

Abstract
The synthesis of novel fluorescent N(9)-alkylated 2-amino-6-triazolylpurine and 7-deazapurine derivatives is described. A new C(2)-regioselectivity in the nucleophilic aromatic substitution reactions of 9-alkylated-2,6-diazidopurines and 7-deazapurines with secondary amines has been disclosed. The obtained intermediates, 9-alkylated-2-amino-6-azido-(7-deaza)purines, were transformed into the title compounds by CuAAC reaction. The designed compounds belong to the push–pull systems and possess promising fluorescence properties with quantum yields in the range from 28% to 60% in acetonitrile solution. Due to electron-withdrawing properties of purine and 7-deazapurine heterocycles, which were additionally extended by triazole moieties, the compounds with electron-donating groups showed intramolecular charge transfer character (ICT/TICT) of the excited states which was proved by solvatochromic dynamics and supported by DFT calculations. In the 7-deazapurine series this led to increased fluorescence quantum yield (74%) in THF solution. The compounds exhibit low cytotoxicity and as such are useful for the cell labelling studies in the future.
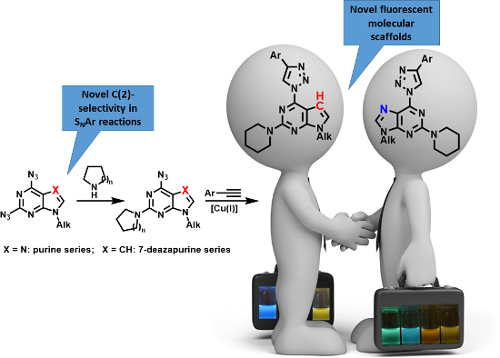
Graphical Abstract
Introduction
Purine [1-7] and 7-deazapurine (IUPAC name: pyrrolo[2,3-d]pyrimidine) [8-11] derivatives have been progressively studied for decades due to their wide range of biological activities and photophysical properties. Currently, the synthesis of push–pull systems is a promising direction in the development of various fluorescent (7-deaza)purine derivatives [12-16]. The push–pull effect arises by adding electron-donating and electron-withdrawing groups at the opposite ends of π-conjugated systems. Large Stokes shifts and high quantum yields are usually characteristic for this type of molecules. Traditionally, fluorescent purine nucleoside analogs were recognized as valuable fluorescent probes for DNA and RNA research [17]. This paved a way for development of various adenosine and guanosine analogs which in many cases contained an additional substituent at C(8) like compounds A and B (Figure 1) [18,19]. The latter attachment does not significantly affect the natural Watson–Crick base-pairing abilities of the modified compounds. In this context, other important structural modifications in such a series include 8-vinyladenosine [20], 8-styryladenosine [13,21] and 8-heteroarylguanosine [22] derivatives which revealed good to excellent quantum yields. Useful levels of fluorescence were reported also for purine nucleos(t)ides bearing azole-type substituents at C(2) or C(6) (e.g., 6-thiazol-2-yl derivative C [23] and 2-(1,2,3-triazol)-1-yladenosines D [24,25] (Figure 1).
![[1860-5397-15-41-1]](/bjoc/content/figures/1860-5397-15-41-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of fluorescent purine/7-deazapurine derivatives.
Figure 1: Examples of fluorescent purine/7-deazapurine derivatives.
7-Deazapurines can be regarded as structurally close congeners to purines with an extra attachment point at their “artificially introduced” C(7). Therefore, it comes as no surprise that also 7-deazapurine derivatives containing heterocyclic substituents at various attachment points [26,27] have been actively studied for their fluorescent properties. Among others, also azole containing compounds of type E [28] and F [29] have been described.
Similarly, to purine derivatives also 7-deazapurines are used in DNA labeling [30]. On the other hand, modified purines have found also applications as cell imaging tools [31,32], biosensors and sensors for detection of heavy metals [33]. The tendency of these structures to form hydrogen bonds, to respond to medium effects (solvatochromism) [34] and to certain impurities (e.g., coordination complexes with metal ions) make these compound classes very attractive for sensor design [33,35]. It is interesting to note, that push–pull type purine non-nucleos(t)ide derivatives have not been actively studied in the context of materials science. The few available examples include the design of methyl 9-benzyl-2-N,N-dimethylamino-9H-purine-8-carboxylate and the corresponding 2,6-bis(dialkylamino) derivatives as fluorescent materials for preparation of organic light-emitting diodes [12,36-38].
In light of the aforementioned facts, we proceeded to design novel and structurally related classes of 2-amino-6-(1,2,3-triazol-1-yl)purines and 7-deazapurines (Figure 1 compounds of type G and H) in order to determine and to compare their fluorescent properties. The synthetic approach towards 7-deazapurine derivatives H has been recently disclosed by us [39], but the photophysical properties of these compounds have not been studied thus far. In this full account we report the newly developed C(2)-regioselective nucleophilic aromatic substitution of 2,6-diazidopurines, transformation of the intermediate 2-amino-6-azidopurines into the title compounds and comparative photophysical study of originally yet identically substituted fluorescent push–pull purines and 7-deazapurines.
Results and Discussion
We have recently observed significant fluorescence in the triazolylpurine and triazolyl-7-deazapurine series [25,29,40,41]. Therefore, we developed synthetic methods that can provide structurally related chromophores of the purine and 7-deazapurine series possessing identical substitution pattern (structural congeners G and H, Figure 1). The easy access to the title compounds opened a possibility for a comparative study of their photophysical properties.
Synthesis
N(9)-Alkylated-2,6-diazidopurine 2a was synthesized in two steps from commercially available 2,6-dichloropurine (1a) by subsequent alkylation of the N(9) position and azido group introduction in C(2) and C(6) positions. The N(9) position of purine was alkylated in two different ways: 1) using alkyl halides in the presence of a strong base, and 2) using alcohols under Mitsunobu conditions.
In the first approach (method A, Scheme 1), the purine was deprotonated with a base (NaH or K2CO3) and alkylated with alkyl halides such as 1-iodoheptane, 1-bromononane and 1-bromododecane. The main disadvantages of this approach are suboptimal yields, which varied from 48% to 52%, and long reaction times. For example, in the case of 1-bromododecane, the reaction required 72 hours to complete. On the other hand, Mitsunobu reaction (method B, Scheme 1) was performed between the corresponding alcohol and 2,6-dichloropurine in anhydrous THF at 0–20 °C for 1–1.5 h, resulting in average yields of 66% for N(9)-alkylated products. Both methodologies produced a mixture of N(9)- and N(7)-alkylated isomers from which the major N(9) product was easily separated by column chromatography [42-47]. Alkylated products were used in SNAr reactions with NaN3 in acetone or DMF, giving good to excellent yields of 2,6-diazidopurines 2a–c in the range from 66 to 93%. In several cases chromatographic separation of the N(9)/(7) isomers was easier at the stage of diazido products 2a and 2a’. Diazido products 2 are light and temperature sensitive [48], but can be stored without degradation in refrigerator at −20 °C for a prolonged period of time. In addition, DSC measurements for compounds 2a–c were done. Diazides 2a–c melted at 67 °C, 75 °C and 82 °C, respectively, and showed an exothermic effect only if heated above 190 °C (Figures S5–S7 in Supporting Information File 1).
![[1860-5397-15-41-i1]](/bjoc/content/inline/1860-5397-15-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General synthetic routes for the compounds 5, 7–9, 10 and 11. Method A: alkyl halogenide, MeCN or DMF, NaH, 1–3 days, 20–55 °C; method B: alcohol, Ph3P, THF, DIAD, 1 h, 0→20 °C.
Scheme 1: General synthetic routes for the compounds 5, 7–9, 10 and 11. Method A: alkyl halogenide, MeCN or D...
In the 7-deaza series deprotonated starting material 1b was methylated with MeI and further submitted to SNAr reaction with NaN3 according to a previously published procedure [39]. The expected 2,6-diazido-7-deazapurine (3) was obtained in 78% yield.
It should be noted that the medium size alkyl chains at N(9) were chosen for several reasons: a) they help to solubilize the target compounds in the organic medium; b) they are considered as a good compromise to achieve both “membrane-like character”, which helps for cell membrane permeability, and sufficient solubility in the aqueous (biological) media [49].
With the key intermediates 2 and 3 in hand, we proceeded with the synthesis and structure elucidation of the designed structures G and H (Figure 1) which are represented by compounds 7–11 in Scheme 1. Firstly, we prepared a regioisomeric compound 5 by repeating the previously elaborated sequence of double CuAAC reaction (2→4) followed by a SNAr process (4→5) which showcases the use of 1,2,3-triazoles as leaving groups. It is well established during our previous research in the purine nucleoside series that such an approach provides 6-amino-2-(1,2,3-triazol-1-yl) derivatives [25]. On the other hand, it was also reported that 2,6-diazidopurine nucleosides exhibit opposite regioselectivity with aliphatic thiols, which do SNAr reactions at C(2) position of purines [50,51]. Taking this information into account, we performed an SNAr reaction between 2,6-diazidopurine 2a and piperidine and obtained the C(2)-substitution product 6b as the major isomer. To the best of our knowledge, this is the first report on such regioselectivity involving combination of 2,6-diazidopurines and N-nucleophiles.
