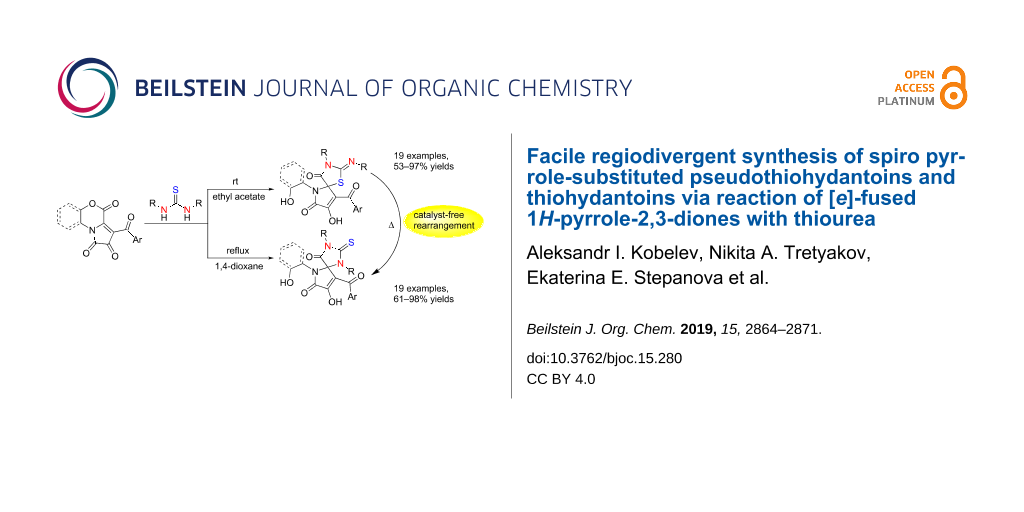
Abstract
A highly divergent synthesis of regioisomeric thiohydantoins and pseudothiohydantoins spiro-fused to a pharmacologically valuable pyrrole-2-one fragment involving the reaction of [e]-fused 1H-pyrrole-2,3-diones with thioureas was developed. The obtained spiro pseudothiohydantoin derivatives were found to undergo a pseudothiohydantoin–thiohydantoin rearrangement. The reactions were shown to proceed under catalyst-free conditions in good yields, and the products were isolated without applying preparative chromatography methods.

Graphical Abstract
Introduction
Hydantoin (imidazolidine-2,4-dione) derivatives are omnipresent among biologically active compounds [1]. Many of them are commercially available drugs, for example, anticonvulsants phenytoin [2] and albutoin [3], the muscle relaxant dantrolene [4], or the nonsteroidal antiandrogen agent enzalutamide [5] (Figure 1).
![[1860-5397-15-280-1]](/bjoc/content/figures/1860-5397-15-280-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Hydantoin-based commercially available drugs.
Figure 1: Hydantoin-based commercially available drugs.
Despite this fact, hydantoin derivatives belong to a special group of scaffolds in medicinal chemistry. Indeed, they are structurally related to rhodanine (2-thioxothiazolidin-4-one), and sometimes are classified as pan-assay interference compounds (PAINS), which are abhorrent in high-throughput screenings [6]. To clarify whether such compounds are privileged scaffolds or promiscuous binders, Mendgen and co-workers performed a systematic comparative study on rhodanines and related structures with respect to their medicinal chemistry properties [7]. As a result, it was shown that such structures could be suitable scaffolds for medicinal chemistry under proper conditions of biological evaluation, while their medicinal chemistry properties dramatically depend on the structure of the five-membered heterocyclic moiety and substituents in the C-5 position [7].
Some pseudothiohydantoins (2-iminothiazolidin-4-ones) are known to undergo a pseudothiohydantoin–thiohydantoin rearrangement (PTR, Scheme 1) [8-11], which is a special case of a quite poorly investigated iminothiolactone–thiolactam rearrangement [12-16]. This reaction offers attractive opportunities for the design of libraries of regioisomeric hydantoin-based compounds for drug discovery.
![[1860-5397-15-280-i1]](/bjoc/content/inline/1860-5397-15-280-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pseudothiohydantoin–thiohydantoin rearrangement.
Scheme 1: Pseudothiohydantoin–thiohydantoin rearrangement.
