Abstract
It was demonstrated that the γ-valerolactone-based ionic liquid, tetrabutylphosphonium 4-ethoxyvalerate as a partially bio-based solvent can be utilized as alternative reaction medium for copper- and auxiliary base-free Pd-catalyzed Sonogashira coupling reactions of aryl iodides and functionalized acetylenes under mild conditions. Twenty-two cross-coupling products were isolated with good to excellent yields (72–99%) and purity (>98%). These results represent an example which proves that biomass-derived safer solvents can be utilized efficiently in common, industrially important transformations exhibiting higher chemical and environmental efficiency.
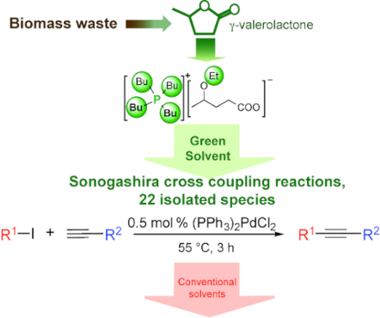
Graphical Abstract
Introduction
In the past few decades, the transition-metal-catalyzed coupling reaction has represented one of the most powerful and atom economical strategies for the efficient assembly of new carbon–carbon bonds. It has therefore become the most attractive approach to the synthesis of a wide range of functionalized organic molecules from laboratory to industrial scale [1-3]. Among these tools, the Pd-catalyzed coupling reactions have received substantial attention, due to the mild operation conditions, excellent functional group tolerance and chemoselectivity as well as wide applicability from syntheses of common building blocks to agrochemicals, just to name a few advantages [4-6]. From the series of palladium-assisted C–C bond formation, the Sonogashira coupling reaction has been identified as a viable synthetic method for the preparation of various alkenyl- and arylacetylenes [7,8] having great importance in organic synthetic schemes of the pharmaceutical industry.
From the environmental point of view, the Sonogashira reactions are usually performed in fossil-based common organic reaction media having high vapor pressure even at higher temperatures, toxicity, flammability, etc., which could result in several serious environmental concerns, especially when they are released into the atmosphere. According to the FDA guidelines [9], the typical solvents of Sonogashira reactions such as toluene [10], THF [11], DMF [12], NMP [13], DMA [14], or MeCN [15] are classified into Class 2, of which applications should be strictly limited, particularly in pharmaceutical industry. To develop an environmentally benign alternative of this useful method, the reaction has been extended to green solvents such as water [16], fluorous solvents [17], supercritical CO2 [18], and very recently γ-valerolactone [19]. The series of these alternative media can implicitly be continued by ionic liquids (ILs) [20], which have attracted considerable attention, due to their extremely low vapor pressure, good solvating properties, reasonable thermal stability, and easily tuneable physical properties [21]. Accordingly, the Sonogashira reactions were also successfully performed in conventional ionic liquids such as [BMIM][PF6] [22-25], [BMIM][BF4] [23], [HMIM][BF4] [24], [EMIM][NTf2] [26], [nBuPy][X] (X = PF6–, BF4–, NO3–) [27], [DectBu3P][BF4] [28]. It was found that some of these systems could operate copper-free [25,27,29] and/or auxiliary base-free conditions [30]. Recently, some “designer" ionic liquids were also developed for this purpose [29-33] from which an imidazolium-based piperidine-appended one could act as task specific compound operating either as a solvent in itself [31] or as an additive to the common ionic liquids [30,33]. It should be noted; however, significant catalyst loadings (5–10 mol %) were necessary to obtain reasonable product yields for latter reactions.
Although the Sonogashira coupling is a well-studied transformation, it has not been carried out in γ-valerolactone-originated ILs, which can act as solvent, ligand and base. Thus, the preparation of various acetylenes in a partially or even biomass-based solvent without any auxiliary material could further control and reduce the environmental impacts of this industrially important transformation.
Herein we report a study on the palladium-catalyzed copper- and added base-free Sonogashira coupling reactions to synthesize various acetylenes in γ-valerolactone-based ionic liquids under mild conditions.
Results and Discussion
We recently demonstrated that copper-catalyzed Ullmann-type N–C coupling reactions could be performed in tetrabutylphosphonium 4-alkoxyvalerate-type ionic liquids, which can easily be synthesized from the renewable platform chemical γ-valerolactone and have negligible vapor pressures even at high temperatures [34]. In order to extend the applicability of valerate-based ionic liquids, the conventional imidazolium-type media and tetrabutylphosphonium 4-ethoxyvalerate ([TBP][4EtOV]) were compared in the coupling of iodobenzene (1a) and phenylacetylene (2a) as a model reaction (Scheme 1) under typically used “Sonogashira conditions” [7,35].
![[1860-5397-15-284-i1]](/bjoc/content/inline/1860-5397-15-284-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Palladium-catalyzed Sonogashira cross-coupling of iodobenzene (1a) and phenylacetylene (2a) in ionic liquids.