It should be noted that in solution, azides 6 exist in equilibrium between the azidopurine and tetrazolopurine forms. This equilibrium was studied in different deuterated solvents by 1H NMR spectroscopy on the example of 6-azido-9-heptyl-2-piperidinopurine 6b. Experiments were performed in CDCl3, THF-d8, CD3CN and DMSO-d6. The 1H NMR spectrum in CD3CN showed the presence of the tetrazole form and has been used for NMR studies in the temperature range from 30 to 60 °C using two different concentrations – 12.5 mg/mL and 25.0 mg/mL. Temperature increase caused the increase of the azido form. These results are summarized in Figure 2. The temperature increase caused the disappearance of signals at 7.92 (s, α), 4.23 (t, β) and 4.01–3.95 (m, γ) and the broadening of signals at 7.69 (s, a), 4.04 (t, b) and 3.84–3.74 (m, c). A similar type of the ring–chain tautomerism is expected to occur in 6-azido-2-amino-7-deazapurines (synthetic intermediates on the way 3→10/11), but it was not studied in detail due to their relatively low stability [39]. Regarding the tautomerism in 2,6-diazido-substituted starting materials, it has been studied previously for both purine [41] and 7-deazapurine [52] derivatives. In both cases practically only the diazido forms are observed in chloroform solution, but the proportion of the tetrazole tautomer increases with the increase of solvent polarity. Additionally, X-ray analysis of 2,6-diazidopurine 2'-deoxyribonucleoside revealed the exclusive existence of the azido tautomer in the solid state [41].
![[1860-5397-15-41-2]](/bjoc/content/figures/1860-5397-15-41-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: 1H NMR spectra of compound 6b in CD3CN at different temperatures (300 MHz, c = 12.5 mg/mL); a, b, c – signals for azide form 6b-A; α, β, γ – signals for tetrazole form 6b-T).
Figure 2: 1H NMR spectra of compound 6b in CD3CN at different temperatures (300 MHz, c = 12.5 mg/mL); a, b, c...
The discovered ability of the azido group to be substituted with amines at C(2) position was used for the synthesis of a novel library of 9-alkyl-2-amino-6-triazolylpurine derivatives. Thus, copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition reaction of compounds 6a–c with different para-substituted phenylacetylenes produced the expected compounds 7a–f, 8a–f and 9 (Scheme 1). The yields of 1,3-dipolar cycloadditions varied from 51% to 76% in the case of pyrrolidinyl purines 7a–f and from 53% to 86% for piperidinyl purines 8a–f (Table 1 and Table 2). To gain an insight into the structure–photophysical properties relationship of the newly obtained structures, various EDG and EWG at the para-position of the phenyl rings were installed. It can be concluded that 2,6-diazidopurines are versatile intermediates for the synthesis of both the 2-amino-6-triazolylpurine and 6-amino-2-triazolylpurine derivatives. Comparison of both synthetic methods (2→4→5 versus 2→6→7/8/9) reveals that the newly developed approach towards 2-amino-6-triazolylpurine derivatives was performed in a more atom economic way. The latter synthesis does not require a double cycloaddition followed by replacement of one of the triazole moieties.
Table 1: Diversity of 9-alkyl-2-pyrrolidino-6-triazolylpurine 7 and 7-deazapurine 10 derivatives obtained in reactions 6a→7a–f and 3→10a–f according to Scheme 1.
| Entry | General structure | Compound | R | Yield, % |
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-41-i2.svg?max-width=637&scale=1.0)
7 |
7a | -H | 51 |
| 2 | 7b | -OMe | 57 | |
| 3 | 7c | -NMe2 | 53 | |
| 4 | 7d | -F | 64 | |
| 5 | 7e | CF3 | 76 | |
| 6 | 7f | -CN | 63 | |
| 7 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-41-i3.svg?max-width=637&scale=1.0)
10 |
10a | -H | 71 |
| 8 | 10b | -OMe | 66 | |
| 9 | 10c | -NMe2 | 68 | |
| 10 | 10d | -F | 80 | |
| 11 | 10e | -CF3 | 76 | |
| 12 | 10f | -CN | 79 | |
Table 2: Diversity of 9-alkyl-2-piperidino-6-triazolylpurine 8 and 7-deazapurine 11 derivatives obtained in reactions 6b→8a–f and 3→11a–f according to Scheme 1.
| Entry | General structure | Compound | R | Yield, % |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-15-41-i4.svg?max-width=637&scale=1.0)
8 |
8a | -H | 71 |
| 2 | 8b | -OMe | 60 | |
| 3 | 8c | -NMe2 | 53 | |
| 4 | 8d | -F | 74 | |
| 5 | 8e | -CF3 | 84 | |
| 6 | 8f | -CN | 86 | |
| 7 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-15-41-i5.svg?max-width=637&scale=1.0)
11 |
11a | -H | 65 |
| 8 | 11b | -OMe | 60 | |
| 9 | 11c | -NMe2 | 58 | |
| 10 | 11d | -F | 68 | |
| 11 | 11e | -CF3 | 63 | |
| 12 | 11f | -CN | 75 | |
The final structural proof for the compounds of the series 7a–f and 8a–f was obtained by spectral comparison between compound 8a and regioisomeric compound 5. The 1H NMR comparison showed two main differences in the spectra (Figure 3): 1) the H–C(triazole) signal of product 8a is shifted downfield from 8.69 to 9.10 ppm and 2) the appearance of a CH2 signal near the N atom is changed from multiplet at 3.96–3.87 ppm to broad singlet at 4.60–3.95 ppm. Furthermore, a significant difference is observed also in the UV spectra. Product 8a has an additional absorption maximum at 370 nm (Figure 4).
![[1860-5397-15-41-3]](/bjoc/content/figures/1860-5397-15-41-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Comparison of 1H NMR spectra of compounds 8a and 5 (300 MHz, CDCl3).
Figure 3: Comparison of 1H NMR spectra of compounds 8a and 5 (300 MHz, CDCl3).
![[1860-5397-15-41-4]](/bjoc/content/figures/1860-5397-15-41-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: a) Experimental UV–vis absorption spectra (lines) with computed theoretical absorption bands (columns) of compounds 5 and 8a (c = 10−4 M). b) Energy diagram and spatial distribution of frontier molecular orbitals (at isosurface value 0.02) of compounds 5 and 8a.
Figure 4: a) Experimental UV–vis absorption spectra (lines) with computed theoretical absorption bands (colum...
In the 7-deazapurine series the SNAr reactions between 2,6-diazido-9-methyl-7-deazapurine (3) and pyrrolidine or piperidine were more regioselective than in the purine series. In the latter case the purine products 6a–c had to be chromatographically separated to remove the 2-azido-6-amino isomer as the purification at the stage of final products was not effective. The observed C(2)-selectivity in the 7-deazapurine series allowed to combine the SNAr and CuAAC reactions into an sequential one-pot process producing target products 10 and 11 directly from diazide 3. The 7-deazapurine structural analogs 10a–f and 11a–f to every purine entry were obtained with 58–80% isolated yields.
UV–vis and fluorescence data
The optical properties of the synthesized compounds were assessed by performing absorption and fluorescence spectroscopy and fluorescence quantum yield measurements. The quantum yields were determined using a fluorescence standard of quinine sulfate in 0.1 M H2SO4 as a reference [53]. Absorption and fluorescence data of the investigated compounds in MeCN solutions are summarized in Table 3. The newly developed purine and 7-deazapurine derivatives exhibit similar photophysical properties to previously reported push–pull systems containing the same central molecular scaffold (see Table 3 versus Figure 1). There are only few other modified purines and 7-deazapurines known for which higher quantum yields were reported. The latter include Castellano’s purines (QY up to 95%) containing electron-withdrawing substituents at C(8) [36,37] and 2-halo-7-deazapurine derivatives (QY up to 83%) reported by Hocek et al. [28].
