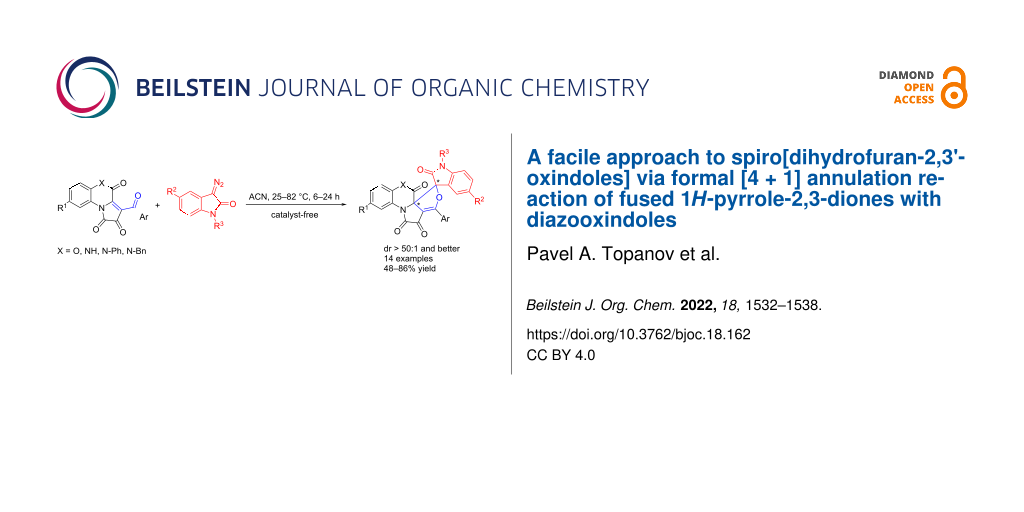
Abstract
There has been developed an easy synthetic approach to spiro[dihydrofuran-2,3'-oxindoles] via a highly diastereoselective formal [4 + 1] cycloaddition reaction of [e]-fused 1H-pyrrole-2,3-diones with diazooxindoles. The described novel heterocyclic systems are heteroanalogues of antimicrobial and antibiofilm fungal metabolites. The developed reaction represents the first example of involving 1H-pyrrole-2,3-diones fused at the [e]-side in a [4 + 1] annulation reaction.

Graphical Abstract
Introduction
Compounds with a spirooxindole scaffold have attracted the attention of researchers, which is demonstrated by the publication of several reviews of both the biological activities of compounds with the spirooxindole moiety [1-5], and methods for constructing spirooxindole systems by employing different approaches [6-12]. Cyclopiazonic acid derivatives such as aspergillins A–E [13] (Figure 1) and speradines C and F [14,15] are secondary metabolites of fungi, and include a furan fragment spiro-fused with 2-oxindole. Cyclopiamides I and J [16] were also isolated from the fungus Penicillium commune and contain a furan fragment spiro-annulated by 2-oxindole. These compounds exhibit anticancer [13] and antimicrobial [17] activities.
![[1860-5397-18-162-1]](/bjoc/content/figures/1860-5397-18-162-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected examples of biologically active natural products bearing a spirofuranoxindole moiety.
Figure 1: Selected examples of biologically active natural products bearing a spirofuranoxindole moiety.
One of the expeditious methods for obtaining dihydrofurans is the cycloaddition reaction of diazo compounds to molecules containing an enone fragment. Cycloaddition reactions involving diazo and enone moieties are usually carried out using transition-metal catalysis [18-20], with catalyst-free reactions being carried out only with the participation of reactive unsubstituted diazomethane [21-23]. With diazooxindoles used as the diazo component, it is possible to obtain the desired spirofuranoxindoles. To date, only one method is known for obtaining spirofuranoxindoles from diazooxindole and an enone, where p-quinone methide acts as the enone, but the reaction requires the use of a catalyst [24] (Scheme 1).
![[1860-5397-18-162-i1]](/bjoc/content/inline/1860-5397-18-162-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of spiro[dihydrofuran-2,3'-oxindoles] from enones and diazooxindoles.
Scheme 1: Synthesis of spiro[dihydrofuran-2,3'-oxindoles] from enones and diazooxindoles.