Scheme 1: Palladium-catalyzed Sonogashira cross-coupling of iodobenzene (1a) and phenylacetylene (2a) in ioni...
Because the role of ionic liquids as coordination agents for transition metal species was demonstrated [36,37] and palladium carboxylate complexes are well-known compounds, it can be proposed that the carboxylate group of the 4-ethoxyvalerate anion could stabilize the catalytically active species and therefore the ligand can be eliminated from the catalytic system.
Complete conversions of 1a to diphenylacetylene (3a) were detected in the presence of Pd, Cu, and triethylamine (Et3N) in all the ILs (Table 1, #1). It should be noted that the typically utilized base, Et3N has a foul smell and could affect the product isolation procedure. In addition, it is well-established that the presence of Cu(I) salt can promote the in situ formation of some Cu(I) acetylides, which can readily undergo oxidative homocoupling of alkynes even in their slight excess in the reaction mixture [15,38,39]. Thus, the elimination of these auxiliary materials could result in an environmental benign alternative protocol. Hence, the scope of the reaction was investigated with a combination of palladium, copper co-catalyst and Et3N as a base. By elimination of toxic Et3N (LD50(rat, oral) = 560 mg/kg) [40], no reactions were detected in imidazolium-type ILs. However, complete conversion of 1a was observed in [TBP][4EtOV], without Et3N, proving that the solvent can act as a base in itself (Table 1, #2) as it was demonstrated for Cu-catalyzed C–N coupling reactions [34].
Table 1: Sonogashira coupling reaction of iodobenzene (1a) and phenylacetylene (2a) in different ionic liquids.a
| Conversion of iodobenzene in the presence of | |||
| Ionic liquid |
Pd, Cu, and Et3N
(#1) |
Pd and Cu
(#2) |
Pd
(#3) |
| [BMIM][BF4] | >99% | <1% | <1% |
| [EMIM][BF4] | >99% | <1% | <1% |
| [BMIM][Octyls]b | >99% | <1% | <1% |
| [BMIM][PF6] | >99% | <1% | <1% |
| [TBP][4EtOV] | >99% | >99% | >99% |
aReaction conditions: 0.8 mL ionic liquid, 0.5 mol % PdCl2(PPh3)2, 1 mol % CuI, 0.75 mmol phenylacetylene, 0.5 mmol iodobenzene, 0.75 mmol Et3N, T = 55 °C, t = 3 h. bOctyls: octylsulfate anion.
When, we attempted to couple 1a and 2a in the absence of any copper salt as cocatalyst, complete formation of 3a was also detected after 3 h revealing that copper can be eliminated from the system without any decrease in the system’s efficiency.
The source of palladium could also have a significant effect on the reaction’s performance [23]. It was found that while Pd(PPh3)4, palladium acetate (Pd(OAc)2), PdCl2(PPh3)2, and tris(dibenzylideneacetone)dipalladium (Pd2(DBA)3) all catalyzed the Sonogashira reaction, the PdCl2(PPh3)2 precursor turned out to have the best activity in the light of reaction rates (Figure 1). In the presence of 0.5 mol % of PdCl2(PPh3)2 the treatment of 0.5 mmol 1a with 0.75 mmol of 2a afforded complete conversion of 1a in 40 min. Similar performance of PdCl2(PPh3)2 were reported by Ryu, when [BMIM][PF6] was used as reaction medium [26]; however, by the use of 10 times higher catalyst loading.
![[1860-5397-15-284-1]](/bjoc/content/figures/1860-5397-15-284-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Effect of catalyst precursors used in Sonogashira coupling reaction of iodobenzene (1a, 0.5 mmol) and phenylacetylene (2a, 0.75 mmol). Reaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % catalyst, T = 55 °C.
Figure 1: Effect of catalyst precursors used in Sonogashira coupling reaction of iodobenzene (1a, 0.5 mmol) a...
The moisture content of the reaction environment could have a significant effect on the efficiency of a transition-metal-catalyzed reaction. Because [TBP][4EtOV] was isolated from an aqueous solution, the investigation of possible influence of the residual water content on the reaction was highly desired. We found that no decrease in the formation of 3a was detected when the moisture content was varied between 0.05 and 5.0 wt % (Table 2). Since the method is hardly sensitive to the residual moisture content, to exclude water from the reaction mixture no special pretreatment or handling of the solvent is necessary.
Table 2: Effect of water content on Sonogashira coupling of iodobenzene (1a) and phenylacetylene (2a).a
| Entry | Water content (wt %) | Isolated yields of 3a (%) |
| 1 | 0.05 | 85 |
| 2 | 1.0 | 86 |
| 2 | 2.5 | 82 |
| 4 | 5.0 | 83 |
aReaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % PdCl2(PPh3)2, 0.75 mmol phenylacetylene, 0.5 mmol iodobenzene, T = 55 °C, t = 3 h.