In our case the lowest-energy absorption band was observed approximately at 360 nm for the purine class compounds (Table 3, entries 1–6 and 13–18), whereas for the 7-deazapurine series (Table 3, entries 7–12 and 19–24) the lowest-energy absorption band was slightly red-shifted by 15 nm to ≈375 nm. A similar trend was observed in the fluorescence spectra of the studied compounds – emission maxima for the purine class compounds was observed at 442–452 nm, whilst it was red-shifted for 7-deazapurine series to 468–472 nm, moreover most of the studied compounds possessed very similar quantum yields 0.56–0.61 for purine and 0.50–0.56 for 7-deazapurine series, with only a few exceptions from a trend (Table 3, entries 3, 9, 12, 15, 21 and 24). Compounds 10f and 11f from the 7-deazapurine class (Table 3, entries 12 and 24), which possess -CN substituents on the para position of the phenyl ring, showed slightly lower quantum yields of 0.36–0.38 and a red shift of the emission maxima by 10–15 nm, whilst similar purine class compounds (Table 3, entries 6 and 18) remained on a previously discussed trend, thus probably indicating a minor change in electronic structure for compounds 10f and 11f. Also, lower quantum yields of 0.28–0.35 and highly red-shifted emission maxima to 528–581 nm were recorded for the compounds possessing strong electron-donating -NMe2 groups (Table 3, entries 3, 9, 15, 21). In addition, it was found that these compounds possess a strong positive solvatochromic effect, which was studied in detail in solvents of different polarity (THF, CHCl3, DMSO, MeCN and MeOH). The study was performed on two model compounds, 8c from the purine class and 11c from the 7-deazapurine class (Figure 5 and Figure 6). As illustrated in Figure 5 and Figure 6 the emission of compounds 8c and 11c showed pronounced changes with solvent polarity in terms of emission maxima and quantum yields, though the absorption spectra showed minor or no changes with increasing solvent polarity indicating small dipole moment in the ground state. A good linear correlation between the fluorescence QY and the Dimroth–Reichardt polarity parameter (ET(30)) [54] was observed (Figure 6) for both classes of compounds. For example, the fluorescence QY of 7-deazapurine 11c in THF were determined to be 0.76 and non-detectable in methanol. Similarly, QY of purine 8c dropped from 0.56 to <0.01 by switching between the same solvents (Table 4). Non-linear dependency in increase of emission maxima is observed (Table 4) with increasing polarity parameter (ET(30)), thus indicating that the sensitivity to solvent polarity is not the only major solute–solvent interaction and the emission maxima are also influenced by other solvent properties such as viscosity or hydrogen bonding. Interestingly, purine class compound 8c showed a dual-fluorescence character in the solvents of higher polarity (DMSO, MeCN) arising from the locally excited and charge transfer excited states, however, dual-fluorescence was not evident for the 7-deazapurine class, which could be explained by a better stabilization of the ICT excited state by the solvatic shell. A decrease of QY accompanied by a high red shift of the emission maxima with increasing polarity of the surrounding media is a typical characteristic for the enhanced ICT of the excited states with a possible character of TICT [55], which could explain a dual-fluorescence and fluorescence quenching in the high polarity media. The charge transfer nature of the transitions for the compounds 8c and 11c, compared to reference compounds 8a and 11a, was confirmed by quantum chemical calculations. Computed spatial distributions of FMO’s and comparison of electronic structures revealed that where is almost no change in energy and distribution of LUMO, which is localized over the extended triazolyl(deaza)purine core (Figure 7). However, a major change in HOMO localization and energy is observed: 1) the HOMO is mostly localized on the central (deaza)purine core and piperidino substituent for the reference compounds 8a and 11a; 2) the HOMO is mostly distributed over the electron-donating Me2N groups, phenyl ring and the 1,2,3-triazolyl moiety for compounds 8c and 11c. Thus, these findings support an enhanced ICT character of the excited states for the compounds possessing strong electron-donating Me2N groups.
![[1860-5397-15-41-5]](/bjoc/content/figures/1860-5397-15-41-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Photos of compound 8c (A and B) and compound 11c (C and D) in THF, CHCl3, DMSO, MeCN and MeOH before excitation (A and C) and under excitation (B and D) of UV (366 nm).
Figure 5: Photos of compound 8c (A and B) and compound 11c (C and D) in THF, CHCl3, DMSO, MeCN and MeOH befor...
Table 3: Photophysical properties of 6-triazolyl compounds 7a–f, 10a–f, 8a–f and 11a–f.a
| Entry | Structure | Compound | R | λabs, nm | λemb, nm | Δλ, nm | Φ |
| 1 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-15-41-i6.svg?max-width=637&scale=1.0)
7 |
7a | -H | 360 | 444 | 84 | 0.59 |
| 2 | 7b | -OMe | 359 | 442 | 83 | 0.61 | |
| 3 | 7c | -NMe2 | 361 | 577 | 216 | 0.32 | |
| 4 | 7d | -F | 360 | 444 | 84 | 0.57 | |
| 5 | 7e | -CF3 | 362 | 452 | 90 | 0.58 | |
| 6 | 7f | -CN | 363 | 453 | 90 | 0.58 | |
| 7 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-15-41-i7.svg?max-width=637&scale=1.0)
10 |
10a | -H | 375 | 472 | 97 | 0.52 |
| 8 | 10b | -OMe | 374 | 466 | 92 | 0.56 | |
| 9 | 10c | -NMe2 | 369 | 528 | 159 | 0.35 | |
| 10 | 10d | -F | 375 | 472 | 97 | 0.53 | |
| 11 | 10e | -CF3 | 377 | 468 | 91 | 0.54 | |
| 12 | 10f | -CN | 378 | 482 | 104 | 0.36 | |
| 13 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-15-41-i8.svg?max-width=637&scale=1.0)
8 |
8a | -H | 357 | 445 | 88 | 0.57 |
| 14 | 8b | -OMe | 357 | 444 | 87 | 0.59 | |
| 15 | 8c | -NMe2 | 360 | 445; 581 | 85; 221 | 0.28c | |
| 16 | 8d | -F | 360 | 449 | 89 | 0.56 | |
| 17 | 8e | -CF3 | 357 | 453 | 96 | 0.57 | |
| 18 | 8f | -CN | 360 | 456 | 96 | 0.60 | |
| 19 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-15-41-i9.svg?max-width=637&scale=1.0)
11 |
11a | -H | 372 | 472 | 100 | 0.54 |
| 20 | 11b | -OMe | 371 | 471 | 100 | 0.52 | |
| 21 | 11c | -NMe2 | 368 | 536 | 168 | 0.35d | |
| 22 | 11d | -F | 373 | 473 | 100 | 0.50 | |
| 23 | 11e | -CF3 | 375 | 481 | 106 | 0.51 | |
| 24 | 11f | -CN | 376 | 485 | 109 | 0.38 | |
aAll spectra were recorded in MeCN solutions (c = 10−4 M) at room temperature; bcompounds 7 and 8 were excited at 360 nm and compounds 10 and 11 were excited at 370 nm; cfluorescence lifetime: 7.79 ± 0.03 ns; dfluorescence lifetime: 8.77 ± 0.03 ns.
![[1860-5397-15-41-6]](/bjoc/content/figures/1860-5397-15-41-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: a) Fluorescence spectra of compounds 8c (λexc = 360 nm) and 11c (λexc = 370 nm) in solvents of different polarity, c = 10−4 M. b) Quantum yield dependence on the Dimroth–Reichardt polarity parameter ET(30) with a linear fit for compounds 8c and 11c.
Figure 6: a) Fluorescence spectra of compounds 8c (λexc = 360 nm) and 11c (λexc = 370 nm) in solvents of diff...
Table 4: Emission maxima and quantum yields of compound 8c and 11c in solvents of different polarity.
| Entry | Compound | Solvent | λem, nma | Δλ | Φ |
| 1 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-15-41-i10.svg?max-width=637&scale=1.0)
8c |
THF | 504 | 147 | 0.56 |
| 2 | CHCl3 | 442 | 76 | 0.51 | |
| 3 | DMSO | 443; 604 | 80; 241 | 0.28 | |
| 4 | MeCN | 445; 581 | 85; 221 | 0.28 | |
| 5 | MeOH | 460; 619 | 96; 255 | <0.01 | |
| 6 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-15-41-i11.svg?max-width=637&scale=1.0)
11c |
THF | 449 | 76 | 0.74 |
| 7 | CHCl3 | 461 | 88 | 0.62 | |
| 8 | DMSO | 572 | 196 | 0.54 | |
| 9 | MeCN | 536 | 166 | 0.35 | |
| 10 | MeOH | -b | -b | 0b | |
aCompound 8c was excited at 360 nm and compound 11c at 370 nm. bFluorescence was lower than the detection limit.
![[1860-5397-15-41-7]](/bjoc/content/figures/1860-5397-15-41-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Energy diagram for the frontier molecular orbitals of compounds 8a, 8c, 11a and 11c.
Figure 7: Energy diagram for the frontier molecular orbitals of compounds 8a, 8c, 11a and 11c.
In summary, moderate to high quantum yields were observed for the studied purine and 7-deazapurine compounds. According to the expectations, the choice of alkyl chains and tertiary amine (piperidine or pyrrolidine) substituents did not significantly influence fluorescent properties. The preliminary study showed that introduction of other aliphatic amines at C(2) does not significantly affect the photophysical properties of the target substances either. On the other hand, the present synthetic development does not allow yet to introduce arylamino moieties at C(2) due to the diminished nucleophilicity of anilines.