Thus, in the present work, we report a simple, catalyst-free diastereoselective method for the synthesis of dihydrofurans spiro-annulated with an oxindole moiety for the first time. The essence of the method is the use of [e]-fused 1H-pyrrole-2,3-diones (FPDs) as the enone component in a formal [4 + 1] cycloaddition (Scheme 1).
Results and Discussion
FPDs are highly reactive compounds [25,26] containing an highly electrophilic enone fragment which facilitates the course of cycloaddition and nucleophilic addition reactions. In recent years, some types of cycloaddition reactions were investigated for FPDs: the [4 + 2] cycloaddition with alkenes resulting in pyran-annulated products [27-34] and the [3 + 2] cycloaddition with nitrones resulting in isoxazole-annulated products [35-37] (Scheme 2). However, formal [4 + 1] cycloaddition reactions for FPDs remain to be unknown.
![[1860-5397-18-162-i2]](/bjoc/content/inline/1860-5397-18-162-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cycloaddition reactions of [e]-fused 1H-pyrrole-2,3-diones.
Scheme 2: Cycloaddition reactions of [e]-fused 1H-pyrrole-2,3-diones.
To evaluate the possibility of synthesizing the target spirooxindole compounds, we initially investigated a reaction of benzoxazine-containing FPD 1a with diazooxindole 2a in anhydrous acetonitrile at room temperature (Scheme 3). The reaction came to an end in 24 hours, with the color of the solution being turned from purple to red. The starting FPD 1a is bright violet; thus, the disappearance of the violet color was used as an indicator of the reaction’s completion. Product 3aa was isolated as yellow crystals in 73% yield and characterized by NMR, IR, and mass spectra, and single crystal X-ray analysis (CCDC 2201614). As evinced by the NMR data, only one diastereomer of product 3aa was obtained. Contrary to the isoxazole-annulated products of a [3 + 2] cycloaddition of nitrones to FPDs [35], product 3aa appears to be stable on storage in solution, which was confirmed by the fact that the NMR data remained unvaried after keeping the product in solution for one day.
![[1860-5397-18-162-i3]](/bjoc/content/inline/1860-5397-18-162-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The model reaction of FPD 1a and diazooxindole 2a.
Scheme 3: The model reaction of FPD 1a and diazooxindole 2a.
Next, the conditions (Table 1) of the model reaction of FPD 1a and diazooxindole 2a were optimized.
Table 1: Reaction of FPD 1a and diazooxindole 2a in different solvents.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-162-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Solvent | Temperature, °C | Yieldb, % |
| 1 | toluene | 25 | 3 |
| 2 | chloroform | 25 | 48 |
| 3 | ethyl acetate | 25 | 29 |
| 4 | 1,4-dioxane | 25 | 9 |
| 5 | DMSO | 25 | 32 |
| 6 | acetonitrile | 25 | 82 |
| 7 | acetonitrile | 25 | 73c |
| 8 | acetonitrile | 83 | 50c |
aA suspension of FPD 1a (100 µmol, 32.0 mg) and diazooxindole 2a (100 µmol, 16.0 mg) in the corresponding solvent (1 mL) was stirred in an oven-dried closed microreaction vial for 24 hours; bUPLC yield (the chromatograms were recorded immediately after sample preparation); cisolated yield.
The best yield of product 3aа (Table 1, entries 6 and 7) was obtained by the reaction performed in acetonitrile at room temperature, therefore, these conditions were taken as a standard for further reactions.
Next, the reagent scope of the reaction was explored by involving diazooxindoles 2a–d into the reaction with FPD 1a (Table 2).
Table 2: Reaction of FPD 1a and diazooxindoles 2a–d.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-162-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Diazooxindole | Product | R1 | R2 | Time, hb | Yieldc, % | drd |
| 1 | 2a | 3aa | H | H | 24 | 73 | >99:1 |
| 2 | 2b | 3ab | H | Br | 168e | –e | – |
| 3 | 2b | 3ab | H | Br | 6f | 60 | >99:1 |
| 4 | 2c | 3ac | H | OMe | 24 | 61 | >99:1 |
| 5 | 2d | 3ad | Bn | H | 24 | 58 | 50:1 |
aA suspension of FPD 1a (500 µmol, 160.0 mg) and diazooxindole 2a–d (500 µmol) in acetonitrile (3 mL) was stirred in an oven-dried closed microreaction vial for the given time; breaction time was monitored by the disappearance of the dark violet color of FPD 1a; cisolated yield; dratio was determined by 1H NMR in isolated product; eno disappearance of dark violet color of FPD 1a, the reaction was monitored by UPLC-MS (the reaction not completed within a week); fthe reaction was carried out in refluxing solvent.
