhaupt, F. M. ChemPhysChem 2018, 19, 1315–1330. doi:10.1002/cphc.201701363
Return to citation in text: [1] -
Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007
Return to citation in text: [1] -
Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009–3066. doi:10.1021/cr9903048
Return to citation in text: [1] -
Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Chem. Rev. 2002, 102, 1359–1470. doi:10.1021/cr000664r
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] -
Surry, D. S.; Buchwald, S. L. Chem. Sci. 2011, 2, 27–50. doi:10.1039/c0sc00331j
Return to citation in text: [1] -
Kambe, N.; Iwasaki, T.; Terao, J. Chem. Soc. Rev. 2011, 40, 4937–4947. doi:10.1039/c1cs15129k
Return to citation in text: [1] -
Choi, J.; Fu, G. C. Science 2017, 356, eaaf7230. doi:10.1126/science.aaf7230
Return to citation in text: [1] -
Koike, T.; Akita, M. Inorg. Chem. Front. 2014, 1, 562–576. doi:10.1039/c4qi00053f
Return to citation in text: [1] -
Chen, Z.-M.; Zhang, X.-M.; Tu, Y.-Q. Chem. Soc. Rev. 2015, 44, 5220–5245. doi:10.1039/c4cs00467a
Return to citation in text: [1] -
Studer, A.; Curran, D. P. Angew. Chem., Int. Ed. 2016, 55, 58–102. doi:10.1002/anie.201505090
Return to citation in text: [1] -
Qiu, G.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 1011–1027. doi:10.1039/c6qo00103c
Return to citation in text: [1] -
Tan, C. K.; Yeung, Y.-Y. Chem. Commun. 2013, 49, 7985–7996. doi:10.1039/c3cc43950j
Return to citation in text: [1] -
Saikia, I.; Borah, A. J.; Phukan, P. Chem. Rev. 2016, 116, 6837–7042. doi:10.1021/acs.chemrev.5b00400
Return to citation in text: [1] -
Petrone, D. A.; Ye, J.; Lautens, M. Chem. Rev. 2016, 116, 8003–8104. doi:10.1021/acs.chemrev.6b00089
Return to citation in text: [1] -
Das, R.; Kapur, M. Asian J. Org. Chem. 2018, 7, 1524–1541. doi:10.1002/ajoc.201800142
Return to citation in text: [1] -
Sabuzi, F.; Pomarico, G.; Floris, B.; Valentini, F.; Galloni, P.; Conte, V. Coord. Chem. Rev. 2019, 385, 100–136. doi:10.1016/j.ccr.2019.01.013
Return to citation in text: [1] -
Phuc Tran, D.; Nomoto, A.; Mita, S.; Dong, C.-p.; Kodama, S.; Mizuno, T.; Ogawa, A. Tetrahedron Lett. 2020, 61, 151959. doi:10.1016/j.tetlet.2020.151959
Return to citation in text: [1] -
Roy, A.; Biswas, B.; Sen, P. K.; Venkateswaran, R. V. Tetrahedron Lett. 2007, 48, 6933–6936. doi:10.1016/j.tetlet.2007.07.177
Return to citation in text: [1] -
Baskar, B.; Dakas, P.-Y.; Kumar, K. Org. Lett. 2011, 13, 1988–1991. doi:10.1021/ol200389p
Return to citation in text: [1] -
Zhao, W.; Lorenz, N.; Jung, K.; Sieber, S. A. Angew. Chem., Int. Ed. 2016, 55, 1187–1191. doi:10.1002/anie.201508052
Return to citation in text: [1] -
Swapnil, N.; Kumar, A. S.; Babu, N. J.; Yadav, J. S. Asian J. Org. Chem. 2017, 6, 1091–1098. doi:10.1002/ajoc.201700131
Return to citation in text: [1] -
Panchal, H.; Clarke, C.; Bell, C.; Karad, S. N.; Lewis, W.; Lam, H. W. Chem. Commun. 2018, 54, 12389–12392. doi:10.1039/c8cc06388e
Return to citation in text: [1] -
Fang, X.-X.; Wang, P.-F.; Yi, W.; Chen, W.; Lou, S.-C.; Liu, G.-Q. J. Org. Chem. 2019, 84, 15677–15684. doi:10.1021/acs.joc.9b02310
Return to citation in text: [1] -