The 7-deazapurine derivatives were characterized with a somewhat larger Stokes shift and bathochromic shift of the lowest energy absorption band in comparison to purine derivatives, but resulted in slightly lower quantum yields. Except for the compounds with strong electron-donating Me2N substituents and ICT/TICT character, where QY were slightly higher for 7-deazapurine derivatives. As the emission properties of compounds (7c, 8c, 10c and 11c) possessing the ICT/TICT character of the excited states are environment-dependent it makes these fluorophores interesting as potential sensors of the surroundings such as polarity or (micro)viscosity sensors. Other purine and 7-deazapurine derivatives, which efficiently emit light in the blue region with respect to the structural biocompatibility could be applied for the cell staining in fluorescence microscopy.
Applications in the cell studies
The newly prepared compound library with purine and 7-deazapurine derivatives was submitted to the screening of their biological activity. The compounds of interest were studied on two cancer cell lines – luminal A breast cancer cell line MCF7 and triple negative breast cancer cell line MDAMB231. The results were compared with those obtained on normal breast epithelial cell line (MCF-10A). All compounds showed low cytotoxicity on all tested cell lines (for Figure S81 see Supporting Information File 1). Bearing in mind fluorescent properties and low cytotoxicity of the newly obtained compounds, we tested their potential application in cell staining. The preliminary experiments revealed that the compounds went through the cell membrane after 1 h or 2 h incubation period and localized uniformly in the cytoplasm. The representative fluorescent image of the labeled MCF-7 cells with compound 9 is shown in Figure 8 (for other examples see Figure S82 in Supporting Information File 1).
![[1860-5397-15-41-8]](/bjoc/content/figures/1860-5397-15-41-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Labeled MCF-7 cells using compound 9 (C,D) and unlabeled MCF-7 cells (A,B) in microscope (2 h, c(9) = 100 μM, B and D passed separately through filter (365 ± 20 nm)).
Figure 8: Labeled MCF-7 cells using compound 9 (C,D) and unlabeled MCF-7 cells (A,B) in microscope (2 h, c(9)...
Based on the recent investigation of metabolic labelling of DNA [30,56] by deaza-7-ethenyl-2'-deoxyguanosine and 7-deaza-7-ethenyl-2'-deoxyadenosine or 5-(azidomethyl)-2'-deoxyuridine, we can assume that nucleoside analogs bearing fluorophores of our type would be susceptible to metabolic phosphorylation and incorporation into DNA in the living cells.
Further studies are required to develop the newly described N(9)-alkylated-2-amino-6-triazolylpurines and 7-deazapurines as cell labeling agents for biological chemistry applications.
Conclusion
A group of novel and structurally related N(9)-alkylated 2-amino-6-triazolylpurines and 7-deazapurines was obtained, using the corresponding 2,6-diazido derivatives as key starting materials. We have reported here for the first time that 9-alkyl-2,6-diazidopurines exhibit C(2)-selectivity in nucleophilic aromatic substitution reactions with amines. A similar selectivity was observed also for 9-alkyl-2,6-diazido-7-deazapurines. We have also demonstrated that the synthetic intermediates obtained in the SNAr reaction (e.g., 6-azido-9-heptyl-2-(piperidin-1-yl)purine (6b)) exists in both tetrazolo- and azido-tautomeric forms in CD3CN solution. The presence of the latter permits the CuAAC reaction with terminal acetylenes and gave a rise to the title compounds.
The obtained compounds possess useful levels of fluorescence in acetonitrile solution with quantum yields ranging from 28% to 60% with emission maxima positioned at 442–581 nm. It should be noted that still the biggest challenge in manufacturing of affordable OLEDs is emission of the color blue. Most of here reported compounds emit blue light and thus are important novel structural entities to be studied in the context of OLEDs in the future. Compounds containing a 4-(4-dimethylaminophenyl)-1,2,3-triazol-1-yl substituent have shown a strong solvatochromic effect including the increase of fluorescence quantum yield to 74% in the case of 7-deazapurine derivative 11c. The solvent change provided a fluorescence shift from dark blue (≈440 nm) to orange (≈620 nm) color.
Finally, the purines and 7-deazapurines were tested in live cell imaging on breast cancer cell lines MCF-7 and MDAMB231. They are biocompatible, cell-permeable, not cytotoxic and do not influence cell proliferation. Thus, one can predict that the developed purine and 7-deazapurine derivatives possessing the novel substitution pattern may find their application also in cell labeling in the future besides the potential use as materials in organic light emitting diodes. The latter study which uses structurally related fluorescent purine derivatives containing sterically bulky amorphousing groups at position N(9) for formation of fluorescent molecular glasses are underway in our laboratories and will be reported in due course.
Experimental
General information
Reactions and purity of the synthesized compounds were monitored by TLC using Silica gel 60 F254 aluminum plates (Merck). Visualization was accomplished by UV light. Column chromatography was performed using silica gel 60 (0.040–0.063 mm) (Merck). Yields of products refer to chromatographically and spectroscopically homogeneous materials. Anhydrous methylene chloride, dimethylformamide and acetonitrile were obtained by distillation over CaH2, tetrahydrofuran was obtained by distillation over sodium. Commercial reagents were used as received.
NMR spectra were recorded on Bruker Avance 300 or Bruker Ascend 400 spectrometers (300 MHz, 400 MHz for 1H and 75 MHz, 100 MHz for 13C, respectively). The proton signals for residual non-deuterated solvents (δ 7.26 ppm for CDCl3, δ 2.50 ppm for DMSO-d6) and carbon signals (δ 77.1 ppm for CDCl3, δ 39.5 ppm for DMSO-d6) were used as an internal reference for 1H NMR and 13C NMR spectra, respectively. Coupling constants are reported in Hz. Chemical shifts of signals are given in ppm and multiplicity assigned as follows: s – singlet, d – doublet, t – triplet, m – multiplet.
The infrared spectra were recorded on a Perkin Elmer Spectrum BX spectrometer. Wavelengths are given in cm−1. The UV–vis absorption spectra of all compounds were acquired using a Perkin-Elmer 35 UV–vis spectrometer. Emission spectra were measured on QuantaMaster 40 steady state spectrofluorometer (Photon Technology International, Inc.). Absolute photoluminescence quantum yields were determined using QuantaMaster 40 steady state spectrofluorometer (Photon Technology International, Inc.) equipped with 6 inch integrating sphere by LabSphere, using a florescence standard of quinine sulfate in 0.1 M H2SO4 as a reference. High-resolution mass spectrometry (HRMS) analyses were carried out on a Dual-ESI Q-TOF 6520 (Agilent Technologies) mass spectrometer and Agilent 1290 Infinity series UPLC system equipped with column Extend C18 RRHD 2.1 × 50 mm, 1.8 µm connected to an Agilent 6230 TOF LC/MS masspectrometer.
For HPLC analysis we used an Agilent Technologies 1200 Series chromatograph equipped with an Agilent XDB-C18 (4.6 × 50 mm, 1.8 µm) column and a Phenomenex Gemini NX (4.6 × 100 mm, 3 µm) column. Eluent A: 0.01 M KH2PO4 solution with 6% v/v MeCN added; eluent B: 0.1% TFA solution with 5% v/v MeCN added; eluent C – MeCN.
General procedures and product characterization
2,6-Bistriazolyl derivative 4 was synthesized using previously reported procedure of Cu(I)-catalyzed azide–alkyne cycloaddition reaction on 2,6-diazidopurine derivatives [25]. Synthesis of 7-deazapurine derivatives 3, 10a, 11a and their characterization are described in our preliminary communication [39].
Synthesis of 9-alkyl-2,6-diazido-9H-purine derivatives 2a–c
Alkylation, method A: A solution of 2,6-dichloropurine (1a, 1.0 g, 5.4 mmol, 1.0 equiv) in anhydrous MeCN or anhydrous DMF (30 mL) was cooled to 0 °C and 57% suspension of NaH (0.3 g, 7.0 mmol, 1.3 equiv) was added in small portions (50 mg). The resulting reaction mixture was stirred for 30 min. After that, the corresponding 1-iodoalkane or 1-bromoalkane (11 mmol, 2.1 equiv) was added and the reaction mixture was stirred for 1–3 days at 20–55 °C. The excess of NaH was neutralized with MeOH or EtOH. The reaction mixture was evaporated under reduced pressure and the residue was dissolved in DCM (30 mL), the organic phase was washed with brine (2 × 15 mL) and subsequently dried over anh. Na2SO4 and evaporated. Silica gel column chromatography (Hex/EtOAc 4:1) provided the desired product.
Alkylation method B: A solution of 2,6-dichloropurine (1a, 5.0 g, 26.5 mmol, 1.0 equiv), corresponding alcohol (31.7 mmol, 1.2 equiv) and Ph3P (9.2 g, 34.9 mmol, 1.3 equiv) in anhydrous THF (30 mL) was cooled to 0 °C. DIAD (6.90 mL, 35.0 mmol, 1.3 equiv) was added dropwise, the mixture was stirred for 1 h at 20 °C, controlled by HPLC, then evaporated to dryness. Subsequently, EtOH (20 mL) was added and the resulting mixture was cooled to −10 °C to form precipitate of Ph3PO, which was filtered as a byproduct and the filtrate was evaporated. The column chromatography (DCM/MeCN 10:1) provided the desired resulting product.