Hereafter, the air-stable and readily available PdCl2(PPh3)2 was selected as a catalyst precursor to facilitate C–C bond couplings involving various iodoaromatic compounds (1a–l) and phenylacetylene (2a) in [TBP][4EtOV] in the absence of any additional ligands and auxiliary base at 55 °C for 3 h. In general, the catalytic system could be applied to various iodoarene substrates and the substrate reactivity was not influenced dramatically by the electronic parameters of the substituents. Both electron-withdrawing (chloro, fluoro and bromo) and electron-donating (methyl, methoxy) groups were tolerated on the aryl iodide (Table 3, entries 2–7). Under identical conditions, 2-iodothiophene, and iodopyridine derivatives could also easily be converted to the corresponding acetylene with good or even excellent isolated yields (3i–n). When 2-amino-3-iodopyridine (1i) was converted no C–N bond formation was detected excluding the copper impurities assisted Ullmann-coupling reactions. When 2-chloro-5-iodopyridine (1l) was reacted with 1 equiv of 2, 2-chloro-5-(2-phenylethynyl)pyridine (3l) was isolated as product with a yield of 72%. By the use of 2.5 equiv of 2, the chloro group also undergoes a coupling reaction to form 2,5-bis(2-phenylethynyl)pyridine (3m) under identical conditions (Table 3, entries 12 and 13).
Table 3: Palladium-catalyzed Sonogashira coupling of various iodoaromatic compounds (1a–l) with phenylacetylene (2a).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-284-i2.svg?max-width=637&scale=1.0)
|
|||||
| # | Iodoaromatic compounds | Product | Yield (%)b | ||
| 1 | 1a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-284-i3.svg?max-width=637&scale=1.0)
|
3a | 85 | |
| 2 | 1b |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-284-i4.svg?max-width=637&scale=1.0)
|
3b | 96 | |
| 3 | 1c |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-284-i5.svg?max-width=637&scale=1.0)
|
3c | 95 | |
| 4 | 1d |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-284-i6.svg?max-width=637&scale=1.0)
|
3d | 82 | |
| 5 | 1e |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-284-i7.svg?max-width=637&scale=1.0)
|
3e | 80 | |
| 6 | 1f |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-284-i8.svg?max-width=637&scale=1.0)
|
3f | 87 | |
| 7 | 1g |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-284-i9.svg?max-width=637&scale=1.0)
|
3g | 52 | |
| 8 | 1h |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-284-i10.svg?max-width=637&scale=1.0)
|
3h | 80 | |
| 9 | 1i |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-284-i11.svg?max-width=637&scale=1.0)
|
3i | 75 | |
| 10 | 1j |
![[Graphic 11]](/bjoc/content/inline/1860-5397-15-284-i12.svg?max-width=637&scale=1.0)
|
3j | 93 | |
| 11 | 1k |
![[Graphic 12]](/bjoc/content/inline/1860-5397-15-284-i13.svg?max-width=637&scale=1.0)
|
3k | 79 | |
| 12 | 1l |
![[Graphic 13]](/bjoc/content/inline/1860-5397-15-284-i14.svg?max-width=637&scale=1.0)
|
3l | 72 | |
| 13c | 1l |
![[Graphic 14]](/bjoc/content/inline/1860-5397-15-284-i15.svg?max-width=637&scale=1.0)
|
3m | 69 | |
aReaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % PdCl2(PPh3)2, 0.75 mmol phenylacetylene, 0.5 mmol iodoaromatic compound, T = 55 °C, t = 3 h; bisolated yields; c1.25 mmol (2.5 equiv) of phenylacetylene, 3m: 2,5-bis(2-phenylethynyl)pyridine.
By comparison with the conversion of 4-chloro-1-iodobenzene (1g), no formation of 1,4-bis(phenylethynyl)benzene was detected. It agrees with the activated substituents of 2-substituted pyridine derivatives. It can be concluded that by varying electronic and steric properties of substituents of the iodoaromatic substrates at all ortho-, meta-, and para- positions, no significant changes in the product yields were achieved according to previous studies [26,41]. Regarding the negligible influence of the 4-substituents, i.e., no Hammett-plot can be obtained for the above reaction, it can be stated that the rate determining step of the Sonogashira coupling is not related to the formation of Pd-aryl species.
Subsequently, a series of different acetylenes, which can readily be dissolved in [TBP][4EtOV] were subjected to the coupling reaction under identical conditions. By comparison of the efficiency of conversion of iodobenzene and its electron-donating 4-methoxy (1d) and electron-withdrawing 4-nitro (1m) derivatives with different aliphatic acetylenes (4, 6, and 8), no significant differences in isolated yields were observed (Table 4, Table 5 and Table 6) verifying the wide range of functional group tolerance by side of acetylenic substrates, as well. Same results were reported by Alper and co-workers, who perform this reaction in [BMIM][PF6] as alternative solvent [22].
