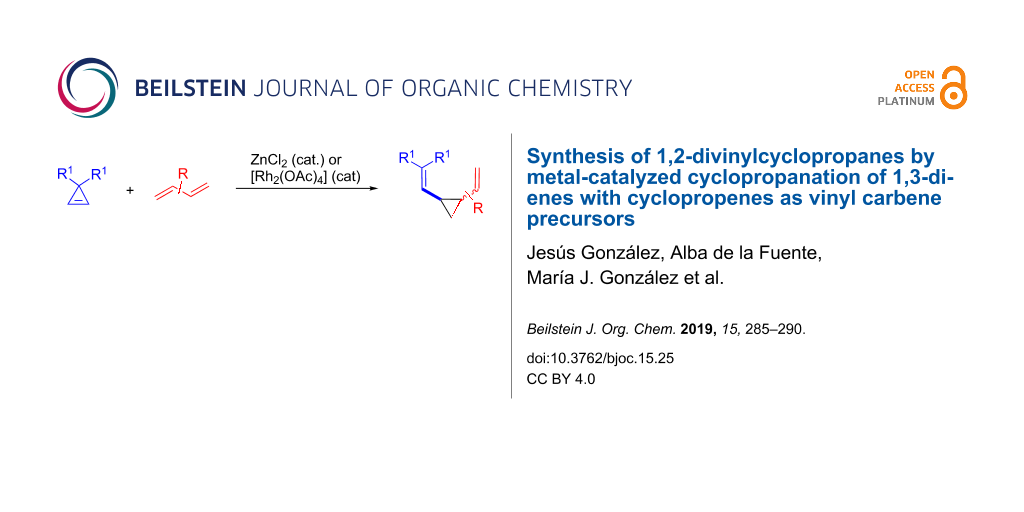
Abstract
The synthesis of 1,2-divinylcyclopropanes by the reaction of cyclopropenes with 1,3-dienes is reported. The process relies on the ability of ZnCl2 or [Rh2(OAc)4] to generate metal–vinyl carbene intermediates from cyclopropenes, which effect cyclopropanation of 1,3-dienes. Most of the reactions proceeded in reasonable yields while the diastereoselectivity strongly depends on the structure of the diene. An example of an intramolecular process as well as the use of furan and 1,4-cyclohexadiene as dienes are also reported.

Graphical Abstract
Introduction
Far from being considered exotic molecules, cyclopropane derivatives constitute an interesting class of compounds. Indeed, far over 4000 natural products bearing a cyclopropane ring have been discovered [1-3], and cyclopropane-containing molecules are recurrent in medicinal chemistry [4-6]. Likewise, due to its unique structure and bond properties, cyclopropanes have exclusive yet useful synthetic utilities [7], which are closely connected with the substitution pattern. For instance, the presence of vinyl groups directly attached to a cyclopropane ring allows sigmatropic rearrangements leading to odd-numbered carbocyclic derivatives [8]. In this sense, seven-membered carbocycles, namely 1,4-cycloheptadienes, can be forthrightly prepared from cis- or trans-1,2-divinylcyclopropanes through a Cope rearrangement [8,9]. The potential of this type of cyclopropanes contrasts with the existence of few straightforward routes for their syntheses. Typical methods rely on the use of reagents containing the required cyclopropane ring, which involve multistep sequences for the installation of adequate functionalization. Thus, Wittig-type olefination with cyclopropanecarboxaldehydes [10] or reactions of metallated vinylcyclopropanes with suitable electrophiles are commonly employed (Scheme 1a) [11-13]. In a more convergent approach where the cyclopropane ring is created at the last stage, divinylcyclopropanes can be prepared by cyclopropanation of 1,3-dienes with metal–vinyl carbenes generated from vinyldiazoacetates (Scheme 1b) [14-16]. This reaction has been fruitfully exploited, although it is inherently limited by the restricted availability of potentially explosive diazo compounds. Consequently, the use of alternative vinyl carbene precursors is highly desirable to expand the accessibility to 1,2-divinylcyclopropanes [17-19]. In this regard, cyclopropenes have demonstrated to be suitable precursors of metal–vinyl carbenes [20,21], which can be easily trapped with alkenes [22-25]. Our recent studies showed that simple ZnCl2 could be used to generate the corresponding zinc–vinyl carbene to efficiently prepare vinylcyclopropane derivatives with a remarkable broad scope (Scheme 1c) [26]. In view of these precedents, we decided to study the feasibility of this reaction in the synthesis of 1,2-divinylcyclopropanes by using 1,3-dienes as trapping reagents (Scheme 1d). Herein, we present the results of this study.