Tran, Q. T. N.; Wong, W. S. F.; Chai, C. L. L. Eur. J. Med. Chem. 2019, 174, 33–44. doi:10.1016/j.ejmech.2019.04.023
Return to citation in text: [1] -
Zhao, B.; Li, Z.; Wu, Y.; Wang, Y.; Qian, J.; Yuan, Y.; Shi, Z. Angew. Chem., Int. Ed. 2019, 58, 9448–9452. doi:10.1002/anie.201903721
Return to citation in text: [1] -
Gu, X.-S.; Yu, N.; Yang, X.-H.; Zhu, A.-T.; Xie, J.-H.; Zhou, Q.-L. Org. Lett. 2019, 21, 4111–4115. doi:10.1021/acs.orglett.9b01290
Return to citation in text: [1] -
Cholewczynski, A. E.; Williams, P. C.; Pierce, J. G. Org. Lett. 2020, 22, 714–717. doi:10.1021/acs.orglett.9b04546
Return to citation in text: [1] -
Zeng, F.; Shen, Y.; Zhu, S.; Pelton, R. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 3821–3827. doi:10.1002/1099-0518(20001015)38:20<3821::aid-pola130>3.0.co;2-g
Return to citation in text: [1] -
Zhu, L.-W.; Yang, W.; Ou, Y.; Wan, L.-S.; Xu, Z.-K. Polym. Chem. 2014, 5, 3666–3672. doi:10.1039/c4py00101j
Return to citation in text: [1] -
Olsén, P.; Undin, J.; Odelius, K.; Albertsson, A.-C. Polym. Chem. 2014, 5, 3847–3854. doi:10.1039/c4py00148f
Return to citation in text: [1] -
Hong, M.; Chen, E. Y.-X. Nat. Chem. 2016, 8, 42–49. doi:10.1038/nchem.2391
Return to citation in text: [1] -
Becker, G.; Wurm, F. R. Chem. Soc. Rev. 2018, 47, 7739–7782. doi:10.1039/c8cs00531a
Return to citation in text: [1] -
Song, Q.; Zhao, J.; Zhang, G.; Peruch, F.; Carlotti, S. Polym. J. 2020, 52, 3–11. doi:10.1038/s41428-019-0265-5
Return to citation in text: [1] -
Price, C. C.; Judge, J. M. Org. Synth. 1965, 45, 22–24. doi:10.15227/orgsyn.045.0022
Return to citation in text: [1] -
Kamibe, S.; Takahashi, S.; Ichikawa, N. Jap. Patent JP2012167020, 2012.
Return to citation in text: [1] -
House, H. O.; Czuba, L. J.; Gall, M.; Olmstead, H. D. J. Org. Chem. 1969, 34, 2324–2336. doi:10.1021/jo01260a018
Return to citation in text: [1] -
Reuss, R. H.; Hassner, A. J. Org. Chem. 1974, 39, 1785–1787. doi:10.1021/jo00925a051
Return to citation in text: [1] -
Carothers, W. H.; Dorough, G. L.; Natta, F. J. v. J. Am. Chem. Soc. 1932, 54, 761–772. doi:10.1021/ja01341a046
Return to citation in text: [1] -
Houk, K. N.; Jabbari, A.; Hall, H. K.; Alemán, C. J. Org. Chem. 2008, 73, 2674–2678. doi:10.1021/jo702567v
Return to citation in text: [1] -
Zhang, X.; Fevre, M.; Jones, G. O.; Waymouth, R. M. Chem. Rev. 2018, 118, 839–885. doi:10.1021/acs.chemrev.7b00329
Return to citation in text: [1] -
Ooi, T.; Sugimoto, H.; Maruoka, K. Heterocycles 2001, 54, 593–596. doi:10.3987/com-00-s(i)84
Return to citation in text: [1] -
Wu, C.-Y.; Brik, A.; Wang, S.-K.; Chen, Y.-H.; Wong, C.-H. ChemBioChem 2005, 6, 2176–2180. doi:10.1002/cbic.200500295
Return to citation in text: [1] -
Although we tried to isolate α-bromo-δ-valerolactone 3a synthesized by using n-Pr4N+OH− as the base (entry 1 in Table 4), 3a was very unstable and some decomposition of 3a was observed during the isolation using a recycling GPC (eluent: CH2Cl2). Finally, we could be isolated pure 3a in 20% yield. The details of the stability of 3a under ambient conditions and the characterization data of 3a are given in Supporting Information File 1.