Table 4: Palladium-catalyzed Sonogashira coupling of various iodoaromatic compounds (1a, d, m) with propargyl alcohol (4).a
![[Graphic 15]](/bjoc/content/inline/1860-5397-15-284-i16.svg?max-width=637&scale=1.0)
|
||||
| # | R | Product | Yield (%)b | |
| 1 | 1a | H | 5a | 80 |
| 2 | 1m | NO2 | 5b | 78 |
| 3 | 1d | OCH3 | 5c | 85 |
aReaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % (PPh3)2PdCl2, 0.75 mmol propargyl alcohol, 0.5 mmol iodoaromatic compounds, T = 55 °C, t = 3 h; bisolated yields.
Table 5: Palladium-catalyzed Sonogashira coupling of various iodoaromatic compounds (1a, d, m) with 1-ethynyl-1-cyclohexanol (6).a
![[Graphic 16]](/bjoc/content/inline/1860-5397-15-284-i17.svg?max-width=637&scale=1.0)
|
||||
| # | R | Product | Yield (%)b | |
| 1 | 1a | H | 7a | 85 |
| 2 | 1m | NO2 | 7b | 99 |
| 3 | 1d | OCH3 | 7c | 99 |
aReaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % (PPh3)2PdCl2, 0.75 mmol 1-ethynyl-1-cyclohexanol, 0.5 mmol iodoaromatic compound, T = 55 °C, t = 3 h; bisolated yields.
Table 6: Palladium-catalyzed Sonogashira coupling of various iodoaromatic compounds (1a, d, m) with 3-ethyl-1-pentyn-3-ol (8).a
![[Graphic 17]](/bjoc/content/inline/1860-5397-15-284-i18.svg?max-width=637&scale=1.0)
|
||||
| # | R | Product | Yield (%)b | |
| 1 | 1a | H | 9a | 87 |
| 2 | 1m | NO2 | 9b | 85 |
| 3 | 1d | OCH3 | 9c | 81 |
aReaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % (PPh3)2PdCl2, 0.75 mmol 3-ethyl-1-pentyn-3-ol, 0.5 mmol iodoaromatic compound, T = 55 °C, t = 3 h; bisolated yields.
The possible reuse of the catalyst was subsequently investigated by the model reaction of 0.5 mmol of 1a and 1.5 equiv of 2a in the presence of 0.5 mol % PdCl2(PPh3)2 at 55 °C for 2 h. After the first extraction of the product from the reaction mixture by the addition of 10 × 5 mL of pentane, 3a was isolated with a yield of 88%. It should be noted that this reaction verified the reproducibility of the experiments (cf. Table 3, entry 1). Same amounts of substrates were added to the Pd catalyst contained in the ionic liquid phase followed by heating to 55 °C. In the second run 12% of decrease in the isolated yield was detected; however, after the 4th cycle, it became significant (Figure 2). The same tendency was reported by Toma for various acetylenes [24]. The 13C NMR investigations of the [4EtOV]− anion throughout the reaction did not indicate any change in its composition. Nevertheless, a reaction of the alkoxyvalerate anion with HI that formed during the reaction could be assumed.
![[1860-5397-15-284-2]](/bjoc/content/figures/1860-5397-15-284-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Re-use of Pd catalyst for Sonogashira coupling of iodobenzene (1a) and phenylacetylene (2a). Reaction conditions: 0.8 mL [TBP][4EtOV], 0.5 mol % catalyst, T = 55 °C, t = 3 h.
Figure 2: Re-use of Pd catalyst for Sonogashira coupling of iodobenzene (1a) and phenylacetylene (2a). Reacti...
Conclusion
In conclusion, we have demonstrated that a γ-valerolactone-based ionic liquid, tetrabutylphosphonium 4-ethoxyvalerate can be utilized as an alternative solvent for Pd-catalyzed Sonogashira coupling reactions of aryl iodides and functionalized acetylenes under mild conditions. The reactions were performed by using 0.5 mol % catalyst loading and we pointed out that both copper and external base could be eliminated from the reaction mixture without any decrease in catalytic activity. The protocol was tested for a wide range of substrates and several products (3a–n, 5a–c, 7a–c, 9a–c) were isolated in good to excellent yields. The Cu- and base-free reaction can be performed under air and are highly tolerant to moisture.
Experimental
The sources of chemicals are listed in Supporting Information File 1. The NMR spectra were recorded on a Brucker Avance 250 MHz spectrometer. The water contents of the ionic liquids were determined by Karl Fischer titration performed by HANNA Instruments 904.
The γ-valerolactone-based ionic liquid ([TBP][4EtOV]) was prepared as described in [34] with details presented in Supporting Information File 1.
Exact mass measurements were performed on a high-resolution Q-Exactive Focus hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a heated electrospray ionization (ESI) source. Samples were dissolved in acetonitrile/water 1:1 (v/v) solvent mixture containing 0.1% (v/v) formic acid. Solutions were directly introduced into the ion source using a syringe pump. Under the applied conditions, the compounds form protonated molecules, [M + H]+ in positive ionization mode (ESI).