2,6-Dichloro-9-heptyl-9H-purine (1a-1): Slightly yellow oil; reaction time (method A) – 1 h; yield 5.0 g, 66%. IR (KBr) ν (cm−1): 2933, 1802, 1733; 1H NMR (300 MHz, CDCl3) δ 8.09 (s, 1H, H-C(8)), 4.23 (t, 3J = 7.2 Hz, 2H, -CH2-), 1.88 (quintet, 3J = 7.2 Hz, 2H, -CH2-), 1.36–1.13 (m, 8H, 4 × -CH2-), 0.82 (t, 3J = 6.8 Hz, 3H, -CH3) ppm; 13C NMR (75.5 MHz, CDCl3) δ 153.3, 152.9, 151.7, 145.9, 130.8, 44.7, 31.6, 29.8, 28.6, 26.6, 22.5, 14.0 ppm; HRMS–ESI (m/z): [M + H]+ calcd for C12H17Cl2N4, 287.0825; found, 287.0826.
Azidation: NaN3 (5.88 g, 90.5 mmol, 3.0 equiv) was added to a solution of 9-alkyl-2,6-dichloro-9H-purine (30 mmol, 1.0 equiv) in acetone (50 mL) and stirred for 14 h at 50 °C, protected from the daylight. Then, the reaction mixture was evaporated and suspended in water (30 mL). The resulting precipitate was filtered and dried in vacuum.
2,6-Diazido-9-heptyl-9H-purine (2a): Colorless solid; reaction time – 14 h; yield 8.4 g, 93%. IR (KBr) ν (cm−1): 2932, 2858, 2170, 2123; 1H NMR (300 MHz, CDCl3) δ 7.87 (s, 1H, H-C(8)), 4.15 (t, 3J = 7.2 Hz, 2H, -CH2-), 1.93–1.77 (m, 2H, -CH2-), 1.39–1.15 (m, 8H, 4 × -CH2-), 0.84 (t, 3J = 6.8 Hz, 3H, -CH3) ppm; 13C NMR (75.5 MHz, CDCl3) δ 155.9, 154.1, 153.7, 143.7, 121.5, 44.2, 31.6, 29.8, 28.7, 26.6, 22.6, 14.1 ppm; HRMS–ESI (m/z): [M + H]+ calcd for C12H17N10, 301.1632; found, 301.1646.
Synthesis of 9-alkyl-6-azido-2-pyrrolidino-9H-purine or 9-alkyl-6-azido-2-piperidino-9H-purine derivatives 6a,b: 9-Alkyl-2,6-diazido-9H-purine 2 (8.3 mmol, 1.0 equiv) was dissolved in DMF (30 mL), pyrrolidine or piperidine (11.7 mmol, 1.4 equiv) was added and the reaction mixture was stirred isolated from the daylight at 30 °C for 5 h. After that, the mixture was evaporated and silica gel column chromatography (DCM/MeCN 50:1) was used to provide the desired product.
6-Azido-9-heptyl-2-pyrrolidino-9H-purine (6a): Slightly brown solid, reaction time – 4 h; yield 0.88 g, 51%. IR (KBr) ν (cm−1): 3062, 2927, 2858, 2148, 2122, 1570, 1254; 1H NMR (300 MHz, CDCl3) δ 7.56 (s, 1H, H-C(8)), 4.03 (t, 3J = 7.0 Hz, 2H, -CH2-), 3.62–3.54 (m, 4H, 2 × -CH2-), 2.00–1.92 (m, 4H, 2 × -CH2-), 1.83 (quintet, 3J = 7.0 Hz, 2H, -CH2-), 1.37–1.16 (m, 8H, 4 × -CH2-), 0.85 (t, 3J = 6.8 Hz, 3H, -CH3) ppm; 13C NMR (75.5 MHz, CDCl3) δ 157.4, 154.8, 152.4, 140.3, 117.0, 47.0 (2C)*, 43.4, 31.7, 29.6, 28.8, 26.6, 25.6 (2C)*, 22.7, 14.1 ppm; HRMS–ESI (m/z): [M + H]+ calcd for C16H25N8, 329.2197; found, 329.2195.
Synthesis of 9-alkyl-2-pyrrolidino-6-(1,2,3-triazol-1-yl)purine derivatives 7a–f and 9-alkyl-2-piperidino-6-(1,2,3-triazol-1-yl)purine derivatives 8a–f (typical procedure): Alkyne (1.2 equiv) and 10% AcOH water solution (1 mL) were added to a solution of compound 6a (200 mg, 0.61 mmol, 1.0 equiv) in THF (7 mL). The flask was isolated from daylight and CuSO4∙5H2O (10 mol %) and sodium ascorbate (20 mol %) were added. The reaction mixture was heated for 20 h at 50 °C. The reaction mixture was cooled to ambient temperature and the precipitated product (bright yellow/green in color) was filtered. The product on the filter was washed with water (5 mL) and MTBE (3 × 5 mL). Then the product was transferred into a flask and dissolved in CHCl3 (7 mL). H2S gas was bubbled through the latter solution until dark brown/black suspension appeared. The resulting mixture was filtered through celite, the filtrate was evaporated under reduced pressure and dried in vacuo. Products can be further purified by silica gel column chromatography, if required.
9-Heptyl-6-(4-phenyl-1H-1,2,3-triazol-1-yl)-2-pyrrolidino-9H-purine (7a): Slightly yellow solid, yield 134 mg, 51%. IR (KBr) ν (cm−1): 2952, 2924, 2857, 1622, 1540; 1H NMR (300 MHz, 70 °C, DMSO-d6 + D2O) δ 9.36 (s, 1H, H-C(triazole)), 8.29 (s, 1H, H-C(8)), 8.01 (d, 3J = 7.2 Hz, 2H, Ar), 7.50 (t, 3J = 7.2 Hz, 2H, Ar), 7.40 (t, 3J = 7.2 Hz, 1H, Ar), 4.15 (t, 3J = 7.0 Hz, 2H, -CH2-), 3.71–3.55 (m, 4H, 2 × -CH2-), 2.08–1.93 (m, 4H, 2 × -CH2-), 1.88 (quintet, 3J = 7.0 Hz, 2H, -CH2-), 1.40–1.16 (m, 8H, 4 × -CH2-), 0.84 (t, 3J = 7.0 Hz, 3H, -CH3) ppm; 13C NMR (75.5 MHz, 70 °C, DMSO-d6 + D2O) δ 156.5, 156.3, 146.4, 143.9, 143.7, 129.6, 128.7, 128.2, 125.5, 120.0, 114.4, 46.6, 42.8, 30.7, 28.4, 27.7, 25.6, 24.7, 21.6, 13.4 ppm; HRMS–ESI (m/z): [M + Na]+ calcd for C24H30N8Na, 453.2478; found, 453.2477.
Synthesis of 2-dialkylamino-9-methyl-6-[4-(4-substituted phenyl)-1,2,3-triazol-1-yl]-7-deazapurines 10a–f, 11a–f (Analogous as described in [39], a typical procedure): A mixture of azide 3 (86 mg, 0.4 mmol) and secondary amine (1.2 mmol) in CH3CN (2 mL) was protected from daylight and stirred for 8–24 h at 40 °C. After completion of the SNAr reaction (TLC control), the reaction mixture was cooled to rt and the corresponding alkyne (0.52 mmol), DIPEA (70 µL, 0.4 mmol), AcOH (23 µL, 0.4 mmol) and CuI (15 mg, 0.08 mmol) were added. The reaction mixture was stirred under argon atmosphere at rt for 8–10 h (TLC control). Then the reaction mixture was poured into aqueous 10% ammonia solution (25 mL), stirred for 10 minutes and extracted with CHCl3 (3 × 20 mL). The combined organic layers were washed with water (2 × 30 mL), dried over anhyd Na2SO4 and filtered. After removal of the solvent in vacuum, the crude products were purified by silica gel column chromatography (CHCl3/EtOAc 6:1) to afford compounds 10a–f, 11a–f.