Spiro heterocycles are relatively new and insufficiently investigated structures in medicinal chemistry [17,18]. The introduction of spiro-fused cyclic moieties in small molecules increases the degree of their structural (shape) complexity, which may lead to the reduced binding promiscuity and improved clinical success [19]. Thus, recent development of 3D modelling methods facilitated investigations on the importance of 3D properties of small-molecular drug candidates [20-22] and inspired chemists to develop diversity-oriented methods for complex 3D molecules [23,24].
Considering that to the best of our knowledge, only a sole report exists [10] that describes the synthesis of regioisomeric 5-spiro-substituted thiohydantoins and pseudothiohydantoins and their PTR (Scheme 2), we were encouraged to develop a divergent synthetic approach to access regioisomeric thiohydantoins and pseudothiohydantoins that are spiro-fused to a pharmacologically valuable pyrrole-2-one fragment and to investigate the scope and conditions of their PTR behavior (Scheme 2).
![[1860-5397-15-280-i2]](/bjoc/content/inline/1860-5397-15-280-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syntheses of regioisomeric 5-spiro-substituted thiohydantoins and pseudothiohydantoins: previous [10] and this work.
Scheme 2: Syntheses of regioisomeric 5-spiro-substituted thiohydantoins and pseudothiohydantoins: previous [10] a...
Results and Discussion
1H-Pyrrole-2,3-diones fused at [e]-side (FPDs) 1 can be viewed as a versatile polyelectrophilic synthetic platform (Figure 2), enabling facile preparation of libraries of heterocyclic molecules with an emphasis on skeletal diversity from a single set of reagents [25-27].
![[1860-5397-15-280-2]](/bjoc/content/figures/1860-5397-15-280-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Electrophilic centers of FPDs 1, starting materials of this study.
Figure 2: Electrophilic centers of FPDs 1, starting materials of this study.
One of the most intriguing chemical properties of FPDs 1 is their ability to undergo a cyclocondensation with 1,3-binucleophilic reagents to form spiro[4.4] heterocycles bearing a pharmacophoric pyrrole-2-one moiety (Scheme 3) [28,29]. We employed this feature as a key step in the development of a straightforward and concise synthetic approach towards regioisomeric thiohydantoins and pseudothiohydantoins spiro-fused to a pyrrole-2-one fragment.
![[1860-5397-15-280-i3]](/bjoc/content/inline/1860-5397-15-280-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclocondensation of FPDs 1 with 1,3-binucleophilic reagents, resulting in pyrrole-2-one-bearing spiro[4.4] heterocycles.
Scheme 3: Cyclocondensation of FPDs 1 with 1,3-binucleophilic reagents, resulting in pyrrole-2-one-bearing sp...
To test the preparation of the target spiro-fused pseudothiohydantoins and thiohydantoins, we examined the reaction of FPD 1a with thiourea by heating equimolar amounts of the reagents in toluene for 5 min (until the disappearance of the dark violet color of FPD 1a). The reaction mixture was examined by UPLC–MS. Two major products with m/z = 396 ([M + H]+, ESI+) were observed in a ratio of ≈1:1, which corresponded to adducts of thiourea with FPD 1a. The adducts were isolated, and their structures were elucidated as the desired spiro-fused thiohydantoin 2a and pseudothiohydantoin 3a (Scheme 4).
![[1860-5397-15-280-i4]](/bjoc/content/inline/1860-5397-15-280-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of FPD 1a with thiourea.
Scheme 4: Reaction of FPD 1a with thiourea.
It should be pointed out that earlier, we have communicated initial data on this transformation [30,31]. However, taking into account more advanced structure elucidation methods employed in the present work (e.g., NMR, single crystal X-ray diffraction), we concluded that the structures of the products were identified incorrectly, and they are revised herein (for a detailed revision see Supporting Information File 1).
Next, to find out whether compounds 2a and 3a were prone to undergo PTR, they were heated in open capillaries at 130 °C for 2 h, and the resulting mixtures were examined by UPLC–MS. As a result, we found that compound 2a did not rearrange, and compound 3a partially converted into its regioisomer 2a due to irreversible PTR (Scheme 5).
![[1860-5397-15-280-i5]](/bjoc/content/inline/1860-5397-15-280-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Possibly, when carrying out the reaction of FPD 1a with thiourea by refluxing the reaction mixture in toluene, part of the product 3a was converted into 2a because of PTR. We assumed that at lower temperatures, the main product of the reaction could be pseudothiohydantoin 3a. To prove this assumption, we examined the reaction of FPD 1a with thiourea at room temperature in various solvents (Table 1).