Return to citation in text: [1] [2] -
Trost, B. M.; Mao, M. K.-T.; Balkovec, J. M.; Buhlmayer, P. J. Am. Chem. Soc. 1986, 108, 4965–4973. doi:10.1021/ja00276a043
Return to citation in text: [1] -
Carretero, J. C.; Rojo, J.; Díaz, N.; Hamdouchi, C.; Poveda, A. Tetrahedron 1995, 51, 8507–8524. doi:10.1016/0040-4020(95)00453-f
Return to citation in text: [1] -
Sabbah, M.; Bernollin, M.; Doutheau, A.; Soulère, L.; Queneau, Y. Med. Chem. Commun. 2013, 4, 363–366. doi:10.1039/c2md20298k
Return to citation in text: [1] -
Zhang, X.; Wu, J.; Liu, Y.; Xie, Y.; Liu, C.; Wang, J.; Zhao, G. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 799–811. doi:10.1080/10426507.2017.1286492
Return to citation in text: [1]
| 1. | Hamlin, T. A.; Swart, M.; Bickelhaupt, F. M. ChemPhysChem 2018, 19, 1315–1330. doi:10.1002/cphc.201701363 |
| 2. | Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. doi:10.1021/cr00039a007 |
| 3. | Beletskaya, I. P.; Cheprakov, A. V. Chem. Rev. 2000, 100, 3009–3066. doi:10.1021/cr9903048 |
| 4. | Hassan, J.; Sévignon, M.; Gozzi, C.; Schulz, E.; Lemaire, M. Chem. Rev. 2002, 102, 1359–1470. doi:10.1021/cr000664r |
| 5. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 6. | Surry, D. S.; Buchwald, S. L. Chem. Sci. 2011, 2, 27–50. doi:10.1039/c0sc00331j |
| 7. | Kambe, N.; Iwasaki, T.; Terao, J. Chem. Soc. Rev. 2011, 40, 4937–4947. doi:10.1039/c1cs15129k |
| 8. | Choi, J.; Fu, G. C. Science 2017, 356, eaaf7230. doi:10.1126/science.aaf7230 |
| 9. | Koike, T.; Akita, M. Inorg. Chem. Front. 2014, 1, 562–576. doi:10.1039/c4qi00053f |
| 10. | Chen, Z.-M.; Zhang, X.-M.; Tu, Y.-Q. Chem. Soc. Rev. 2015, 44, 5220–5245. doi:10.1039/c4cs00467a |
| 11. | Studer, A.; Curran, D. P. Angew. Chem., Int. Ed. 2016, 55, 58–102. doi:10.1002/anie.201505090 |
| 12. | Qiu, G.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 1011–1027. doi:10.1039/c6qo00103c |
| 13. | Tan, C. K.; Yeung, Y.-Y. Chem. Commun. 2013, 49, 7985–7996. doi:10.1039/c3cc43950j |
| 14. | Saikia, I.; Borah, A. J.; Phukan, P. Chem. Rev. 2016, 116, 6837–7042. doi:10.1021/acs.chemrev.5b00400 |
| 15. | Petrone, D. A.; Ye, J.; Lautens, M. Chem. Rev. 2016, 116, 8003–8104. doi:10.1021/acs.chemrev.6b00089 |
| 16. | Das, R.; Kapur, M. Asian J. Org. Chem. 2018, 7, 1524–1541. doi:10.1002/ajoc.201800142 |
| 17. | Sabuzi, F.; Pomarico, G.; Floris, B.; Valentini, F.; Galloni, P.; Conte, V. Coord. Chem. Rev. 2019, 385, 100–136. doi:10.1016/j.ccr.2019.01.013 |