Detailed experimental procedures, characterization data are reported in Supporting Information File 1.
General procedure for Sonogashira coupling reactions
In a 4 mL screw-cap vial, 0.5 mmol of corresponding iodoarene compound, 1.5 equiv of phenylacetylene or propargyl alcohol, 0.005 equiv PdCl2(PPh3)2, and 0.8 mL of ionic liquid were mixed and stirred at 55 °C for 3 h. After cooling, the mixture was partitioned between 5 mL of water and 5 mL of pentane. After separation, the aqueous phase was extracted subsequently with 2 × 5 mL of pentane. The combined organic phase was washed with brine, dried over MgSO4, filtered, and the solvent was evaporated under reduced pressure (ca. 10 mmHg). The oily residue was purified by chromatography on silica gel (Merck Silicagel 60 (0.063−0.200 mm) for column chromatography (70−230 mesh ASTM)) eluted with n-pentane/EtOAc. The purity of the isolated products was >98%. The detailed experimental procedure as well as the characterization of isolated compounds are provided in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Source of chemicals, the detailed experimental procedure as well as characterization data of isolated compounds. | ||
| Format: PDF | Size: 524.3 KB | Download |
Acknowledgements
This work was funded by the National Research, Development and Innovation Office – NKFIH (KH 129508 and K113177) and the Higher Education Excellence Program of the Ministry of Human Capacities in the frame of Biotechnology research area of Budapest University of Technology and Economics (BME FIKP-BIO). L.T. Mika is grateful for the support of a Scholarship of József Varga Foundation, Budapest University of Technology and Economics, Budapest, Hungary.
References
-
Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787–8863. doi:10.1021/acs.chemrev.6b00567
Return to citation in text: [1] -
Shi, W.; Liu, C.; Lei, A. Chem. Soc. Rev. 2011, 40, 2761–2776. doi:10.1039/c0cs00125b
Return to citation in text: [1] -
de Meijere, A.; Diederich, F., Eds. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/9783527619535
Return to citation in text: [1] -
Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. doi:10.1002/anie.201107017
Return to citation in text: [1] -
Devendar, P.; Qu, R.-Y.; Kang, W.-M.; He, B.; Yang, G.-F. J. Agric. Food Chem. 2018, 66, 8914–8934. doi:10.1021/acs.jafc.8b03792
Return to citation in text: [1] -
Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417–1492. doi:10.1021/cr100327p
Return to citation in text: [1] -
Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x
Return to citation in text: [1] [2] -
Sonogashira, K. Palladium‐Catalyzed Alkynylation: Sonogashira Alkyne Synthesis. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; John Wiley & Sons: New York, NY, USA; pp 493–529. doi:10.1002/0471212466.ch22
Return to citation in text: [1] -
https://www.fda.gov/media/71737/download (accessed Oct 2, 2019).
Return to citation in text: [1] -
Severin, R.; Reimer, J.; Doye, S. J. Org. Chem. 2010, 75, 3518–3521. doi:10.1021/jo100460v
Return to citation in text: [1] -
Karpov, A. S.; Müller, T. J. J. Synthesis 2003, 2815–2826. doi:10.1055/s-2003-42480
Return to citation in text: [1] -
Panda, B.; Sarkar, T. K. Synthesis 2013, 45, 817–829. doi:10.1055/s-0032-1318119
Return to citation in text: [1] -
Moon, J.; Jeong, M.; Nam, H.; Ju, J.; Moon, J. H.; Jung, H. M.; Lee, S. Org. Lett. 2008, 10, 945–948. doi:10.1021/ol703130y
Return to citation in text: [1] -
Nagy, A.; Novák, Z.; Kotschy, A. J. Organomet. Chem. 2005, 690, 4453–4461. doi:10.1016/j.jorganchem.2004.12.036
Return to citation in text: [1] -
Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993–5996. doi:10.1002/anie.200353015