![[1860-5397-15-25-i1]](/bjoc/content/inline/1860-5397-15-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Typical syntheses of 1,2-divinylcyclopropanes and rationale hypothesis for their syntheses from cyclopropenes and 1,3-dienes.
Scheme 1: Typical syntheses of 1,2-divinylcyclopropanes and rationale hypothesis for their syntheses from cyc...
Results and Discussion
At the outset, the reaction of cyclopropene 1a and freshly distilled 1,3-cyclohexadiene (2a, 5.0 equiv) in the presence of ZnCl2 as catalyst was performed under the reaction conditions previously employed for simple alkenes, namely ZnCl2 10 mol %, CH2Cl2, at ambient temperature (Scheme 2) [26]. Pleasantly, 1,2-divinylcyclopropane 3a was obtained in good yield (81%) with moderate endo (syn) selectivity (endo/exo = 6:1). It should be indicated that the preference for the endo isomer has already been observed in related reactions [17]. Other metals capable of promoting both carbene generation from cyclopropenes and cyclopropanation reactions were also evaluated. Interestingly, when [Rh2(OAc)4] (1.0 mol %) was employed, compound 3a was isolated in 71% yield, a slightly lower value when compared to ZnCl2, but more importantly, with complete endo (syn) selectivity. The use of gold, platinum or ruthenium catalysts showed poorer results with respect to reaction yield and endo/exo (syn/anti) selectivity, as indicated in Scheme 2.
![[1860-5397-15-25-i2]](/bjoc/content/inline/1860-5397-15-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1,2-divinylcyclopropane 3a: Optimization studies. aIsolated yield. bDetermined by 1H NMR on the reaction crude mixture (unaltered after purification). cEstimated by 1H NMR. (IPr = 1,3-bis(2,6-diisopropylphenylimidazol-2-ylidene).
Scheme 2: Synthesis of 1,2-divinylcyclopropane 3a: Optimization studies. aIsolated yield. bDetermined by 1H N...
Considering these results, the scope of the reaction with unbiased 1,3-dienes was next investigated using both ZnCl2, as it provided the best yield being the most inexpensive catalyst, and [Rh2(OAc)4], since it delivered the best result in terms of selectivity. The results are summarized in Scheme 3.
![[1860-5397-15-25-i3]](/bjoc/content/inline/1860-5397-15-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1,2-divinylcyclopropanes 3 from cyclopropenes 1 and unbiased 1,3-dienes 2: Scope. (Yields of isolated products, diastereoisomeric ratios were determined by 1H NMR). aAt 50 °C. b1 was completely consumed. c1,3-Butadiene was used in large excess (ca. 0.5 mL condensed prior the reaction). dcis relationship is referred to substituents labelled with an asterisk.
Scheme 3: Synthesis of 1,2-divinylcyclopropanes 3 from cyclopropenes 1 and unbiased 1,3-dienes 2: Scope. (Yie...