6-[4-(4-Methoxyphenyl)-1,2,3-triazol-1-yl]-9-methyl-2-pyrrolidino-7-deazapurine (10b): Yellow solid, mp 258 °C dec, yield 99 mg, 66%. 1H NMR (400 MHz, CDCl3) δ 2.01–2.11 (m, 4H, piperidine 2 × CH2), 3.65–3.72 (m, 4H, piepridine 2 × CH2), 3.74 (s, 3H, CH3), 3.88 (s, 3H, OCH3), 6.88 (d, J = 3.6 Hz, 1H, 6-H), 7.02 (d, J = 8.8 Hz, 2H, ArH), 7.08 (d, J = 3.6 Hz, 5-H, 1H), 7.92 ( d, J = 8.8 Hz, 2H,ArH), 8.79 ( s, 1H,triazole-H); 13C NMR (100 MHz, CDCl3) δ 25.6, 30.7, 46.9, 55.3, 99.5, 101.6, 114.3, 116.2, 123.1, 126.7, 127.3, 146.7, 147.0, 156.6, 156.8, 159.8; HRMS–ESI (m/z): [M + H]+ calcd for C20H22N7O, 376.1880; found, 376.1887.
Quantum chemical calculations
The initial molecular geometries were generated by using a molecular mechanics method (force field MMF94, steepest descent algorithm) and systematic conformational analysis as implemented in Avogadro 1.1.1 software. The minimum energy conformers found by molecular mechanics were further optimized with the Gaussian 09 program package [57] by the means of DFT using B3LYP exchange–correlation hybrid functional together with the 6-31G** basis set including the polarizable continuum model in MeCN. Further, harmonic vibrational frequencies were calculated to verify the stability of optimized geometries. All the calculated vibrational frequencies were real (positive), which indicates the true minimum of the total energy on the potential energy hypersurface. The theoretical absorption bands were obtained by the means of time-dependent extension of DFT (TDDFT). Spatial distributions of electron density of calculated HOMOs and LUMOs were obtained at an isosurface value of 0.02 where red and green colors stand for the positive and negative signs of the molecular orbital wave function, respectively.
Experimental procedure for cell studies
The Human breast adenocarcinoma cell line MCF-7, and normal mammary epithelial cell line MCF10A were ordered from Food Industry Research and Development Institute (Taiwan). MCF-7 cells were cultured in RPMI medium supplemented with fetal bovine serum (10%), antibiotic–antimycotic (1.0%). MCF-10A cells were cultured in α-MEM supplemented with FBS (10%), and antibiotic–antimycotic (1.0%). All cells were cultured in an environment equilibrated with 5% CO2 at 37 °C. The cell number and viability of the cells were determined by applying the trypan blue exclusion method and Alamar Blue assay, respectively. Following the separated incubation of MCF-7 and MCF-10A cells (7.0 × 104 cells per well) in a culture medium for 24 h at 37 °C containing 5% CO2, each of the culture media were replaced with 500 μL of cell culture medium containing the compound, and then further cultured for an additional 1 h. Those cells were washed twice with 1× PBS and fixed to the membrane using 4% paraformaldehyde in 5.0 mM sodium phosphate buffer (pH 7.4; 1.0 mL) for 10 min. The fluorescence images of cancer cells were acquired with a fluorescence microscope (Olympus IX 71, Center Valley, PA, USA). Light from an Hg lamp (100 W) passed separately through filter (365 ± 20 nm) before it was used to excite the compound. The emission filters used in this study are long pass filters, with cut-on wavelengths at 420 nm (Semrock, Rochester, NY, USA).
Supporting Information
| Supporting Information File 1: Full experimental procedures, emission spectra, DSC data, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 5.3 MB | Download |
Acknowledgements
This work is supported by the ERDF 1.1.1.1 activity project Nr. 1.1.1.1/16/A/131 "Design and Investigation of Light Emitting and Solution Processable Organic Molecular Glasses" (Latvia) and by a grant (No. TAP-LLT-01/2015) from the Research Council of Lithuania. The authors thank Ms. I. Māliņa for absorption and emission spectra and Dr. J. Zicāns and R. Merijs Meri for DSC measurements. Copyrights to the picture “business people greet” used in the graphical abstract belong to Publitek, Inc. dba GoGraph (https://www.gograph.com/).
References
-
Skalski, B.; Steer, R. P.; Verrall, R. E. J. Am. Chem. Soc. 1991, 113, 1756–1762. doi:10.1021/ja00005a045
Return to citation in text: [1] -
Legraverend, M. Tetrahedron 2008, 64, 8585–8603. doi:10.1016/j.tet.2008.05.115
Return to citation in text: [1] -
Wierzchowski, J. Curr. Top. Biophys. 2010, 33, 9–16.
Return to citation in text: [1] -
Manvar, A.; Shah, A. Tetrahedron 2013, 69, 8105–8127. doi:10.1016/j.tet.2013.06.034
Return to citation in text: [1] -
Matarazzo, A.; Hudson, R. H. E. Tetrahedron 2015, 71, 1627–1657. doi:10.1016/j.tet.2014.12.066
Return to citation in text: [1] -
Sharma, S.; Singh, J.; Ojha, R.; Singh, H.; Kaur, M.; Bedi, P. M. S.; Nepali, K. Eur. J. Med. Chem. 2016, 112, 298–346. doi:10.1016/j.ejmech.2016.02.018
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527
Return to citation in text: [1] -
Perlíková, P.; Hocek, M. Med. Res. Rev. 2017, 37, 1429–1460. doi:10.1002/med.21465
Return to citation in text: [1] -
De Coen, L. M.; Heugebaert, T. S. A.; García, D.; Stevens, C. V. Chem. Rev. 2016, 116, 80–139. doi:10.1021/acs.chemrev.5b00483
Return to citation in text: [1] -
Tumkevicius, S.; Dodonova, J. Chem. Heterocycl. Compd. 2012, 48, 258–279. doi:10.1007/s10593-012-0986-2
Return to citation in text: [1] -
De Ornellas, S.; Slattery, J. M.; Edkins, R. M.; Beeby, A.; Baumann, C. G.; Fairlamb, I. J. S. Org. Biomol. Chem. 2015, 13, 68–72. doi:10.1039/c4ob02081b
Return to citation in text: [1] -
Butler, R. S.; Myers, A. K.; Bellarmine, P.; Abboud, K. A.; Castellano, R. K. J. Mater. Chem. 2007, 17, 1863–1865. doi:10.1039/b618171f
Return to citation in text: [1] [2] -
Vabre, R.; Legraverend, M.; Piguel, S. Dyes Pigm. 2014, 105, 145–151. doi:10.1016/j.dyepig.2014.01.025
Return to citation in text: [1] [2] -
Saito, Y.; Suzuki, A.; Imai, K.; Nemoto, N.; Saito, I. Tetrahedron Lett. 2010, 51, 2606–2609. doi:10.1016/j.tetlet.2010.03.012
Return to citation in text: [1] -
Seela, F.; Schweinberger, E.; Xu, K.; Sirivolu, V. R.; Rosemeyer, H.; Becker, E.-M. Tetrahedron 2007, 63, 3471–3482. doi:10.1016/j.tet.2006.09.114
Return to citation in text: [1] -
Hudson, R. H. E.; Dambenieks, A. K.; Viirre, R. D. Synlett 2004, 2400–2402. doi:10.1055/s-2004-832851