Return to citation in text: [1] [2] -
Bakherad, M. Appl. Organomet. Chem. 2013, 27, 125–140. doi:10.1002/aoc.2931
Return to citation in text: [1] -
Markert, C.; Bannwarth, W. Helv. Chim. Acta 2002, 85, 1877–1882. doi:10.1002/1522-2675(200207)85:7<1877::aid-hlca1877>3.0.co;2-5
Return to citation in text: [1] -
Akiyama, Y.; Meng, X.; Fujita, S.; Chen, Y.-C.; Lu, N.; Cheng, H.; Zhao, F.; Arai, M. J. Supercrit. Fluids 2009, 51, 209–216. doi:10.1016/j.supflu.2009.08.006
Return to citation in text: [1] -
Strappaveccia, G.; Luciani, L.; Bartollini, E.; Marrocchi, A.; Pizzo, F.; Vaccaro, L. Green Chem. 2015, 17, 1071–1076. doi:10.1039/c4gc01728e
Return to citation in text: [1] -
Li, J.; Yang, S.; Wu, W.; Jiang, H. Eur. J. Org. Chem. 2018, 1284–1306. doi:10.1002/ejoc.201701509
Return to citation in text: [1] -
Lei, Z.; Chen, B.; Koo, Y.-M.; MacFarlane, D. R. Chem. Rev. 2017, 117, 6633–6635. doi:10.1021/acs.chemrev.7b00246
Return to citation in text: [1] -
Bong Park, S.; Alper, H. Chem. Commun. 2004, 1306–1307. doi:10.1039/b402477j
Return to citation in text: [1] [2] -
Błaszczyk, I.; Trzeciak, A. M.; Ziółkowski, J. J. Catal. Lett. 2009, 133, 262–266. doi:10.1007/s10562-009-0181-y
Return to citation in text: [1] [2] [3] -
Kmentová, I.; Gotov, B.; Gajda, V.; Toma, T. Monatsh. Chem. 2003, 134, 545–549. doi:10.1007/s00706-002-0558-8
Return to citation in text: [1] [2] [3] -
Fukuyama, T.; Shinmen, M.; Nishitani, S.; Sato, M.; Ryu, I. Org. Lett. 2002, 4, 1691–1694. doi:10.1021/ol0257732
Return to citation in text: [1] [2] -
Fukuyama, T.; Rahman, M. T.; Sumino, Y.; Ryu, I. Synlett 2012, 23, 2279–2283. doi:10.1055/s-0031-1290456
Return to citation in text: [1] [2] [3] -
de Lima, P. G.; Antunes, O. A. C. Tetrahedron Lett. 2008, 49, 2506–2509. doi:10.1016/j.tetlet.2008.02.110
Return to citation in text: [1] [2] -
Ermolaev, V.; Miluykov, V.; Arkhipova, D.; Zvereva, E.; Sinyashin, O. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 168–170. doi:10.1080/10426507.2012.744013
Return to citation in text: [1] -
Harjani, J. R.; Abraham, T. J.; Gomez, A. T.; Garcia, M. T.; Singer, R. D.; Scammells, P. J. Green Chem. 2010, 12, 650–655. doi:10.1039/b919394d
Return to citation in text: [1] [2] -
Prabhala, P.; Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2019, 2061–2064. doi:10.1002/ejoc.201900093
Return to citation in text: [1] [2] [3] -
Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2018, 5285–5288. doi:10.1002/ejoc.201800804
Return to citation in text: [1] [2] -
Iranpoor, N.; Firouzabadi, H.; Ahmadi, Y. Eur. J. Org. Chem. 2012, 305–311. doi:10.1002/ejoc.201100701
Return to citation in text: [1] -
Reddy, A. S.; Laali, K. K. Tetrahedron Lett. 2015, 56, 4807–4810. doi:10.1016/j.tetlet.2015.06.067
Return to citation in text: [1] [2] -
Orha, L.; Tukacs, J. M.; Gyarmati, B.; Szilágyi, A.; Kollár, L.; Mika, L. T. ACS Sustainable Chem. Eng. 2018, 6, 5097–5104. doi:10.1021/acssuschemeng.7b04775
Return to citation in text: [1] [2] [3] -
Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084–5121. doi:10.1039/c1cs15071e
Return to citation in text: [1] -
Hallett, J. P.; Welton, T. Chem. Rev. 2011, 111, 3508–3576. doi:10.1021/cr1003248
Return to citation in text: [1] -
McLachlan, F.; Mathews, C. J.; Smith, P. J.; Welton, T. Organometallics 2003, 22, 5350–5357. doi:10.1021/om034075y
Return to citation in text: [1] -
Siemsen, P.; Livingston, R. C.; Diederich, F. Angew. Chem., Int. Ed. 2000, 39, 2632–2657. doi:10.1002/1521-3773(20000804)39:15<2632::aid-anie2632>3.0.co;2-f
Return to citation in text: [1] -
Li, J.-H.; Liang, Y.; Zhang, X.-D. Tetrahedron 2005, 61, 1903–1907. doi:10.1016/j.tet.2004.12.026