Using 1,3-cyclohexadiene (2a), the analogous reaction was accomplished with 3,3-dihexyl- (1b) and 3,3-dibenzylcyclopropene (1c). Thus, divinylcyclopropane 3b was obtained in reasonable yields with both catalysts (69% Zn and 75% Rh), albeit better selectivity was obtained again with rhodium(II) catalyst (endo/exo = 17:1). In contrast, the reaction with 1c led to the corresponding cyclopropane 3c only when ZnCl2 was used as catalyst (85%, endo/exo = 4.5:1) at slightly higher temperature (50 °C). In this case, [Rh2(OAc)4] completely failed under different reaction conditions leading inevitably to degradation of starting cyclopropene. Other cyclic 1,3-dienes were then evaluated. For instance, the use of cyclopentadiene in the reaction with 1a and 1b enabled the preparation of divinylcyclopropanes 3d,e in good yields. Both catalysts provided similar yields, while complete endo selectivity was only reached with [Rh2(OAc)4]. Interestingly, the use of 1,3-cyclooctadiene led to the formation of compound 3f (Zn: 73%, endo/exo = 11:1; Rh: 71%, only endo), in which the 1,2-divinylcyclopropane moiety is embedded within a bicyclo[6.1.0]nonane core. Then, we studied the reaction with various representative unbiased acyclic 1,3-dienes. Unsurprisingly, these substrates could be easily converted into the corresponding 1,2-divinylcyclopropanes but with very low selectivities. For instance, when 2,3-dimethyl-1,3-butadiene was employed with cyclopropenes 1a,b, the corresponding 1,2-divinylcyclopropanes 3g,h were prepared in good yields regardless of the catalyst employed. However, 3g,h were obtained as an almost equimolar mixture of cis/trans diastereoisomers (cis refers to both vinyl substituents). Even though the lack of selectivity was already noticed by Uemura and co-workers in related reactions [17], we attempted the reaction with other zinc salts or rhodium(II) carboxylates as catalysts. Unfortunately, these experiments were futile and led to similar low selectivities. Besides, gaseous 1,3-butadiene could be also employed, as demonstrated by the preparation of compounds 3i,j, which were obtained in moderate yields and low cis selectivities. The reaction with isoprene showed a remarkable selectivity for the most substituted alkene, allowing the synthesis of 1,2-divinylcyclopropane 3k within the typical range of yields and cis/trans selectivities. Finally, 1,2,3-trisubstituted divinylcyclopropane 3l was prepared in 50% (cis/trans = 2.5:1) from cyclopropene 1a and (1E,3E)-1,4-diphenylbuta-1,3-diene through a stereoselective reaction. Once more, simple ZnCl2 was particularly effective for this reaction in sharp contrast to the incompetence of [Rh2(OAc)4].
It should be noticed that 1,2-divinylcyclopropanes 3a–l did not undergo [3,3]-Cope rearrangements under the reaction conditions. Moreover, when pure endo-3e was refluxed in toluene (24 h) partial isomerization was observed (endo/exo = 1:1.6). This behavior can be attributed to the high substitution at one of the alkenes, which might lead to sterically overcrowded transition states required for the rearrangement [27]. Besides, the diastereoisomeric mixture of 3j suffered complete degradation under the same reaction conditions.
In recent years, Cossy and co-workers elegantly prepared various bicyclo[n.1.0] derivatives through intramolecular cyclopropanation reactions using adequately decorated 1,n-cyclopropenenes [24]. Hence, we preliminarily explored the feasibility of an analogous reaction with 1,3-dienes preparing dienylcyclopropene 4. While treatment of 4 with ZnCl2 led to complex mixtures, likely due to the Lewis acid sensitivity of the benzylic cyclopropenylcarbinol moiety, the use of [Rh2(OAc)4] (1.0 mol %, CH2Cl2, rt) led to the tricyclic compound 5 in good yield (77%) in a stereoctive manner (Scheme 4).
![[1860-5397-15-25-i4]](/bjoc/content/inline/1860-5397-15-25-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Rh-catalyzed intramolecular cyclopropanation with dienylcyclopropene 4 (the trans/cis ratio is related to the C-atom labelled with an asterisk).
Scheme 4: Rh-catalyzed intramolecular cyclopropanation with dienylcyclopropene 4 (the trans/cis ratio is rela...
Finally, we were curious to study the reactivity of metal–vinyl carbenes generated from 3,3-disubstituted cyclopropenes with some particular dienes (Scheme 5). Interestingly, we found that the reaction of cyclopropenes 1a,b with furan (6) using ZnCl2 as catalyst allowed the isolation of diastereomerically pure endo-7a,b oxabicycles showing the 1,2-divinylcyclopropane moiety. In contrast, [Rh2(OAc)4] afforded complex reaction mixtures. In spite of the modest yields, this zinc-catalyzed reaction deserves some comments as it constitutes a rare example of isolation of these structures. Indeed, Lee and co-worker found that the corresponding gold-catalyzed reaction leads to ring-opened products through a facile oxy-Cope-rearrangement [28]. Moreover, these structures are also not accessible with metal–vinyl carbenes generated from vinyldiazo compounds, which led again to oxy-Cope rearranged or [4 + 3]-cycloaddition products using rhodium catalysts [14,28-30], or to a C2-allylation of furan with gold catalysts [31]. Finally, to compare the reactivity of cyclopropenes and vinyldiazo compounds, we probed the reaction of 1a with 1,4-cyclohexadiene (8). Under otherwise identical reaction conditions, both ZnCl2 and [Rh2(OAc)4] yielded exclusively cyclopropane 9 in practical yields and endo selectivity. This reaction outcome differs from the comparable reactions with vinyldiazo compounds and rhodium(II) catalysts, which preferentially undergo C–H allylic insertions [31-35].