Compared to substrate 2a, the presence of substituents in diazo compounds 2b–d led to decreased reaction yields (Table 2, entries 2–5). The reaction with diazooxindole 2b having an electron-withdrawing group (-Br) in the C(5) position (Table 2, entries 2 and 3) required additional heating to obtain the product 3ab. On the other hand, the reaction of diazooxindoles 2c and 2d (Table 2, entries 4 and 5) bearing an EDG (electron-donating group) in positions N(1) (Bn-) or C(5) (MeO-) did not require heating and proceeded under conditions similar to the ones with unsubstituted diazooxindole 2a.
Next, we investigated the substrate scope using different FPDs 2 (Table 3).
Table 3: Reaction of FPDs 1a–j and diazooxindole 2a.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-162-i8.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Product | R1 | R2 | X | Time, hb | Yieldc, % | drd |
| 1 | 3aa | H | Ph | O | 24 | 73 | >99:1 |
| 2 | 3ba | H | C6H4Cl-4 | O | 24 | 71 | >99:1 |
| 3 | 3ca | H | C6H4Br-4 | O | 24 | 62 | >99:1 |
| 4 | 3da | H | C6H4Me-4 | O | 24 | 73 | >99:1 |
| 5 | 3ea | H | C6H4OMe-4 | O | 24 | 78 | >99:1 |
| 6 | 3fa | H | C6H4NO2-4 | O | 24 | 48 | >99:1 |
| 7 | 3ga | Cl | Ph | O | 24 | 86 | >99:1 |
| 8 | 3ha | H | C6H4Cl-4 | NH | 6e | 70 | 50:1 |
| 9 | 3ia | H | Ph | N–Ph | 6e | 85 | >99:1 |
| 10 | 3ja | H | C6H4Cl-4 | N–Bn | 6e | 63 | >99:1 |
aA suspension of FPD 1a–j (500 µmol) and diazooxindole 2a (500 µmol, 80.0 mg) in acetonitrile (3 mL) was stirred in an oven-dried closed microreaction vial; breaction time was monitored by the disappearance of the dark violet color of FPD 1; cisolated yield; dratio was determined by 1H NMR in isolated product; ethe reaction was carried out in refluxing solvent.
FPDs 1а–f successfully reacted with diazooxindole 2а under the previously developed conditions and gave good product yields (Table 3, entries 1–6). Neither the yield, nor the reaction rate were observed as being markedly affected by electron-donating or weak electron-withdrawing groups present in the aroyl substituent of FPDs 1. However, the presence of a strong electron-withdrawing group (–NO2) in the aroyl fragment of the FPD 1f, significantly decreased the yield of the target product 3fa (Table 3, entry 6). The introduction of an electron-withdrawing group (–Cl) into the benzoxazine fragment of FPD 1g increased the yield of the target reaction product, without affecting the reaction rate (Table 3, entry 7). Quinoxaline-annulated FPDs 1h–j required heating, as these compounds reacted too slowly at room temperature (Table 3, entries 8–10). It should be noted that FPDs 1h–j gave yields of the target products close to that of the products obtained from FPDs annulated with a benzoxazine fragment. The structures of products 3aa, 3ab, and 3ha were approved by single crystal X-ray analysis (CCDC 2201614, CCDC 2201616, CCDC 2201615).
We also decided to study the effect the benzo-annulated moiety in FPDs has on inducing the reaction. Under the same conditions, FPD 1k containing a morpholine fragment (Scheme 4) was involved in the reaction with 2a which gave the expected product 3ka in a fairly good yield of 56% and dr 99:1. The characteristic signals in the NMR spectra of the products 3aa and 3ka appeared to be the same; thus, the structure of product 3ka was ascertained to be similar to that of product 3aa.