| 18. | Phuc Tran, D.; Nomoto, A.; Mita, S.; Dong, C.-p.; Kodama, S.; Mizuno, T.; Ogawa, A. Tetrahedron Lett. 2020, 61, 151959. doi:10.1016/j.tetlet.2020.151959 |
| 37. | House, H. O.; Czuba, L. J.; Gall, M.; Olmstead, H. D. J. Org. Chem. 1969, 34, 2324–2336. doi:10.1021/jo01260a018 |
| 38. | Reuss, R. H.; Hassner, A. J. Org. Chem. 1974, 39, 1785–1787. doi:10.1021/jo00925a051 |
| 35. | Price, C. C.; Judge, J. M. Org. Synth. 1965, 45, 22–24. doi:10.15227/orgsyn.045.0022 |
| 36. | Kamibe, S.; Takahashi, S.; Ichikawa, N. Jap. Patent JP2012167020, 2012. |
| 29. | Zeng, F.; Shen, Y.; Zhu, S.; Pelton, R. J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 3821–3827. doi:10.1002/1099-0518(20001015)38:20<3821::aid-pola130>3.0.co;2-g |
| 30. | Zhu, L.-W.; Yang, W.; Ou, Y.; Wan, L.-S.; Xu, Z.-K. Polym. Chem. 2014, 5, 3666–3672. doi:10.1039/c4py00101j |
| 31. | Olsén, P.; Undin, J.; Odelius, K.; Albertsson, A.-C. Polym. Chem. 2014, 5, 3847–3854. doi:10.1039/c4py00148f |
| 32. | Hong, M.; Chen, E. Y.-X. Nat. Chem. 2016, 8, 42–49. doi:10.1038/nchem.2391 |
| 33. | Becker, G.; Wurm, F. R. Chem. Soc. Rev. 2018, 47, 7739–7782. doi:10.1039/c8cs00531a |
| 34. | Song, Q.; Zhao, J.; Zhang, G.; Peruch, F.; Carlotti, S. Polym. J. 2020, 52, 3–11. doi:10.1038/s41428-019-0265-5 |
| 19. | Roy, A.; Biswas, B.; Sen, P. K.; Venkateswaran, R. V. Tetrahedron Lett. 2007, 48, 6933–6936. doi:10.1016/j.tetlet.2007.07.177 |
| 20. | Baskar, B.; Dakas, P.-Y.; Kumar, K. Org. Lett. 2011, 13, 1988–1991. doi:10.1021/ol200389p |
| 21. | Zhao, W.; Lorenz, N.; Jung, K.; Sieber, S. A. Angew. Chem., Int. Ed. 2016, 55, 1187–1191. doi:10.1002/anie.201508052 |
| 22. | Swapnil, N.; Kumar, A. S.; Babu, N. J.; Yadav, J. S. Asian J. Org. Chem. 2017, 6, 1091–1098. doi:10.1002/ajoc.201700131 |
| 23. | Panchal, H.; Clarke, C.; Bell, C.; Karad, S. N.; Lewis, W.; Lam, H. W. Chem. Commun. 2018, 54, 12389–12392. doi:10.1039/c8cc06388e |
| 24. | Fang, X.-X.; Wang, P.-F.; Yi, W.; Chen, W.; Lou, S.-C.; Liu, G.-Q. J. Org. Chem. 2019, 84, 15677–15684. doi:10.1021/acs.joc.9b02310 |
| 25. | Tran, Q. T. N.; Wong, W. S. F.; Chai, C. L. L. Eur. J. Med. Chem. 2019, 174, 33–44. doi:10.1016/j.ejmech.2019.04.023 |
| 26. | Zhao, B.; Li, Z.; Wu, Y.; Wang, Y.; Qian, J.; Yuan, Y.; Shi, Z. Angew. Chem., Int. Ed. 2019, 58, 9448–9452. doi:10.1002/anie.201903721 |