![[1860-5397-15-25-i5]](/bjoc/content/inline/1860-5397-15-25-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Zn- or Rh-catalyzed reactions of cyclopropenes 1 with furan (6) and 1,4-cyclohexadiene (8) and comparison with related processes (1b, R1 = n-Hex).
Scheme 5: Zn- or Rh-catalyzed reactions of cyclopropenes 1 with furan (6) and 1,4-cyclohexadiene (8) and comp...
Conclusion
In summary, we have described a straightforward method for the synthesis of 1,2-divinylcyclopropane derivatives by cyclopropanation reactions using unbiased 1,3-dienes and cyclopropenes, which served as metal–vinyl carbene precursors. The use of cyclic dienes allowed the synthesis of the corresponding 1,2-divinylcyclopropanes with good-to-complete endo selectivity. In contrast, acyclic dienes were also efficiently converted into the expected cyclopropanes but with low cis/trans selectivities. In general, simple ZnCl2 and [Rh2(OAc)4] proved to be adequate catalysts. An intramolecular version of this reaction is also reported. Finally, the use of other dienes, such as furan and 1,4-cyclohexadiene was explored showing interesting reaction outcomes that are complementary to previous related reports. Subsequent studies to expand the scope of this cyclopropanation reaction to biased 1,3-dienes as well as to increase selectivities are currently ongoing.
Supporting Information
| Supporting Information File 1: Experimental details as well as 1H and 13C NMR spectra of new compounds. | ||
| Format: PDF | Size: 2.0 MB | Download |
Acknowledgements
Financial support from Ministerio de Economía y Competitividad (MINECO), Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER) (CTQ2016-76840-R) and Principado de Asturias (grant GRUPIN14-013) is gratefully acknowledged. We thank Prof. J. M. González for his unrestricted support.
References
-
Chen, D. Y.-K.; Pouwer, R. H.; Richard, J.-A. Chem. Soc. Rev. 2012, 41, 4631–4642. doi:10.1039/c2cs35067j
Return to citation in text: [1] -
Gnad, F.; Reiser, O. Chem. Rev. 2003, 103, 1603–1624. doi:10.1021/cr010015v
Return to citation in text: [1] -
Donaldson, W. A. Tetrahedron 2001, 57, 8589–8627. doi:10.1016/s0040-4020(01)00777-3
Return to citation in text: [1] -
Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625
Return to citation in text: [1] -
Talele, T. T. J. Med. Chem. 2016, 59, 8712–8756. doi:10.1021/acs.jmedchem.6b00472
Return to citation in text: [1] -
Gagnon, A.; Duplessis, M.; Fader, L. Org. Prep. Proced. Int. 2010, 42, 1–69. doi:10.1080/00304940903507788
Return to citation in text: [1] -
Kulinkovich, O. G. Cyclopropanes in Organic Synthesis; John Wiley & Sons, Inc: Hoboken, NJ, 2015. doi:10.1002/9781118978429
Return to citation in text: [1] -
Vshyvenko, S.; Reed, J. W.; Hudlicky, T.; Piers, E. Rearrangements of Vinylcyclopropanes, Divinylcyclopropanes, and Related Systems. In Comprehensive Organic Synthesis II; Knochel, P., Ed.; Elsevier, 2014; Vol. 5, pp 999–1076. doi:10.1016/b978-0-08-097742-3.00523-1