Return to citation in text: [1] -
Triethylamine. https://onlinelibrary.wiley.com/doi/pdf/10.1002/3527600418.mb12144e0013 (accessed Oct 1, 2019). doi:10.22233/20412495.1019.1
Return to citation in text: [1] -
Liang, Y.; Xie, Y.-X.; Li, J.-H. J. Org. Chem. 2006, 71, 379–381. doi:10.1021/jo051882t
Return to citation in text: [1]
| 34. | Orha, L.; Tukacs, J. M.; Gyarmati, B.; Szilágyi, A.; Kollár, L.; Mika, L. T. ACS Sustainable Chem. Eng. 2018, 6, 5097–5104. doi:10.1021/acssuschemeng.7b04775 |
| 7. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 35. | Chinchilla, R.; Nájera, C. Chem. Soc. Rev. 2011, 40, 5084–5121. doi:10.1039/c1cs15071e |
| 36. | Hallett, J. P.; Welton, T. Chem. Rev. 2011, 111, 3508–3576. doi:10.1021/cr1003248 |
| 37. | McLachlan, F.; Mathews, C. J.; Smith, P. J.; Welton, T. Organometallics 2003, 22, 5350–5357. doi:10.1021/om034075y |
| 1. | Yang, Y.; Lan, J.; You, J. Chem. Rev. 2017, 117, 8787–8863. doi:10.1021/acs.chemrev.6b00567 |
| 2. | Shi, W.; Liu, C.; Lei, A. Chem. Soc. Rev. 2011, 40, 2761–2776. doi:10.1039/c0cs00125b |
| 3. | de Meijere, A.; Diederich, F., Eds. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/9783527619535 |
| 10. | Severin, R.; Reimer, J.; Doye, S. J. Org. Chem. 2010, 75, 3518–3521. doi:10.1021/jo100460v |
| 20. | Li, J.; Yang, S.; Wu, W.; Jiang, H. Eur. J. Org. Chem. 2018, 1284–1306. doi:10.1002/ejoc.201701509 |
| 21. | Lei, Z.; Chen, B.; Koo, Y.-M.; MacFarlane, D. R. Chem. Rev. 2017, 117, 6633–6635. doi:10.1021/acs.chemrev.7b00246 |
| 24. | Kmentová, I.; Gotov, B.; Gajda, V.; Toma, T. Monatsh. Chem. 2003, 134, 545–549. doi:10.1007/s00706-002-0558-8 |
| 7. | Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874–922. doi:10.1021/cr050992x |
| 8. | Sonogashira, K. Palladium‐Catalyzed Alkynylation: Sonogashira Alkyne Synthesis. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; John Wiley & Sons: New York, NY, USA; pp 493–529. doi:10.1002/0471212466.ch22 |
| 18. | Akiyama, Y.; Meng, X.; Fujita, S.; Chen, Y.-C.; Lu, N.; Cheng, H.; Zhao, F.; Arai, M. J. Supercrit. Fluids 2009, 51, 209–216. doi:10.1016/j.supflu.2009.08.006 |
| 26. | Fukuyama, T.; Rahman, M. T.; Sumino, Y.; Ryu, I. Synlett 2012, 23, 2279–2283. doi:10.1055/s-0031-1290456 |
| 4. | Johansson Seechurn, C. C. C.; Kitching, M. O.; Colacot, T. J.; Snieckus, V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. doi:10.1002/anie.201107017 |
| 5. | Devendar, P.; Qu, R.-Y.; Kang, W.-M.; He, B.; Yang, G.-F. J. Agric. Food Chem. 2018, 66, 8914–8934. doi:10.1021/acs.jafc.8b03792 |
| 6. | Jana, R.; Pathak, T. P.; Sigman, M. S. Chem. Rev. 2011, 111, 1417–1492. doi:10.1021/cr100327p |
| 19. | Strappaveccia, G.; Luciani, L.; Bartollini, E.; Marrocchi, A.; Pizzo, F.; Vaccaro, L. Green Chem. 2015, 17, 1071–1076. doi:10.1039/c4gc01728e |
| 26. | Fukuyama, T.; Rahman, M. T.; Sumino, Y.; Ryu, I. Synlett 2012, 23, 2279–2283. doi:10.1055/s-0031-1290456 |
| 41. | Liang, Y.; Xie, Y.-X.; Li, J.-H. J. Org. Chem. 2006, 71, 379–381. doi:10.1021/jo051882t |
| 14. | Nagy, A.; Novák, Z.; Kotschy, A. J. Organomet. Chem. 2005, 690, 4453–4461. doi:10.1016/j.jorganchem.2004.12.036 |
| 34. | Orha, L.; Tukacs, J. M.; Gyarmati, B.; Szilágyi, A.; Kollár, L.; Mika, L. T. ACS Sustainable Chem. Eng. 2018, 6, 5097–5104. doi:10.1021/acssuschemeng.7b04775 |
| 13. | Moon, J.; Jeong, M.; Nam, H.; Ju, J.; Moon, J. H.; Jung, H. M.; Lee, S. Org. Lett. 2008, 10, 945–948. doi:10.1021/ol703130y |
| 17. | Markert, C.; Bannwarth, W. Helv. Chim. Acta 2002, 85, 1877–1882. doi:10.1002/1522-2675(200207)85:7<1877::aid-hlca1877>3.0.co;2-5 |
| 23. | Błaszczyk, I.; Trzeciak, A. M.; Ziółkowski, J. J. Catal. Lett. 2009, 133, 262–266. doi:10.1007/s10562-009-0181-y |