| 27. | Gu, X.-S.; Yu, N.; Yang, X.-H.; Zhu, A.-T.; Xie, J.-H.; Zhou, Q.-L. Org. Lett. 2019, 21, 4111–4115. doi:10.1021/acs.orglett.9b01290 |
| 28. | Cholewczynski, A. E.; Williams, P. C.; Pierce, J. G. Org. Lett. 2020, 22, 714–717. doi:10.1021/acs.orglett.9b04546 |
| 44. | Although we tried to isolate α-bromo-δ-valerolactone 3a synthesized by using n-Pr4N+OH− as the base (entry 1 in Table 4), 3a was very unstable and some decomposition of 3a was observed during the isolation using a recycling GPC (eluent: CH2Cl2). Finally, we could be isolated pure 3a in 20% yield. The details of the stability of 3a under ambient conditions and the characterization data of 3a are given in Supporting Information File 1. |
| 44. | Although we tried to isolate α-bromo-δ-valerolactone 3a synthesized by using n-Pr4N+OH− as the base (entry 1 in Table 4), 3a was very unstable and some decomposition of 3a was observed during the isolation using a recycling GPC (eluent: CH2Cl2). Finally, we could be isolated pure 3a in 20% yield. The details of the stability of 3a under ambient conditions and the characterization data of 3a are given in Supporting Information File 1. |
| 42. | Ooi, T.; Sugimoto, H.; Maruoka, K. Heterocycles 2001, 54, 593–596. doi:10.3987/com-00-s(i)84 |
| 43. | Wu, C.-Y.; Brik, A.; Wang, S.-K.; Chen, Y.-H.; Wong, C.-H. ChemBioChem 2005, 6, 2176–2180. doi:10.1002/cbic.200500295 |
| 39. | Carothers, W. H.; Dorough, G. L.; Natta, F. J. v. J. Am. Chem. Soc. 1932, 54, 761–772. doi:10.1021/ja01341a046 |
| 40. | Houk, K. N.; Jabbari, A.; Hall, H. K.; Alemán, C. J. Org. Chem. 2008, 73, 2674–2678. doi:10.1021/jo702567v |
| 41. | Zhang, X.; Fevre, M.; Jones, G. O.; Waymouth, R. M. Chem. Rev. 2018, 118, 839–885. doi:10.1021/acs.chemrev.7b00329 |
| 45. | Trost, B. M.; Mao, M. K.-T.; Balkovec, J. M.; Buhlmayer, P. J. Am. Chem. Soc. 1986, 108, 4965–4973. doi:10.1021/ja00276a043 |
| 46. | Carretero, J. C.; Rojo, J.; Díaz, N.; Hamdouchi, C.; Poveda, A. Tetrahedron 1995, 51, 8507–8524. doi:10.1016/0040-4020(95)00453-f |
| 47. | Sabbah, M.; Bernollin, M.; Doutheau, A.; Soulère, L.; Queneau, Y. Med. Chem. Commun. 2013, 4, 363–366. doi:10.1039/c2md20298k |
| 48. | Zhang, X.; Wu, J.; Liu, Y.; Xie, Y.; Liu, C.; Wang, J.; Zhao, G. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 799–811. doi:10.1080/10426507.2017.1286492 |
© 2021 Yamamoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.