Return to citation in text: [1] [2] -
Krüger, S.; Gaich, T. Beilstein J. Org. Chem. 2014, 10, 163–193. doi:10.3762/bjoc.10.14
Return to citation in text: [1] -
Marino, J. P.; Kaneko, T. Tetrahedron Lett. 1973, 14, 3975–3978. doi:10.1016/s0040-4039(01)87087-8
Return to citation in text: [1] -
Marino, J. P.; Browne, L. J. Tetrahedron Lett. 1976, 17, 3245–3248. doi:10.1016/s0040-4039(00)93892-9
Return to citation in text: [1] -
Wender, P. A.; Filosa, M. P. J. Org. Chem. 1976, 41, 3490–3491. doi:10.1021/jo00883a043
Return to citation in text: [1] -
Wender, P. A.; Hillemann, C. L.; Szymonifka, M. J. Tetrahedron Lett. 1980, 21, 2205–2208. doi:10.1016/0040-4039(80)80003-7
Return to citation in text: [1] -
Davies, H. M. L.; McAfee, M. J.; Oldenburg, C. E. M. J. Org. Chem. 1989, 54, 930–936. doi:10.1021/jo00265a037
Return to citation in text: [1] [2] -
Cantrell, W. R., Jr.; Davies, H. M. L. J. Org. Chem. 1991, 56, 723–727. doi:10.1021/jo00002a044
Return to citation in text: [1] -
Davies, H. M. L.; Clark, T. J.; Smith, H. D. J. Org. Chem. 1991, 56, 3817–3824. doi:10.1021/jo00012a011
Return to citation in text: [1] -
Miki, K.; Ohe, K.; Uemura, S. J. Org. Chem. 2003, 68, 8505–8513. doi:10.1021/jo034841a
Return to citation in text: [1] [2] [3] -
Takeda, T.; Mori, A.; Fujii, T.; Tsubouchi, A. Tetrahedron 2014, 70, 5776–5786. doi:10.1016/j.tet.2014.06.043
Return to citation in text: [1] -
Barluenga, J.; Quiñones, N.; Tomás-Gamasa, M.; Cabal, M.-P. Eur. J. Org. Chem. 2012, 2312–2317. doi:10.1002/ejoc.201200149
Return to citation in text: [1] -
Archambeau, A.; Miege, F.; Meyer, C.; Cossy, J. Acc. Chem. Res. 2015, 48, 1021–1031. doi:10.1021/acs.accounts.5b00016
Return to citation in text: [1] -
Vicente, R. Synthesis 2016, 48, 2343–2360. doi:10.1055/s-0035-1561644
Return to citation in text: [1] -
Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f
Return to citation in text: [1] -
Miege, F.; Meyer, C.; Cossy, J. Chem. – Eur. J. 2012, 18, 7810–7822. doi:10.1002/chem.201200566
Return to citation in text: [1] -
Miege, F.; Meyer, C.; Cossy, J. Angew. Chem., Int. Ed. 2011, 50, 5932–5937. doi:10.1002/anie.201101220
Return to citation in text: [1] [2] -
Tomilov, Y. V.; Bordakov, V. G.; Tsvetkova, N. M.; Dolgii, I. E.; Nefedov, O. M. Russ. Chem. Bull. 1982, 31, 2129. doi:10.1007/bf00950678
Return to citation in text: [1] -
González, M. J.; González, J.; López, L. A.; Vicente, R. Angew. Chem., Int. Ed. 2015, 54, 12139–12143. doi:10.1002/anie.201505954
Return to citation in text: [1] [2] -
Sasaki, T.; Eguchi, S.; Ohno, M. J. Org. Chem. 1972, 37, 466–469. doi:10.1021/jo00968a030
Return to citation in text: [1] -
Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j
Return to citation in text: [1] [2] -
Davies, H. M. L.; Clark, D. M.; Alligood, D. B.; Eiband, G. R. Tetrahedron 1987, 43, 4265–4270. doi:10.1016/s0040-4020(01)90301-1
Return to citation in text: [1] -