| 12. | Panda, B.; Sarkar, T. K. Synthesis 2013, 45, 817–829. doi:10.1055/s-0032-1318119 |
| 15. | Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993–5996. doi:10.1002/anie.200353015 |
| 38. | Siemsen, P.; Livingston, R. C.; Diederich, F. Angew. Chem., Int. Ed. 2000, 39, 2632–2657. doi:10.1002/1521-3773(20000804)39:15<2632::aid-anie2632>3.0.co;2-f |
| 39. | Li, J.-H.; Liang, Y.; Zhang, X.-D. Tetrahedron 2005, 61, 1903–1907. doi:10.1016/j.tet.2004.12.026 |
| 11. | Karpov, A. S.; Müller, T. J. J. Synthesis 2003, 2815–2826. doi:10.1055/s-2003-42480 |
| 15. | Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993–5996. doi:10.1002/anie.200353015 |
| 40. | Triethylamine. https://onlinelibrary.wiley.com/doi/pdf/10.1002/3527600418.mb12144e0013 (accessed Oct 1, 2019). doi:10.22233/20412495.1019.1 |
| 24. | Kmentová, I.; Gotov, B.; Gajda, V.; Toma, T. Monatsh. Chem. 2003, 134, 545–549. doi:10.1007/s00706-002-0558-8 |
| 22. | Bong Park, S.; Alper, H. Chem. Commun. 2004, 1306–1307. doi:10.1039/b402477j |
| 23. | Błaszczyk, I.; Trzeciak, A. M.; Ziółkowski, J. J. Catal. Lett. 2009, 133, 262–266. doi:10.1007/s10562-009-0181-y |
| 24. | Kmentová, I.; Gotov, B.; Gajda, V.; Toma, T. Monatsh. Chem. 2003, 134, 545–549. doi:10.1007/s00706-002-0558-8 |
| 25. | Fukuyama, T.; Shinmen, M.; Nishitani, S.; Sato, M.; Ryu, I. Org. Lett. 2002, 4, 1691–1694. doi:10.1021/ol0257732 |
| 34. | Orha, L.; Tukacs, J. M.; Gyarmati, B.; Szilágyi, A.; Kollár, L.; Mika, L. T. ACS Sustainable Chem. Eng. 2018, 6, 5097–5104. doi:10.1021/acssuschemeng.7b04775 |
| 23. | Błaszczyk, I.; Trzeciak, A. M.; Ziółkowski, J. J. Catal. Lett. 2009, 133, 262–266. doi:10.1007/s10562-009-0181-y |
| 31. | Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2018, 5285–5288. doi:10.1002/ejoc.201800804 |
| 30. | Prabhala, P.; Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2019, 2061–2064. doi:10.1002/ejoc.201900093 |
| 33. | Reddy, A. S.; Laali, K. K. Tetrahedron Lett. 2015, 56, 4807–4810. doi:10.1016/j.tetlet.2015.06.067 |
| 30. | Prabhala, P.; Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2019, 2061–2064. doi:10.1002/ejoc.201900093 |
| 29. | Harjani, J. R.; Abraham, T. J.; Gomez, A. T.; Garcia, M. T.; Singer, R. D.; Scammells, P. J. Green Chem. 2010, 12, 650–655. doi:10.1039/b919394d |
| 30. | Prabhala, P.; Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2019, 2061–2064. doi:10.1002/ejoc.201900093 |
| 31. | Savanur, H. M.; Kalkhambkar, R. G.; Laali, K. K. Eur. J. Org. Chem. 2018, 5285–5288. doi:10.1002/ejoc.201800804 |
| 32. | Iranpoor, N.; Firouzabadi, H.; Ahmadi, Y. Eur. J. Org. Chem. 2012, 305–311. doi:10.1002/ejoc.201100701 |
| 33. | Reddy, A. S.; Laali, K. K. Tetrahedron Lett. 2015, 56, 4807–4810. doi:10.1016/j.tetlet.2015.06.067 |
| 28. | Ermolaev, V.; Miluykov, V.; Arkhipova, D.; Zvereva, E.; Sinyashin, O. Phosphorus, Sulfur Silicon Relat. Elem. 2013, 188, 168–170. doi:10.1080/10426507.2012.744013 |
| 25. | Fukuyama, T.; Shinmen, M.; Nishitani, S.; Sato, M.; Ryu, I. Org. Lett. 2002, 4, 1691–1694. doi:10.1021/ol0257732 |
| 27. | de Lima, P. G.; Antunes, O. A. C. Tetrahedron Lett. 2008, 49, 2506–2509. doi:10.1016/j.tetlet.2008.02.110 |
| 29. | Harjani, J. R.; Abraham, T. J.; Gomez, A. T.; Garcia, M. T.; Singer, R. D.; Scammells, P. J. Green Chem. 2010, 12, 650–655. doi:10.1039/b919394d |
| 26. | Fukuyama, T.; Rahman, M. T.; Sumino, Y.; Ryu, I. Synlett 2012, 23, 2279–2283. doi:10.1055/s-0031-1290456 |
| 27. | de Lima, P. G.; Antunes, O. A. C. Tetrahedron Lett. 2008, 49, 2506–2509. doi:10.1016/j.tetlet.2008.02.110 |
© 2019 Orha et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)