Table 1: Reaction of FPD 1a with thiourea at room temperature in various solvents.a
| Entry | Solvent | 2a:3ab |
|---|---|---|
| 1 | acetone | 30:70 |
| 2 | ethyl acetate | 5:95 |
| 3 | 1,4-dioxane | 50:50 |
| 4 | acetic acid | 30:70 |
| 5 | acetonitrile | 50:50 |
| 6 | butyl acetate | 5:95 |
| 7 | toluene | tracesc |
aReaction mixtures were stirred until the disappearance of the dark violet color of FPD 1a (about 2–4 h). bAccording to UPLC–MS data. cThe reaction proceeded too slowly, and FPD 1a underwent hydrolysis faster than reaction with thiourea (the reaction vessel was contaminated with water during samplings for analyses).
Obviously, the ratio of products 2a and 3a was dependent not only on the temperature of the reaction, but also on the polarity of the solvent. We found that optimal conditions for dominant formation of pseudothiohydantoin 3a were applied by stirring the reaction mixture at room temperature in ethyl or butyl acetate (Table 1, entries 2 and 6).
For the development of a convenient procedure for thiohydantoin 2a without isolation of its regioisomer 3a being required, we carried out the reaction of FPD 1a with thiourea in various solvents under reflux (Table 2).
Table 2: Reaction of FPD 1a with thiourea at reflux in various solvents.a
| Entry | Solvent | 2a:3ab |
|---|---|---|
| 1 | acetone | 30:70c |
| 2 | ethyl acetate | 22:78c |
| 3 | 1,4-dioxane | 90:10c |
| 4 | 1,4-dioxane | 99:1d |
| 5 | acetic acid | 30:70c |
| 6 | acetonitrile | 25:75c |
| 7 | butyl acetate | 95:5c |
| 8 | toluene | 50:50c |
aReaction mixtures were refluxed until the disappearance of the dark violet color of FPD 1a (about 5–15 min). bAccording to UPLC–MS data. cThe reagents were mixed prior the heating. dThiourea was added to a boiling solution of FPD 1a.
As a result of this optimization, we established that thiohydantoin 2a was formed as major product upon refluxing the reaction mixture in butyl acetate or 1,4-dioxane (Table 2, entries 3, 4, and 7). Interestingly, at different reagent ratios, the yield of compound 2a was affected as well (Table 2, entries 3 and 4). When the reagents were mixed prior to heating (Table 2, entry 3), the yield of compound 2a was lower than when the reagents were mixed in the boiling solvent (Table 2, entry 4). Probably, compound 2a was formed not only as a result of the corresponding PTR, but as a result of a concurrent attack of the amino group of thiourea on the C-3a atom of FPD 1a (Scheme 6).
![[1860-5397-15-280-i6]](/bjoc/content/inline/1860-5397-15-280-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Pathways for the formation of compound 2a.
Scheme 6: Pathways for the formation of compound 2a.
Using the optimization data, we obtained a series of spiro-fused thiohydantoins 2a–m and pseudothiohydantoins 3a–m by utilizing various FPDs 1a–n (Table 3). The aryl group-bearing FPDs 1a–m reacted facilely to yield the desired compounds 2 and 3, but ethoxycarbonyl group-bearing FPD 1n reacted unselectively, giving a complex mixture of hardly identifiable products due to the occurrence of multiple side reactions involving the ethoxycarbonyl group.