Davies, H. M. L.; Calvo, R. L.; Townsend, R. J.; Ren, P.; Churchill, R. M. J. Org. Chem. 2000, 65, 4261–4268. doi:10.1021/jo991959b
Return to citation in text: [1] -
Barluenga, J.; Lonzi, G.; Tomás, M.; López, L. A. Chem. – Eur. J. 2013, 19, 1573–1576. doi:10.1002/chem.201203217
Return to citation in text: [1] [2] -
Bykowski, D.; Wu, K.-H.; Doyle, M. P. J. Am. Chem. Soc. 2006, 128, 16038–16039. doi:10.1021/ja066452e
Return to citation in text: [1] -
Davies, H. M. L.; Stafford, D. G.; Hansen, T. Org. Lett. 1999, 1, 233–236. doi:10.1021/ol9905699
Return to citation in text: [1] -
Davies, H. M. L.; Antoulinakis, E. G. J. Organomet. Chem. 2001, 617–618, 47–55. doi:10.1016/s0022-328x(00)00599-4
Return to citation in text: [1] -
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n
Return to citation in text: [1]
| 14. | Davies, H. M. L.; McAfee, M. J.; Oldenburg, C. E. M. J. Org. Chem. 1989, 54, 930–936. doi:10.1021/jo00265a037 |
| 28. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 29. | Davies, H. M. L.; Clark, D. M.; Alligood, D. B.; Eiband, G. R. Tetrahedron 1987, 43, 4265–4270. doi:10.1016/s0040-4020(01)90301-1 |
| 30. | Davies, H. M. L.; Calvo, R. L.; Townsend, R. J.; Ren, P.; Churchill, R. M. J. Org. Chem. 2000, 65, 4261–4268. doi:10.1021/jo991959b |
| 24. | Miege, F.; Meyer, C.; Cossy, J. Angew. Chem., Int. Ed. 2011, 50, 5932–5937. doi:10.1002/anie.201101220 |
| 28. | Hadfield, M. S.; Lee, A.-L. Chem. Commun. 2011, 47, 1333–1335. doi:10.1039/c0cc04217j |
| 1. | Chen, D. Y.-K.; Pouwer, R. H.; Richard, J.-A. Chem. Soc. Rev. 2012, 41, 4631–4642. doi:10.1039/c2cs35067j |
| 2. | Gnad, F.; Reiser, O. Chem. Rev. 2003, 103, 1603–1624. doi:10.1021/cr010015v |
| 3. | Donaldson, W. A. Tetrahedron 2001, 57, 8589–8627. doi:10.1016/s0040-4020(01)00777-3 |
| 8. | Vshyvenko, S.; Reed, J. W.; Hudlicky, T.; Piers, E. Rearrangements of Vinylcyclopropanes, Divinylcyclopropanes, and Related Systems. In Comprehensive Organic Synthesis II; Knochel, P., Ed.; Elsevier, 2014; Vol. 5, pp 999–1076. doi:10.1016/b978-0-08-097742-3.00523-1 |
| 9. | Krüger, S.; Gaich, T. Beilstein J. Org. Chem. 2014, 10, 163–193. doi:10.3762/bjoc.10.14 |
| 17. | Miki, K.; Ohe, K.; Uemura, S. J. Org. Chem. 2003, 68, 8505–8513. doi:10.1021/jo034841a |
| 8. | Vshyvenko, S.; Reed, J. W.; Hudlicky, T.; Piers, E. Rearrangements of Vinylcyclopropanes, Divinylcyclopropanes, and Related Systems. In Comprehensive Organic Synthesis II; Knochel, P., Ed.; Elsevier, 2014; Vol. 5, pp 999–1076. doi:10.1016/b978-0-08-097742-3.00523-1 |
| 27. | Sasaki, T.; Eguchi, S.; Ohno, M. J. Org. Chem. 1972, 37, 466–469. doi:10.1021/jo00968a030 |
| 7. | Kulinkovich, O. G. Cyclopropanes in Organic Synthesis; John Wiley & Sons, Inc: Hoboken, NJ, 2015. doi:10.1002/9781118978429 |
| 26. | González, M. J.; González, J.; López, L. A.; Vicente, R. Angew. Chem., Int. Ed. 2015, 54, 12139–12143. doi:10.1002/anie.201505954 |
| 4. | Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625 |