![[1860-5397-18-162-i4]](/bjoc/content/inline/1860-5397-18-162-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The reaction of FPD 1k with diazooxindole 2a.
Scheme 4: The reaction of FPD 1k with diazooxindole 2a.
With the above observations and the reported literature [24] as a basis, the formation of spirofuranoxindoles 3aa–ka was assumed as proceeding via two stages: (a) the nucleophilic Michael attack of the negatively charged [38] C(3) atom of diazooxindoles 2 at the C(3a) atom of FPDs 1 (Scheme 5), and (b) further intramolecular SN2 attack by the oxygen of the aroyl group with ensuing elimination of a nitrogen molecule. To verify our assumption, 3-bromooxindole (4) was involved in the reaction with FPD 1i in the presence of 1.1 equiv of TEA. In this case, the base-promoted deprotonation of 3-bromooxindole (4) affords a highly nucleophilic intermediate, which undergoes Michael addition to FPD 1i, followed by intramolecular SN2 attack [39-43] by the oxygen of the aroyl group (Scheme 5) to give the same diastereomer 3ia with a good 54% yield.
![[1860-5397-18-162-i5]](/bjoc/content/inline/1860-5397-18-162-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A) Plausible mechanism of formal [4 + 1] cycloaddition of FPDs 1 with diazooxindoles 2 (negative charge delocalization is colored in blue); B) plausible base-promoted reaction mechanism of FPD 1i and 3-bromooxindole (4, negative charge delocalization is colored blue).
Scheme 5: A) Plausible mechanism of formal [4 + 1] cycloaddition of FPDs 1 with diazooxindoles 2 (negative ch...
Conclusion
To conclude, we have developed a facile synthetic approach to spirofuranoxindoles 3 via the highly diastereoselective formal [4 + 1] cycloaddition reaction of FPDs 1 with diazooxindoles 2. The obtained compounds 3 were found to be stable. Benzoxazine, quinoxaline, and morpholine FPDs were successfully involved into the reaction, with the modification of diazo compounds decreasing the reaction yield. The reaction time of the cycloaddition was found to be independent on substituents in the aroyl moiety of FPDs 1. The described reaction is the first example of a catalyst-free formal [4 + 1] cycloaddition reaction of enones and complex diazo compounds. The synthesized compounds 3 have a pharmaceutically interesting fungal metabolites-like structure with a spiro[dihydrofuran-2,3'-oxindole] moiety.
Supporting Information
| Supporting Information File 1: Experimental part, compound characterization, and copies of NMR spectra. | ||
| Format: PDF | Size: 4.6 MB | Download |
References
-
Yu, B.; Zheng, Y.-C.; Shi, X.-J.; Qi, P.-P.; Liu, H.-M. Anti-Cancer Agents Med. Chem. 2016, 16, 1315–1324. doi:10.2174/1871520615666151102093825
Return to citation in text: [1] -
Riedinger, C.; McDonnell, J. M. Future Med. Chem. 2009, 1, 1075–1094. doi:10.4155/fmc.09.75
Return to citation in text: [1] -
Zak, K.; Pecak, A.; Rys, B.; Wladyka, B.; Dömling, A.; Weber, L.; Holak, T. A.; Dubin, G. Expert Opin. Ther. Pat. 2013, 23, 425–448. doi:10.1517/13543776.2013.765405