Table 3: Series of spiro-fused thiohydantoins 2a–m and pseudothiohydantoins 3a–m from various FPDs 1a–n.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-280-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry | R | R' | Yield of 2 (%)a,b | Yield of 3 (%)a,c |
| a | 2-OH-C6H4 | Ph |
97
(CCDC 1952743) |
79 |
| b | 5-Cl-2-OH-C6H3 | Ph | 78 | 87 |
| c | 2-OH-C6H4 | 4-OMe-C6H4 | 79 | 91 |
| d | 2-OH-C6H4 | 4-OEt-C6H4 | 90 | 79 |
| e | 2-OH-C6H4 | 4-Cl-C6H4 | 78 |
61
(CCDC 1952745) |
| f | 2-OH-C6H4 | 4-Br-C6H4 | 87 | 61 |
| g | 2-OH-C6H4 | 4-Me-C6H4 | 89 | 89 |
| h | 5-NO2-2-OH-C6H3 | Ph | 95 | 58 |
| i | 2-OH-C6H4 | 4-NO2-C6H4 | 61 | 53 |
| j | 5-Br-2-OH-C6H3 | Ph | 65 | 59 |
| k | 2-OH-C6H4 | 4-F-C6H4 | 81 | 82 |
| l | CH2CH2OH | 4-Cl-C6H4 |
90
(CCDC 1952798) |
97 |
| m | CH2CH2OH | 4-Me-C6H4 | 98 | 75 |
| n | 2-OH-C6H4 | OEt | – | – |
aIsolated yields. bConditions A: a mixture of FPD 1a–n (1 mmol) and thiourea (1 mmol) was refluxed in 1,4-dioxane (5 mL) for 4 h. cConditions B: a mixture of FPD 1a–n (1 mmol) and thiourea (1 mmol) was stirred in ethyl acetate (5 mL) for 12 h at room temperature.
Next, we examined the influence of substituents in the thiourea motif on its reaction with FPDs 1. It was found that monosubstituted thioureas (N-methylthiourea, N-phenylthiourea, N-(4-chlorophenyl)thiourea, N-(3,5-dimethylphenyl)thiourea, N-(4-nitrophenyl)thiourea, and N-1-naphtylthiourea) reacted with FPD 1a unselectively, forming a mixture of four inseparable adducts (the reaction mixtures were analyzed by UPLC–MS) (Scheme 7). Unfortunately, we did not succeed to find any conditions for a selective formation of either of them.
![[1860-5397-15-280-i7]](/bjoc/content/inline/1860-5397-15-280-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction of FPD 1a with monosubstituted thioureas.
Scheme 7: Reaction of FPD 1a with monosubstituted thioureas.
Unexpectedly, the implementation of N-acetylthiourea in the reaction with FPDs 1, both under conditions A and B (see Table 3), afforded only one type of adducts, pseudothiohydantoins 4 (Table 4). Boiling compounds 4 in various solvents and heating them in open capillaries at 130 °C for 12 h did not provoke PTR, and compounds 4 remained unconverted. This phenomenon could be explained by the influence of the electron-withdrawing effect of the acetyl group, which reduced the nucleophilic properties of the acetyl-substituted nitrogen atom in N-acetylthiourea and decreased its reactivity, preventing the formation of regioisomeric compounds.
Table 4: bReaction of FPDs 1 with N-acetylthiourea.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-280-i11.svg?max-width=637&scale=1.0)
|
|||
| Product | Ar | R | Yield of 4 (%)a |
| 4ab | Ph | H | 80 |
| 4b | 4-Cl-C6H4 | H | 75 |
| 4c | Ph | Cl | 79 |
aIsolated yields. bCCDC 1952746.
1,3-Dibutylthiourea reacted smoothly at room temperature in ethyl acetate with FPDs 1 to form mixtures of products 5 and 6 (Scheme 8). Moreover, it was observed that the percentage of compounds 5 in reaction mixtures increased over time upon storage of the solutions. As such, 6 readily underwent PTR, affording the corresponding compounds 5, but unfortunately, we did not succeed to isolate pseudothiohydantoins 6.
![[1860-5397-15-280-i8]](/bjoc/content/inline/1860-5397-15-280-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reaction of FPDs 1 with 1,3-dibutylthiourea at room temperature.
Scheme 8: Reaction of FPDs 1 with 1,3-dibutylthiourea at room temperature.
The reaction of FPDs 1 with 1,3-dibutylthiourea in 1,4-dioxane resulted in exclusive formation of thiohydantoins 5 upon heating at reflux for 2–4 h (Table 5).
Table 5: Reaction of FPDs 1 with 1,3-dibutylthiourea at reflux.
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-280-i12.svg?max-width=637&scale=1.0)
|
|||
| Product | Ar | R |
Yield of
5 (%)a |
| 5a | Ph | 2-OH-C6H4 | 75 |
| 5bb | Ph | 5-Cl-2-OH-C6H3 | 76 |
| 5c | 4-Cl-C6H4 | CH2CH2OH | 76 |
aIsolated yields. bCCDC 1952744.