Return to citation in text: [1] -
Sinkeldam, R. W.; Greco, N. J.; Tor, Y. Chem. Rev. 2010, 110, 2579–2619. doi:10.1021/cr900301e
Return to citation in text: [1] -
Dyrager, C.; Börjesson, K.; Dinér, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L. M.; Grøtli, M. Eur. J. Org. Chem. 2009, 1515–1521. doi:10.1002/ejoc.200900018
Return to citation in text: [1] -
Greco, N. J.; Tor, Y. Tetrahedron 2007, 63, 3515–3527. doi:10.1016/j.tet.2007.01.073
Return to citation in text: [1] -
Gaied, N. B.; Glasser, N.; Ramalanjaona, N.; Beltz, H.; Wolff, P.; Marquet, R.; Burger, A.; Mély, Y. Nucleic Acids Res. 2005, 33, 1031–1039. doi:10.1093/nar/gki253
Return to citation in text: [1] -
Zilbershtein-Shklanovsky, L.; Weitman, M.; Major, D. T.; Fischer, B. J. Org. Chem. 2013, 78, 11999–12008. doi:10.1021/jo402050x
Return to citation in text: [1] -
Rankin, K. M.; Sproviero, M.; Rankin, K.; Sharma, P.; Wetmore, S. D.; Manderville, R. A. J. Org. Chem. 2012, 77, 10498–10508. doi:10.1021/jo302164c
Return to citation in text: [1] -
Mitsui, T.; Kimoto, M.; Kawai, R.; Yokoyama, S.; Hirao, I. Tetrahedron 2007, 63, 3528–3537. doi:10.1016/j.tet.2006.11.096
Return to citation in text: [1] -
Zayas, J.; Annoual, M.; Das, J. K.; Felty, Q.; Gonzalez, W. G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S. F. Bioconjugate Chem. 2015, 26, 1519–1532. doi:10.1021/acs.bioconjchem.5b00300
Return to citation in text: [1] -
Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095
Return to citation in text: [1] [2] [3] [4] -
Kavoosi, S.; Rayala, R.; Walsh, B.; Barrios, M.; Gonzalez, W. G.; Miksovska, J.; Mathivathanan, L.; Raptis, R. G.; Wnuk, S. F. Tetrahedron Lett. 2016, 57, 4364–4367. doi:10.1016/j.tetlet.2016.08.053
Return to citation in text: [1] -
Tokugawa, M.; Kaneko, K.; Saito, M.; Kanamori, T.; Masaki, Y.; Ohkubo, A.; Sekine, M.; Seio, K. Chem. Lett. 2015, 44, 64–66. doi:10.1246/cl.140879
Return to citation in text: [1] -
Sabat, N.; Nauš, P.; Matyašovský, J.; Dziuba, D.; Slavětínská, L. P.; Hocek, M. Synthesis 2016, 48, 1029–1045. doi:10.1055/s-0035-1561287
Return to citation in text: [1] [2] -
Bucevicius, J.; Skardziute, L.; Dodonova, J.; Kazlauskas, K.; Bagdziunas, G.; Jursenas, S.; Tumkevicius, S. RSC Adv. 2015, 5, 38610–38622. doi:10.1039/c5ra05482f
Return to citation in text: [1] [2] -
Neef, A. B.; Samain, F.; Luedtke, N. W. ChemBioChem 2012, 13, 1750–1753. doi:10.1002/cbic.201200253
Return to citation in text: [1] [2] -
Venkatesh, V.; Shukla, A.; Sivakumar, S.; Verma, S. ACS Appl. Mater. Interfaces 2014, 6, 2185–2191. doi:10.1021/am405345h
Return to citation in text: [1] -
Li, J.; Zhang, Y.; Zhang, H.; Xuan, X.; Xie, M.; Xia, S.; Qu, G.; Guo, H. Anal. Chem. (Washington, DC, U. S.) 2016, 88, 5554–5560. doi:10.1021/acs.analchem.6b01395
Return to citation in text: [1] -
Li, J.-P.; Wang, H.-X.; Wang, H.-X.; Xie, M.-S.; Qu, G.-R.; Niu, H.-Y.; Guo, H.-M. Eur. J. Org. Chem. 2014, 2225–2230. doi:10.1002/ejoc.201301897
Return to citation in text: [1] [2] -
Saito, Y.; Suzuki, A.; Ishioroshi, S.; Saito, I. Tetrahedron Lett. 2011, 52, 4726–4729. doi:10.1016/j.tetlet.2011.06.089
Return to citation in text: [1] -
Pratibha; Singh, S.; Sivakumar, S.; Verma, S. Eur. J. Inorg. Chem. 2017, 4202–4209. doi:10.1002/ejic.201700806
Return to citation in text: [1] -
Butler, R. S.; Cohn, P.; Tenzel, P.; Abboud, K. A.; Castellano, R. K. J. Am. Chem. Soc. 2009, 131, 623–633. doi:10.1021/ja806348z
Return to citation in text: [1] [2] -
Yang, Y.; Cohn, P.; Eom, S.-H.; Abboud, K. A.; Castellano, R. K.; Xue, J. J. Mater. Chem. C 2013, 1, 2867–2874. doi:10.1039/c3tc00734k
Return to citation in text: [1] [2] -
Yang, Y.; Cohn, P.; Dyer, A. L.; Eom, S.-H.; Reynolds, J. R.; Castellano, R. K.; Xue, J. Chem. Mater. 2010, 22, 3580–3582. doi:10.1021/cm100407n
Return to citation in text: [1] -
Bucevicius, J.; Turks, M.; Tumkevicius, S. Synlett 2018, 29, 525–529. doi:10.1055/s-0036-1590942
Return to citation in text: [1] [2] [3] [4] [5] -
Cīrule, D.; Ozols, K.; Platnieks, O.; Bizdēna, Ē.; Māliņa, I.; Turks, M. Tetrahedron 2016, 72, 4177–4185. doi:10.1016/j.tet.2016.05.043
Return to citation in text: [1] -
Ozols, K.; Cīrule, D.; Novosjolova, I.; Stepanovs, D.; Liepinsh, E.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2016, 57, 1174–1178. doi:10.1016/j.tetlet.2016.02.003
Return to citation in text: [1] [2] [3] -
Lu, W.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Org. Chem. 2007, 72, 5012–5015. doi:10.1021/jo070515+
Return to citation in text: [1] -
Zatloukal, M.; Jorda, R.; Gucký, T.; Řezníčková, E.; Voller, J.; Pospíšil, T.; Malínková, V.; Adamcová, H.; Kryštof, V.; Strnad, M. Eur. J. Med. Chem. 2013, 61, 61–72. doi:10.1016/j.ejmech.2012.06.036
Return to citation in text: [1] -
Gucký, T.; Jorda, R.; Zatloukal, M.; Bazgier, V.; Berka, K.; Řezníčková, E.; Béres, T.; Strnad, M.; Kryštof, V. J. Med. Chem. 2013, 56, 6234–6247. doi:10.1021/jm4006884
Return to citation in text: [1] -
Morales, F.; Ramírez, A.; Conejo-García, A.; Morata, C.; Marchal, J. A.; Campos, J. M. Eur. J. Med. Chem. 2014, 76, 118–124. doi:10.1016/j.ejmech.2014.02.012
Return to citation in text: [1] -
Calderón-Arancibia, J.; Espinosa-Bustos, C.; Cañete-Molina, Á.; Tapia, R. A.; Faúndez, M.; Torres, M. J.; Aguirre, A.; Paulino, M.; Salas, C. O. Molecules 2015, 20, 6808–6826. doi:10.3390/molecules20046808
Return to citation in text: [1] -
Malínková, V.; Řezníčková, E.; Jorda, R.; Gucký, T.; Kryštof, V. Bioorg. Med. Chem. 2017, 25, 6523–6535. doi:10.1016/j.bmc.2017.10.032
Return to citation in text: [1] -
Komodziński, K.; Lepczyńska, J.; Gdaniec, Z.; Bartolotti, L.; Delley, B.; Franzen, S.; Skalski, B. Photochem. Photobiol. Sci. 2014, 13, 563–573. doi:10.1039/c3pp50385b
Return to citation in text: [1] -
Pignatello, R.; Puleo, A.; Acquaviva, R.; Di Giacomo, C.; Vanella, A.; Puglisi, G. J. Drug Delivery Sci. Technol. 2004, 14, 181–186. doi:10.1016/s1773-2247(04)50098-9
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095
Return to citation in text: [1] -
Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435
Return to citation in text: [1] -
Bucevičius, J.; Tumkevičius, S. Chemija 2015, 26, 126–131.