Return to citation in text: [1] -
Arulananda Babu, S.; Padmavathi, R.; Ahmad Aslam, N.; Rajkumar, V. Stud. Nat. Prod. Chem. 2015, 46, 227–339. doi:10.1016/b978-0-444-63462-7.00008-7
Return to citation in text: [1] -
Zhou, L.-M.; Qu, R.-Y.; Yang, G.-F. Expert Opin. Drug Discovery 2020, 15, 603–625. doi:10.1080/17460441.2020.1733526
Return to citation in text: [1] -
Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8
Return to citation in text: [1] -
Nasri, S.; Bayat, M.; Mirzaei, F. Top. Curr. Chem. 2021, 379, 25. doi:10.1007/s41061-021-00337-7
Return to citation in text: [1] -
Gasperi, T.; Miceli, M.; Campagne, J.-M.; Marcia de Figueiredo, R. Molecules 2017, 22, 1636. doi:10.3390/molecules22101636
Return to citation in text: [1] -
Xia, M.; Ma, R.-Z. J. Heterocycl. Chem. 2014, 51, 539–554. doi:10.1002/jhet.1114
Return to citation in text: [1] -
Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r
Return to citation in text: [1] -
Mei, G.-J.; Shi, F. Chem. Commun. 2018, 54, 6607–6621. doi:10.1039/c8cc02364f
Return to citation in text: [1] -
Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Rev. 2015, 115, 5301–5365. doi:10.1021/cr5006974
Return to citation in text: [1] -
Zhou, M.; Miao, M.-M.; Du, G.; Li, X.-N.; Shang, S.-Z.; Zhao, W.; Liu, Z.-H.; Yang, G.-Y.; Che, C.-T.; Hu, Q.-F.; Gao, X.-M. Org. Lett. 2014, 16, 5016–5019. doi:10.1021/ol502307u
Return to citation in text: [1] [2] -
Ma, X.; Peng, J.; Wu, G.; Zhu, T.; Li, G.; Gu, Q.; Li, D. Tetrahedron 2015, 71, 3522–3527. doi:10.1016/j.tet.2015.03.050
Return to citation in text: [1] -
Hu, X.; Xia, Q.-W.; Zhao, Y.-Y.; Zheng, Q.-H.; Liu, Q.-Y.; Chen, L.; Zhang, Q.-Q. Chem. Pharm. Bull. 2014, 62, 942–946. doi:10.1248/cpb.c14-00312
Return to citation in text: [1] -
Xu, X.; Zhang, X.; Nong, X.; Wei, X.; Qi, S. Tetrahedron 2015, 71, 610–615. doi:10.1016/j.tet.2014.12.031
Return to citation in text: [1] -
Gomes, N. M.; Bessa, L. J.; Buttachon, S.; Costa, P. M.; Buaruang, J.; Dethoup, T.; Silva, A. M. S.; Kijjoa, A. Mar. Drugs 2014, 12, 822–839. doi:10.3390/md12020822
Return to citation in text: [1] -
Zhou, J.-L.; Wang, L.-J.; Xu, H.; Sun, X.-L.; Tang, Y. ACS Catal. 2013, 3, 685–688. doi:10.1021/cs400019u
Return to citation in text: [1] -
Liu, C.-R.; Zhu, B.-H.; Zheng, J.-C.; Sun, X.-L.; Xie, Z.; Tang, Y. Chem. Commun. 2011, 47, 1342–1344. doi:10.1039/c0cc02347g
Return to citation in text: [1] -
Şeyma Güngör, F.; Hancıoğlu, N.; Anaç, O. Helv. Chim. Acta 2013, 96, 488–493. doi:10.1002/hlca.201200233
Return to citation in text: [1] -
Avolio, F.; Rimando, A. M.; Cimmino, A.; Andolfi, A.; Jain, S.; Tekwani, B. L.; Evidente, A. J. Antibiot. 2014, 67, 597–601. doi:10.1038/ja.2014.47
Return to citation in text: [1] -
Hadi, V.; Hotard, M.; Ling, T.; Salinas, Y. G.; Palacios, G.; Connelly, M.; Rivas, F. Eur. J. Med. Chem. 2013, 65, 376–380. doi:10.1016/j.ejmech.2013.04.030
Return to citation in text: [1] -
Mustafa, A.; Hilmy, M. K. J. Chem. Soc. 1952, 1434–1436. doi:10.1039/jr9520001434