The facility of thiohydantoins 5 formation could be explained by the electron-donating effect of the butyl substituents, which increased the nucleophilicity of the butyl-substituted nitrogen atoms and promoted its attack on the electrophilic center C-3a of FPDs 1.
The reaction of FPDs 1 with 1,3-diphenylthiourea proceeded in a similar manner. Thiohydantoins 7 were predominantly formed when the reaction was conducted at reflux in 1,4-dioxane, and pseudothiohydantoins 8 were formed as main products at room temperature (Table 6).
Table 6: Reaction of FPDs 1 with 1,3-diphenylthiourea.
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-280-i13.svg?max-width=637&scale=1.0)
|
||||
| Entry | Ar | R |
Yield of
7 (%)a,b |
Yield of
8 (%)a,c |
| a | Ph | 2-OH-C6H4 | 71 | 78 |
| b | 4-Cl-C6H4 | CH2CH2OH | 63 | 82 |
| c | 4-Me-C6H4 | CH2CH2OH | 72 | 76 |
aIsolated yields. bConditions A: a mixture FPD 1 (1 mmol) and 1,3-diphenylthiourea (1 mmol) was refluxed in 1,4-dioxane (5 mL) for 8–12 h. cConditions B: a mixture FPD 1 (1 mmol) and 1,3-diphenylthiourea (1 mmol) was stirred in ethyl acetate (5 mL) for 12 h at room temperature.
Notably, the formation of thiohydantoins 7 required longer time in comparison with unsubstituted thiourea and 1,3-dibutylthiourea. Possible reasons for this are the influence of the steric hindrance induced by the phenyl substituents and weakening of the nucleophilic properties of the phenyl-substituted nitrogen atom, which prevented a nucleophilic attack of 1,3-diphenylthiourea on the electrophilic center C-3a of FPDs 1.
The observed PTRs could have proceed through two alternative pathways (Scheme 9), with the first stage being dissociation of the Cspiro–S bond [32] in pseudothiohydantoins PThH, affording intermediate A. Then, A could have further undergone either an intramolecular cyclization by NH attack, forming thiohydantoin ThH (path A), or further dissociation to FPD 1 and thiourea. The latter would subsequently attack FPD 1 with both nucleophilic NH centers, forming thiohydantoin ThH (path B).
![[1860-5397-15-280-i9]](/bjoc/content/inline/1860-5397-15-280-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have developed a divergent approach to pharmaceutically interesting regiomeric thiohydantoins and pseudothiohydantoins spiro-fused to a pyrrole-2-one fragment via the reaction of [e]-fused 1H-pyrrole-2,3-diones with thioureas. The obtained pseudothiohydantoins were found to be prone to undergo a pseudothiohydantoin–thiohydantoin rearrangement. Therein, the substituents of the thiourea moiety were found to be crucial for tuning the conditions of this rearrangement. Notably, the reactions proceeded under catalyst-free conditions with good yields.
Supporting Information
| Supporting Information File 1: Experimental details, copies of 1H and 13C NMR spectra, X-ray crystallographic details, references to antimicrobial assay results, and a detailed revision of previously published structures. | ||
| Format: PDF | Size: 4.7 MB | Download |
References
-
Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Chem. Rev. 2017, 117, 13757–13809. doi:10.1021/acs.chemrev.7b00067
Return to citation in text: [1] -
World Health Organization Model List of Essential Medicines, 21st List. World Health Organization: Geneva, 2019; https://www.who.int/medicines/publications/essentialmedicines/ (accessed Sept 11, 2019).