| 5. | Talele, T. T. J. Med. Chem. 2016, 59, 8712–8756. doi:10.1021/acs.jmedchem.6b00472 |
| 6. | Gagnon, A.; Duplessis, M.; Fader, L. Org. Prep. Proced. Int. 2010, 42, 1–69. doi:10.1080/00304940903507788 |
| 17. | Miki, K.; Ohe, K.; Uemura, S. J. Org. Chem. 2003, 68, 8505–8513. doi:10.1021/jo034841a |
| 17. | Miki, K.; Ohe, K.; Uemura, S. J. Org. Chem. 2003, 68, 8505–8513. doi:10.1021/jo034841a |
| 18. | Takeda, T.; Mori, A.; Fujii, T.; Tsubouchi, A. Tetrahedron 2014, 70, 5776–5786. doi:10.1016/j.tet.2014.06.043 |
| 19. | Barluenga, J.; Quiñones, N.; Tomás-Gamasa, M.; Cabal, M.-P. Eur. J. Org. Chem. 2012, 2312–2317. doi:10.1002/ejoc.201200149 |
| 22. | Miege, F.; Meyer, C.; Cossy, J. Org. Lett. 2010, 12, 4144–4147. doi:10.1021/ol101741f |
| 23. | Miege, F.; Meyer, C.; Cossy, J. Chem. – Eur. J. 2012, 18, 7810–7822. doi:10.1002/chem.201200566 |
| 24. | Miege, F.; Meyer, C.; Cossy, J. Angew. Chem., Int. Ed. 2011, 50, 5932–5937. doi:10.1002/anie.201101220 |
| 25. | Tomilov, Y. V.; Bordakov, V. G.; Tsvetkova, N. M.; Dolgii, I. E.; Nefedov, O. M. Russ. Chem. Bull. 1982, 31, 2129. doi:10.1007/bf00950678 |
| 14. | Davies, H. M. L.; McAfee, M. J.; Oldenburg, C. E. M. J. Org. Chem. 1989, 54, 930–936. doi:10.1021/jo00265a037 |
| 15. | Cantrell, W. R., Jr.; Davies, H. M. L. J. Org. Chem. 1991, 56, 723–727. doi:10.1021/jo00002a044 |
| 16. | Davies, H. M. L.; Clark, T. J.; Smith, H. D. J. Org. Chem. 1991, 56, 3817–3824. doi:10.1021/jo00012a011 |
| 26. | González, M. J.; González, J.; López, L. A.; Vicente, R. Angew. Chem., Int. Ed. 2015, 54, 12139–12143. doi:10.1002/anie.201505954 |
| 11. | Marino, J. P.; Browne, L. J. Tetrahedron Lett. 1976, 17, 3245–3248. doi:10.1016/s0040-4039(00)93892-9 |
| 12. | Wender, P. A.; Filosa, M. P. J. Org. Chem. 1976, 41, 3490–3491. doi:10.1021/jo00883a043 |
| 13. | Wender, P. A.; Hillemann, C. L.; Szymonifka, M. J. Tetrahedron Lett. 1980, 21, 2205–2208. doi:10.1016/0040-4039(80)80003-7 |
| 31. | Barluenga, J.; Lonzi, G.; Tomás, M.; López, L. A. Chem. – Eur. J. 2013, 19, 1573–1576. doi:10.1002/chem.201203217 |
| 10. | Marino, J. P.; Kaneko, T. Tetrahedron Lett. 1973, 14, 3975–3978. doi:10.1016/s0040-4039(01)87087-8 |
| 20. | Archambeau, A.; Miege, F.; Meyer, C.; Cossy, J. Acc. Chem. Res. 2015, 48, 1021–1031. doi:10.1021/acs.accounts.5b00016 |
| 21. | Vicente, R. Synthesis 2016, 48, 2343–2360. doi:10.1055/s-0035-1561644 |
| 31. | Barluenga, J.; Lonzi, G.; Tomás, M.; López, L. A. Chem. – Eur. J. 2013, 19, 1573–1576. doi:10.1002/chem.201203217 |
| 32. | Bykowski, D.; Wu, K.-H.; Doyle, M. P. J. Am. Chem. Soc. 2006, 128, 16038–16039. doi:10.1021/ja066452e |
| 33. | Davies, H. M. L.; Stafford, D. G.; Hansen, T. Org. Lett. 1999, 1, 233–236. doi:10.1021/ol9905699 |
| 34. | Davies, H. M. L.; Antoulinakis, E. G. J. Organomet. Chem. 2001, 617–618, 47–55. doi:10.1016/s0022-328x(00)00599-4 |
| 35. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
© 2019 González et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)