Return to citation in text: [1] -
Wu, Y.-C.; Cui, B.-D.; Long, Y.; Han, W.-Y.; Wan, N.-W.; Yuan, W.-C.; Chen, Y.-Z. Adv. Synth. Catal. 2021, 363, 1702–1713. doi:10.1002/adsc.202001309
Return to citation in text: [1] [2] -
Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889
Return to citation in text: [1] -
Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204
Return to citation in text: [1] -
Stepanova, E. E.; Babenysheva, A. V.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 937–940. doi:10.1134/s1070428011060182
Return to citation in text: [1] -
Stepanova, E. E.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2013, 49, 1762–1767. doi:10.1134/s1070428013120105
Return to citation in text: [1] -
Stepanova, E. E.; Maslivets, A. N. Russ. J. Org. Chem. 2014, 50, 1382–1383. doi:10.1134/s1070428014090279
Return to citation in text: [1] -
Stepanova, E. E.; Maslivets, A. N. Russ. J. Org. Chem. 2016, 52, 879–882. doi:10.1134/s1070428016060191
Return to citation in text: [1] -
Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2017, 53, 1851–1856. doi:10.1134/s1070428017120119
Return to citation in text: [1] -
Kasatkina, S. O.; Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1515–1518. doi:10.1134/s1070428018100135
Return to citation in text: [1] -
Kasatkina, S. O.; Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1055–1060. doi:10.1134/s107042801807014x
Return to citation in text: [1] -
Khramtsova, E. E.; Lystsova, E. A.; Dmitriev, M. V.; Maslivets, A. N.; Jasiński, R. ChemistrySelect 2021, 6, 6295–6301. doi:10.1002/slct.202101990
Return to citation in text: [1] -
Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Tetrahedron Lett. 2020, 61, 151595. doi:10.1016/j.tetlet.2020.151595
Return to citation in text: [1] [2] -
Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2021, 57, 32–37. doi:10.1134/s107042802101005x
Return to citation in text: [1] -
Sabitov, A. A.; Khramtsova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2022, 58, 287–294. doi:10.1134/s1070428022030058
Return to citation in text: [1] -
Murphy, G. K.; Abbas, F. Z.; Poulton, A. V. Adv. Synth. Catal. 2014, 356, 2919–2923. doi:10.1002/adsc.201400581
Return to citation in text: [1] -
Chuang, C.-P.; Tsai, A.-I. Synthesis 2006, 675–679. doi:10.1055/s-2006-926300
Return to citation in text: [1] -
Chen, R.; Fan, X.; Xu, Z.; He, Z. Tetrahedron Lett. 2017, 58, 3722–3726. doi:10.1016/j.tetlet.2017.08.027
Return to citation in text: [1] -
Liu, Y.-Q.; Li, Q.-Z.; Zhu, H.-P.; Feng, X.; Peng, C.; Huang, W.; Li, J.-L.; Han, B. J. Org. Chem. 2018, 83, 12753–12762. doi:10.1021/acs.joc.8b02189
Return to citation in text: [1] -
Jiang, X.-L.; Liu, S.-J.; Gu, Y.-Q.; Mei, G.-J.; Shi, F. Adv. Synth. Catal. 2017, 359, 3341–3346. doi:10.1002/adsc.201700487
Return to citation in text: [1] -
Lu, Z.; Zhang, Q.; Ke, M.; Hu, S.; Xiao, X.; Chen, F. J. Org. Chem. 2021, 86, 7625–7635. doi:10.1021/acs.joc.1c00672
Return to citation in text: [1]
| 39. | Chuang, C.-P.; Tsai, A.-I. Synthesis 2006, 675–679. doi:10.1055/s-2006-926300 |