Return to citation in text: [1] -
Krause, T.; Gerbershagen, M. U.; Fiege, M.; Weißhorn, R.; Wappler, F. Anaesthesia 2004, 59, 364–373. doi:10.1111/j.1365-2044.2004.03658.x
Return to citation in text: [1] -
Cereghino, J. J.; Brock, J. T.; Van Meter, J. C.; Kiffin Penry, J.; Smith, L. D.; Fisher, P.; Ellenberg, J. Clin. Pharmacol. Ther. (Hoboken, NJ, U. S.) 1974, 15, 406–416. doi:10.1002/cpt1974154406
Return to citation in text: [1] -
Bianchini, D.; Lorente, D.; Rodriguez-Vida, A.; Omlin, A.; Pezaro, C.; Ferraldeschi, R.; Zivi, A.; Attard, G.; Chowdhury, S.; de Bono, J. S. Eur. J. Cancer 2014, 50, 78–84. doi:10.1016/j.ejca.2013.08.020
Return to citation in text: [1] -
Baell, J. B.; Holloway, G. A. J. Med. Chem. 2010, 53, 2719–2740. doi:10.1021/jm901137j
Return to citation in text: [1] -
Mendgen, T.; Steuer, C.; Klein, C. D. J. Med. Chem. 2012, 55, 743–753. doi:10.1021/jm201243p
Return to citation in text: [1] [2] -
Ulrich, H.; Sayigh, A. A. R. J. Org. Chem. 1965, 30, 2781–2783. doi:10.1021/jo01019a067
Return to citation in text: [1] -
Schaumann, E.; Grabley, S. Justus Liebigs Ann. Chem. 1978, 1568–1585. doi:10.1002/jlac.197819781004
Return to citation in text: [1] -
Yamagishi, M.; Ozaki, K.-i.; Yamada, Y.; Da-Te, T.; Okamura, K.; Suzuki, M. Chem. Pharm. Bull. 1991, 39, 1694–1698. doi:10.1248/cpb.39.1694
Return to citation in text: [1] [2] [3] -
El-Aasar, N. K.; Saied, K. F. J. Heterocycl. Chem. 2008, 45, 645–652. doi:10.1002/jhet.5570450302
Return to citation in text: [1] -
Makarenko, A. G.; Parkhomenko, P. I.; Rozhenko, A. B.; Grigor'ev, A. A.; Rybakova, M. V.; Bezuglyi, Y. V. Chem. Heterocycl. Compd. 1994, 30, 1106–1109. doi:10.1007/bf01171175
Return to citation in text: [1] -
Fathalla, W. M.; Pazdera, P. Molecules 2002, 7, 96–103. doi:10.3390/70100096
Return to citation in text: [1] -
Briel, D.; Aurich, R.; Egerland, U.; Unverferth, K. Pharmazie 2005, 60, 732–735.
Return to citation in text: [1] -
Hilmy, K. M. H.; Soliman, D. H.; Shahin, E. B. A.; El-Deeb, H. S.; El-Kousy, S. M. Eur. J. Med. Chem. 2014, 78, 419–424. doi:10.1016/j.ejmech.2014.03.068
Return to citation in text: [1] -
Belikov, M. Y.; Fedoseev, S. V.; Ievlev, M. Y.; Ershov, O. V. Chem. Heterocycl. Compd. 2017, 53, 1057–1060. doi:10.1007/s10593-017-2170-1
Return to citation in text: [1] -
Marson, C. M. Chem. Soc. Rev. 2011, 40, 5514–5533. doi:10.1039/c1cs15119c
Return to citation in text: [1] -
Zheng, Y.; Tice, C. M.; Singh, S. B. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. doi:10.1016/j.bmcl.2014.06.081
Return to citation in text: [1] -
Lovering, F. MedChemComm 2013, 4, 515–519. doi:10.1039/c2md20347b
Return to citation in text: [1] -
Sauer, W. H. B.; Schwarz, M. K. J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. doi:10.1021/ci025599w
Return to citation in text: [1] -
Aldeghi, M.; Malhotra, S.; Selwood, D. L.; Chan, A. W. E. Chem. Biol. Drug Des. 2014, 83, 450–461. doi:10.1111/cbdd.12260
Return to citation in text: [1] -
Mohammad, H. B.; Khurshid, A.; Sudeep, R.; Jalaluddin, M. A.; Mohd, A.; Mohammad, H. S.; Saif, K.; Mohammad, A. K.; Ivo, P.; Inho, C. Curr. Pharm. Des. 2016, 22, 572–581. doi:10.2174/1381612822666151125000550
Return to citation in text: [1] -
Hung, A. W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J. A.; Clemons, P. A.; Young, D. W. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6799–6804. doi:10.1073/pnas.1015271108
Return to citation in text: [1] -
Murray, C. W.; Rees, D. C. Angew. Chem., Int. Ed. 2016, 55, 488–492. doi:10.1002/anie.201506783
Return to citation in text: [1] -
Stepanova, E. E.; Kasatkina, S. O.; Dmitriev, M. V.; Maslivets, A. N. Synthesis 2018, 50, 4897–4904. doi:10.1055/s-0037-1610647
Return to citation in text: [1] -
Kobelev, A. I.; Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Beilstein J. Org. Chem. 2019, 15, 364–370. doi:10.3762/bjoc.15.32
10.3762/bjoc.15.32.
Return to citation in text: [1] -
Kasatkina, S.; Stepanova, E.; Dmitriev, M.; Mokrushin, I.; Maslivets, A. Tetrahedron Lett. 2019, 60, No. 151088. doi:10.1016/j.tetlet.2019.151088
Return to citation in text: [1] -
Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC (Gainesville, FL, U. S.) 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889
Return to citation in text: [1] -
Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204
Return to citation in text: [1] -
Mashevskaya, I. V.; Kol'tsova, S. V.; Voronina, E. V.; Odegova, T. F.; Maslivets, A. N. Pharm. Chem. J. 2001, 35, 18–21. doi:10.1023/a:1010494525001
Return to citation in text: [1] -
Babenysheva, A. V.; Maslivets, V. A.; Maslivets, A. N. Russ. J. Org. Chem. 2007, 43, 1577–1578. doi:10.1134/s107042800710034x
Return to citation in text: [1] -
Kobelev, A. I.; Stepanova, E. E.; Dmitriev, M. V.; Denislamova, E. S.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 766–770. doi:10.1134/s1070428018050159
Return to citation in text: [1]
| 30. | Mashevskaya, I. V.; Kol'tsova, S. V.; Voronina, E. V.; Odegova, T. F.; Maslivets, A. N. Pharm. Chem. J. 2001, 35, 18–21. doi:10.1023/a:1010494525001 |
| 31. | Babenysheva, A. V.; Maslivets, V. A.; Maslivets, A. N. Russ. J. Org. Chem. 2007, 43, 1577–1578. doi:10.1134/s107042800710034x |
| 25. | Stepanova, E. E.; Kasatkina, S. O.; Dmitriev, M. V.; Maslivets, A. N. Synthesis 2018, 50, 4897–4904. doi:10.1055/s-0037-1610647 |
| 26. |
Kobelev, A. I.; Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Beilstein J. Org. Chem. 2019, 15, 364–370. doi:10.3762/bjoc.15.32
10.3762/bjoc.15.32. |
| 27. | Kasatkina, S.; Stepanova, E.; Dmitriev, M.; Mokrushin, I.; Maslivets, A. Tetrahedron Lett. 2019, 60, No. 151088. doi:10.1016/j.tetlet.2019.151088 |
| 28. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC (Gainesville, FL, U. S.) 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 29. | Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204 |
| 1. | Konnert, L.; Lamaty, F.; Martinez, J.; Colacino, E. Chem. Rev. 2017, 117, 13757–13809. doi:10.1021/acs.chemrev.7b00067 |
| 5. | Bianchini, D.; Lorente, D.; Rodriguez-Vida, A.; Omlin, A.; Pezaro, C.; Ferraldeschi, R.; Zivi, A.; Attard, G.; Chowdhury, S.; de Bono, J. S. Eur. J. Cancer 2014, 50, 78–84. doi:10.1016/j.ejca.2013.08.020 |
| 10. | Yamagishi, M.; Ozaki, K.-i.; Yamada, Y.; Da-Te, T.; Okamura, K.; Suzuki, M. Chem. Pharm. Bull. 1991, 39, 1694–1698. doi:10.1248/cpb.39.1694 |
| 4. | Cereghino, J. J.; Brock, J. T.; Van Meter, J. C.; Kiffin Penry, J.; Smith, L. D.; Fisher, P.; Ellenberg, J. Clin. Pharmacol. Ther. (Hoboken, NJ, U. S.) 1974, 15, 406–416. doi:10.1002/cpt1974154406 |
| 10. | Yamagishi, M.; Ozaki, K.-i.; Yamada, Y.; Da-Te, T.; Okamura, K.; Suzuki, M. Chem. Pharm. Bull. 1991, 39, 1694–1698. doi:10.1248/cpb.39.1694 |
| 3. | Krause, T.; Gerbershagen, M. U.; Fiege, M.; Weißhorn, R.; Wappler, F. Anaesthesia 2004, 59, 364–373. doi:10.1111/j.1365-2044.2004.03658.x |