| 40. | Chen, R.; Fan, X.; Xu, Z.; He, Z. Tetrahedron Lett. 2017, 58, 3722–3726. doi:10.1016/j.tetlet.2017.08.027 |
| 41. | Liu, Y.-Q.; Li, Q.-Z.; Zhu, H.-P.; Feng, X.; Peng, C.; Huang, W.; Li, J.-L.; Han, B. J. Org. Chem. 2018, 83, 12753–12762. doi:10.1021/acs.joc.8b02189 |
| 42. | Jiang, X.-L.; Liu, S.-J.; Gu, Y.-Q.; Mei, G.-J.; Shi, F. Adv. Synth. Catal. 2017, 359, 3341–3346. doi:10.1002/adsc.201700487 |
| 43. | Lu, Z.; Zhang, Q.; Ke, M.; Hu, S.; Xiao, X.; Chen, F. J. Org. Chem. 2021, 86, 7625–7635. doi:10.1021/acs.joc.1c00672 |
| 1. | Yu, B.; Zheng, Y.-C.; Shi, X.-J.; Qi, P.-P.; Liu, H.-M. Anti-Cancer Agents Med. Chem. 2016, 16, 1315–1324. doi:10.2174/1871520615666151102093825 |
| 2. | Riedinger, C.; McDonnell, J. M. Future Med. Chem. 2009, 1, 1075–1094. doi:10.4155/fmc.09.75 |
| 3. | Zak, K.; Pecak, A.; Rys, B.; Wladyka, B.; Dömling, A.; Weber, L.; Holak, T. A.; Dubin, G. Expert Opin. Ther. Pat. 2013, 23, 425–448. doi:10.1517/13543776.2013.765405 |
| 4. | Arulananda Babu, S.; Padmavathi, R.; Ahmad Aslam, N.; Rajkumar, V. Stud. Nat. Prod. Chem. 2015, 46, 227–339. doi:10.1016/b978-0-444-63462-7.00008-7 |
| 5. | Zhou, L.-M.; Qu, R.-Y.; Yang, G.-F. Expert Opin. Drug Discovery 2020, 15, 603–625. doi:10.1080/17460441.2020.1733526 |
| 16. | Xu, X.; Zhang, X.; Nong, X.; Wei, X.; Qi, S. Tetrahedron 2015, 71, 610–615. doi:10.1016/j.tet.2014.12.031 |
| 24. | Wu, Y.-C.; Cui, B.-D.; Long, Y.; Han, W.-Y.; Wan, N.-W.; Yuan, W.-C.; Chen, Y.-Z. Adv. Synth. Catal. 2021, 363, 1702–1713. doi:10.1002/adsc.202001309 |
| 14. | Ma, X.; Peng, J.; Wu, G.; Zhu, T.; Li, G.; Gu, Q.; Li, D. Tetrahedron 2015, 71, 3522–3527. doi:10.1016/j.tet.2015.03.050 |
| 15. | Hu, X.; Xia, Q.-W.; Zhao, Y.-Y.; Zheng, Q.-H.; Liu, Q.-Y.; Chen, L.; Zhang, Q.-Q. Chem. Pharm. Bull. 2014, 62, 942–946. doi:10.1248/cpb.c14-00312 |
| 38. | Murphy, G. K.; Abbas, F. Z.; Poulton, A. V. Adv. Synth. Catal. 2014, 356, 2919–2923. doi:10.1002/adsc.201400581 |
| 13. | Zhou, M.; Miao, M.-M.; Du, G.; Li, X.-N.; Shang, S.-Z.; Zhao, W.; Liu, Z.-H.; Yang, G.-Y.; Che, C.-T.; Hu, Q.-F.; Gao, X.-M. Org. Lett. 2014, 16, 5016–5019. doi:10.1021/ol502307u |
| 35. | Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Tetrahedron Lett. 2020, 61, 151595. doi:10.1016/j.tetlet.2020.151595 |
| 36. | Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2021, 57, 32–37. doi:10.1134/s107042802101005x |
| 37. | Sabitov, A. A.; Khramtsova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2022, 58, 287–294. doi:10.1134/s1070428022030058 |
| 6. | Pavlovska, T. L.; Redkin, R. G.; Lipson, V. V.; Atamanuk, D. V. Mol. Diversity 2016, 20, 299–344. doi:10.1007/s11030-015-9629-8 |
| 7. | Nasri, S.; Bayat, M.; Mirzaei, F. Top. Curr. Chem. 2021, 379, 25. doi:10.1007/s41061-021-00337-7 |
| 8. | Gasperi, T.; Miceli, M.; Campagne, J.-M.; Marcia de Figueiredo, R. Molecules 2017, 22, 1636. doi:10.3390/molecules22101636 |
| 9. | Xia, M.; Ma, R.-Z. J. Heterocycl. Chem. 2014, 51, 539–554. doi:10.1002/jhet.1114 |
| 10. | Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r |