| 20. | Sauer, W. H. B.; Schwarz, M. K. J. Chem. Inf. Comput. Sci. 2003, 43, 987–1003. doi:10.1021/ci025599w |
| 21. | Aldeghi, M.; Malhotra, S.; Selwood, D. L.; Chan, A. W. E. Chem. Biol. Drug Des. 2014, 83, 450–461. doi:10.1111/cbdd.12260 |
| 22. | Mohammad, H. B.; Khurshid, A.; Sudeep, R.; Jalaluddin, M. A.; Mohd, A.; Mohammad, H. S.; Saif, K.; Mohammad, A. K.; Ivo, P.; Inho, C. Curr. Pharm. Des. 2016, 22, 572–581. doi:10.2174/1381612822666151125000550 |
| 2. | World Health Organization Model List of Essential Medicines, 21st List. World Health Organization: Geneva, 2019; https://www.who.int/medicines/publications/essentialmedicines/ (accessed Sept 11, 2019). |
| 23. | Hung, A. W.; Ramek, A.; Wang, Y.; Kaya, T.; Wilson, J. A.; Clemons, P. A.; Young, D. W. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6799–6804. doi:10.1073/pnas.1015271108 |
| 24. | Murray, C. W.; Rees, D. C. Angew. Chem., Int. Ed. 2016, 55, 488–492. doi:10.1002/anie.201506783 |
| 8. | Ulrich, H.; Sayigh, A. A. R. J. Org. Chem. 1965, 30, 2781–2783. doi:10.1021/jo01019a067 |
| 9. | Schaumann, E.; Grabley, S. Justus Liebigs Ann. Chem. 1978, 1568–1585. doi:10.1002/jlac.197819781004 |
| 10. | Yamagishi, M.; Ozaki, K.-i.; Yamada, Y.; Da-Te, T.; Okamura, K.; Suzuki, M. Chem. Pharm. Bull. 1991, 39, 1694–1698. doi:10.1248/cpb.39.1694 |
| 11. | El-Aasar, N. K.; Saied, K. F. J. Heterocycl. Chem. 2008, 45, 645–652. doi:10.1002/jhet.5570450302 |
| 17. | Marson, C. M. Chem. Soc. Rev. 2011, 40, 5514–5533. doi:10.1039/c1cs15119c |
| 18. | Zheng, Y.; Tice, C. M.; Singh, S. B. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. doi:10.1016/j.bmcl.2014.06.081 |
| 7. | Mendgen, T.; Steuer, C.; Klein, C. D. J. Med. Chem. 2012, 55, 743–753. doi:10.1021/jm201243p |
| 7. | Mendgen, T.; Steuer, C.; Klein, C. D. J. Med. Chem. 2012, 55, 743–753. doi:10.1021/jm201243p |
| 32. | Kobelev, A. I.; Stepanova, E. E.; Dmitriev, M. V.; Denislamova, E. S.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 766–770. doi:10.1134/s1070428018050159 |
| 6. | Baell, J. B.; Holloway, G. A. J. Med. Chem. 2010, 53, 2719–2740. doi:10.1021/jm901137j |
| 12. | Makarenko, A. G.; Parkhomenko, P. I.; Rozhenko, A. B.; Grigor'ev, A. A.; Rybakova, M. V.; Bezuglyi, Y. V. Chem. Heterocycl. Compd. 1994, 30, 1106–1109. doi:10.1007/bf01171175 |
| 13. | Fathalla, W. M.; Pazdera, P. Molecules 2002, 7, 96–103. doi:10.3390/70100096 |
| 14. | Briel, D.; Aurich, R.; Egerland, U.; Unverferth, K. Pharmazie 2005, 60, 732–735. |
| 15. | Hilmy, K. M. H.; Soliman, D. H.; Shahin, E. B. A.; El-Deeb, H. S.; El-Kousy, S. M. Eur. J. Med. Chem. 2014, 78, 419–424. doi:10.1016/j.ejmech.2014.03.068 |
| 16. | Belikov, M. Y.; Fedoseev, S. V.; Ievlev, M. Y.; Ershov, O. V. Chem. Heterocycl. Compd. 2017, 53, 1057–1060. doi:10.1007/s10593-017-2170-1 |
© 2019 Kobelev et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)