| 11. | Mei, G.-J.; Shi, F. Chem. Commun. 2018, 54, 6607–6621. doi:10.1039/c8cc02364f |
| 12. | Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Rev. 2015, 115, 5301–5365. doi:10.1021/cr5006974 |
| 35. | Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Tetrahedron Lett. 2020, 61, 151595. doi:10.1016/j.tetlet.2020.151595 |
| 21. | Avolio, F.; Rimando, A. M.; Cimmino, A.; Andolfi, A.; Jain, S.; Tekwani, B. L.; Evidente, A. J. Antibiot. 2014, 67, 597–601. doi:10.1038/ja.2014.47 |
| 22. | Hadi, V.; Hotard, M.; Ling, T.; Salinas, Y. G.; Palacios, G.; Connelly, M.; Rivas, F. Eur. J. Med. Chem. 2013, 65, 376–380. doi:10.1016/j.ejmech.2013.04.030 |
| 23. | Mustafa, A.; Hilmy, M. K. J. Chem. Soc. 1952, 1434–1436. doi:10.1039/jr9520001434 |
| 25. | Konovalova, V. V.; Shklyaev, Y. V.; Maslivets, A. N. ARKIVOC 2015, No. i, 48–69. doi:10.3998/ark.5550190.p008.889 |
| 26. | Konovalova, V. V.; Maslivets, A. N. Mini-Rev. Org. Chem. 2019, 16, 173–192. doi:10.2174/1570193x15666180712115204 |
| 18. | Zhou, J.-L.; Wang, L.-J.; Xu, H.; Sun, X.-L.; Tang, Y. ACS Catal. 2013, 3, 685–688. doi:10.1021/cs400019u |
| 19. | Liu, C.-R.; Zhu, B.-H.; Zheng, J.-C.; Sun, X.-L.; Xie, Z.; Tang, Y. Chem. Commun. 2011, 47, 1342–1344. doi:10.1039/c0cc02347g |
| 20. | Şeyma Güngör, F.; Hancıoğlu, N.; Anaç, O. Helv. Chim. Acta 2013, 96, 488–493. doi:10.1002/hlca.201200233 |
| 27. | Stepanova, E. E.; Babenysheva, A. V.; Maslivets, A. N. Russ. J. Org. Chem. 2011, 47, 937–940. doi:10.1134/s1070428011060182 |
| 28. | Stepanova, E. E.; Aliev, Z. G.; Maslivets, A. N. Russ. J. Org. Chem. 2013, 49, 1762–1767. doi:10.1134/s1070428013120105 |
| 29. | Stepanova, E. E.; Maslivets, A. N. Russ. J. Org. Chem. 2014, 50, 1382–1383. doi:10.1134/s1070428014090279 |
| 30. | Stepanova, E. E.; Maslivets, A. N. Russ. J. Org. Chem. 2016, 52, 879–882. doi:10.1134/s1070428016060191 |
| 31. | Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2017, 53, 1851–1856. doi:10.1134/s1070428017120119 |
| 32. | Kasatkina, S. O.; Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1515–1518. doi:10.1134/s1070428018100135 |
| 33. | Kasatkina, S. O.; Stepanova, E. E.; Dmitriev, M. V.; Maslivets, A. N. Russ. J. Org. Chem. 2018, 54, 1055–1060. doi:10.1134/s107042801807014x |
| 34. | Khramtsova, E. E.; Lystsova, E. A.; Dmitriev, M. V.; Maslivets, A. N.; Jasiński, R. ChemistrySelect 2021, 6, 6295–6301. doi:10.1002/slct.202101990 |
| 17. | Gomes, N. M.; Bessa, L. J.; Buttachon, S.; Costa, P. M.; Buaruang, J.; Dethoup, T.; Silva, A. M. S.; Kijjoa, A. Mar. Drugs 2014, 12, 822–839. doi:10.3390/md12020822 |
| 13. | Zhou, M.; Miao, M.-M.; Du, G.; Li, X.-N.; Shang, S.-Z.; Zhao, W.; Liu, Z.-H.; Yang, G.-Y.; Che, C.-T.; Hu, Q.-F.; Gao, X.-M. Org. Lett. 2014, 16, 5016–5019. doi:10.1021/ol502307u |
| 24. | Wu, Y.-C.; Cui, B.-D.; Long, Y.; Han, W.-Y.; Wan, N.-W.; Yuan, W.-C.; Chen, Y.-Z. Adv. Synth. Catal. 2021, 363, 1702–1713. doi:10.1002/adsc.202001309 |
© 2022 Topanov et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.