Return to citation in text: [1] -
Williams, A. T. R.; Winfield, S. A.; Miller, J. N. Analyst 1983, 108, 1067–1071. doi:10.1039/an9830801067
Return to citation in text: [1] -
Reichardt, C. Chem. Rev. 1994, 94, 2319–2358. doi:10.1021/cr00032a005
Return to citation in text: [1] -
Grabowski, Z. R.; Rotkiewicz, K.; Rettig, W. Chem. Rev. 2003, 103, 3899–4032. doi:10.1021/cr940745l
Return to citation in text: [1] -
Neef, A. B.; Luedtke, N. W. ChemBioChem 2014, 15, 789–793. doi:10.1002/cbic.201400037
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1]
| 39. | Bucevicius, J.; Turks, M.; Tumkevicius, S. Synlett 2018, 29, 525–529. doi:10.1055/s-0036-1590942 |
| 41. | Ozols, K.; Cīrule, D.; Novosjolova, I.; Stepanovs, D.; Liepinsh, E.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2016, 57, 1174–1178. doi:10.1016/j.tetlet.2016.02.003 |
| 1. | Skalski, B.; Steer, R. P.; Verrall, R. E. J. Am. Chem. Soc. 1991, 113, 1756–1762. doi:10.1021/ja00005a045 |
| 2. | Legraverend, M. Tetrahedron 2008, 64, 8585–8603. doi:10.1016/j.tet.2008.05.115 |
| 3. | Wierzchowski, J. Curr. Top. Biophys. 2010, 33, 9–16. |
| 4. | Manvar, A.; Shah, A. Tetrahedron 2013, 69, 8105–8127. doi:10.1016/j.tet.2013.06.034 |
| 5. | Matarazzo, A.; Hudson, R. H. E. Tetrahedron 2015, 71, 1627–1657. doi:10.1016/j.tet.2014.12.066 |
| 6. | Sharma, S.; Singh, J.; Ojha, R.; Singh, H.; Kaur, M.; Bedi, P. M. S.; Nepali, K. Eur. J. Med. Chem. 2016, 112, 298–346. doi:10.1016/j.ejmech.2016.02.018 |
| 7. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Eur. J. Org. Chem. 2015, 3629–3649. doi:10.1002/ejoc.201403527 |
| 18. | Dyrager, C.; Börjesson, K.; Dinér, P.; Elf, A.; Albinsson, B.; Wilhelmsson, L. M.; Grøtli, M. Eur. J. Org. Chem. 2009, 1515–1521. doi:10.1002/ejoc.200900018 |
| 19. | Greco, N. J.; Tor, Y. Tetrahedron 2007, 63, 3515–3527. doi:10.1016/j.tet.2007.01.073 |
| 31. | Venkatesh, V.; Shukla, A.; Sivakumar, S.; Verma, S. ACS Appl. Mater. Interfaces 2014, 6, 2185–2191. doi:10.1021/am405345h |
| 32. | Li, J.; Zhang, Y.; Zhang, H.; Xuan, X.; Xie, M.; Xia, S.; Qu, G.; Guo, H. Anal. Chem. (Washington, DC, U. S.) 2016, 88, 5554–5560. doi:10.1021/acs.analchem.6b01395 |
| 30. | Neef, A. B.; Samain, F.; Luedtke, N. W. ChemBioChem 2012, 13, 1750–1753. doi:10.1002/cbic.201200253 |
| 56. | Neef, A. B.; Luedtke, N. W. ChemBioChem 2014, 15, 789–793. doi:10.1002/cbic.201400037 |
| 17. | Sinkeldam, R. W.; Greco, N. J.; Tor, Y. Chem. Rev. 2010, 110, 2579–2619. doi:10.1021/cr900301e |
| 33. | Li, J.-P.; Wang, H.-X.; Wang, H.-X.; Xie, M.-S.; Qu, G.-R.; Niu, H.-Y.; Guo, H.-M. Eur. J. Org. Chem. 2014, 2225–2230. doi:10.1002/ejoc.201301897 |
| 25. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 12. | Butler, R. S.; Myers, A. K.; Bellarmine, P.; Abboud, K. A.; Castellano, R. K. J. Mater. Chem. 2007, 17, 1863–1865. doi:10.1039/b618171f |
| 13. | Vabre, R.; Legraverend, M.; Piguel, S. Dyes Pigm. 2014, 105, 145–151. doi:10.1016/j.dyepig.2014.01.025 |
| 14. | Saito, Y.; Suzuki, A.; Imai, K.; Nemoto, N.; Saito, I. Tetrahedron Lett. 2010, 51, 2606–2609. doi:10.1016/j.tetlet.2010.03.012 |
| 15. | Seela, F.; Schweinberger, E.; Xu, K.; Sirivolu, V. R.; Rosemeyer, H.; Becker, E.-M. Tetrahedron 2007, 63, 3471–3482. doi:10.1016/j.tet.2006.09.114 |
| 16. | Hudson, R. H. E.; Dambenieks, A. K.; Viirre, R. D. Synlett 2004, 2400–2402. doi:10.1055/s-2004-832851 |
| 29. | Bucevicius, J.; Skardziute, L.; Dodonova, J.; Kazlauskas, K.; Bagdziunas, G.; Jursenas, S.; Tumkevicius, S. RSC Adv. 2015, 5, 38610–38622. doi:10.1039/c5ra05482f |
| 8. | Perlíková, P.; Hocek, M. Med. Res. Rev. 2017, 37, 1429–1460. doi:10.1002/med.21465 |
| 9. | De Coen, L. M.; Heugebaert, T. S. A.; García, D.; Stevens, C. V. Chem. Rev. 2016, 116, 80–139. doi:10.1021/acs.chemrev.5b00483 |
| 10. | Tumkevicius, S.; Dodonova, J. Chem. Heterocycl. Compd. 2012, 48, 258–279. doi:10.1007/s10593-012-0986-2 |
| 11. | De Ornellas, S.; Slattery, J. M.; Edkins, R. M.; Beeby, A.; Baumann, C. G.; Fairlamb, I. J. S. Org. Biomol. Chem. 2015, 13, 68–72. doi:10.1039/c4ob02081b |
| 30. | Neef, A. B.; Samain, F.; Luedtke, N. W. ChemBioChem 2012, 13, 1750–1753. doi:10.1002/cbic.201200253 |
| 55. | Grabowski, Z. R.; Rotkiewicz, K.; Rettig, W. Chem. Rev. 2003, 103, 3899–4032. doi:10.1021/cr940745l |
| 23. | Mitsui, T.; Kimoto, M.; Kawai, R.; Yokoyama, S.; Hirao, I. Tetrahedron 2007, 63, 3528–3537. doi:10.1016/j.tet.2006.11.096 |
| 26. | Kavoosi, S.; Rayala, R.; Walsh, B.; Barrios, M.; Gonzalez, W. G.; Miksovska, J.; Mathivathanan, L.; Raptis, R. G.; Wnuk, S. F. Tetrahedron Lett. 2016, 57, 4364–4367. doi:10.1016/j.tetlet.2016.08.053 |
| 27. | Tokugawa, M.; Kaneko, K.; Saito, M.; Kanamori, T.; Masaki, Y.; Ohkubo, A.; Sekine, M.; Seio, K. Chem. Lett. 2015, 44, 64–66. doi:10.1246/cl.140879 |
| 36. | Butler, R. S.; Cohn, P.; Tenzel, P.; Abboud, K. A.; Castellano, R. K. J. Am. Chem. Soc. 2009, 131, 623–633. doi:10.1021/ja806348z |
| 37. | Yang, Y.; Cohn, P.; Eom, S.-H.; Abboud, K. A.; Castellano, R. K.; Xue, J. J. Mater. Chem. C 2013, 1, 2867–2874. doi:10.1039/c3tc00734k |
| 22. | Rankin, K. M.; Sproviero, M.; Rankin, K.; Sharma, P.; Wetmore, S. D.; Manderville, R. A. J. Org. Chem. 2012, 77, 10498–10508. doi:10.1021/jo302164c |
| 28. | Sabat, N.; Nauš, P.; Matyašovský, J.; Dziuba, D.; Slavětínská, L. P.; Hocek, M. Synthesis 2016, 48, 1029–1045. doi:10.1055/s-0035-1561287 |
| 28. | Sabat, N.; Nauš, P.; Matyašovský, J.; Dziuba, D.; Slavětínská, L. P.; Hocek, M. Synthesis 2016, 48, 1029–1045. doi:10.1055/s-0035-1561287 |
| 13. | Vabre, R.; Legraverend, M.; Piguel, S. Dyes Pigm. 2014, 105, 145–151. doi:10.1016/j.dyepig.2014.01.025 |
| 21. | Zilbershtein-Shklanovsky, L.; Weitman, M.; Major, D. T.; Fischer, B. J. Org. Chem. 2013, 78, 11999–12008. doi:10.1021/jo402050x |
| 41. | Ozols, K.; Cīrule, D.; Novosjolova, I.; Stepanovs, D.; Liepinsh, E.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2016, 57, 1174–1178. doi:10.1016/j.tetlet.2016.02.003 |
| 20. | Gaied, N. B.; Glasser, N.; Ramalanjaona, N.; Beltz, H.; Wolff, P.; Marquet, R.; Burger, A.; Mély, Y. Nucleic Acids Res. 2005, 33, 1031–1039. doi:10.1093/nar/gki253 |
| 24. | Zayas, J.; Annoual, M.; Das, J. K.; Felty, Q.; Gonzalez, W. G.; Miksovska, J.; Sharifai, N.; Chiba, A.; Wnuk, S. F. Bioconjugate Chem. 2015, 26, 1519–1532. doi:10.1021/acs.bioconjchem.5b00300 |
| 25. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 53. | Williams, A. T. R.; Winfield, S. A.; Miller, J. N. Analyst 1983, 108, 1067–1071. doi:10.1039/an9830801067 |
| 12. | Butler, R. S.; Myers, A. K.; Bellarmine, P.; Abboud, K. A.; Castellano, R. K. J. Mater. Chem. 2007, 17, 1863–1865. doi:10.1039/b618171f |
| 36. | Butler, R. S.; Cohn, P.; Tenzel, P.; Abboud, K. A.; Castellano, R. K. J. Am. Chem. Soc. 2009, 131, 623–633. doi:10.1021/ja806348z |
| 37. | Yang, Y.; Cohn, P.; Eom, S.-H.; Abboud, K. A.; Castellano, R. K.; Xue, J. J. Mater. Chem. C 2013, 1, 2867–2874. doi:10.1039/c3tc00734k |
| 38. | Yang, Y.; Cohn, P.; Dyer, A. L.; Eom, S.-H.; Reynolds, J. R.; Castellano, R. K.; Xue, J. Chem. Mater. 2010, 22, 3580–3582. doi:10.1021/cm100407n |
| 34. | Saito, Y.; Suzuki, A.; Ishioroshi, S.; Saito, I. Tetrahedron Lett. 2011, 52, 4726–4729. doi:10.1016/j.tetlet.2011.06.089 |
| 39. | Bucevicius, J.; Turks, M.; Tumkevicius, S. Synlett 2018, 29, 525–529. doi:10.1055/s-0036-1590942 |
| 33. | Li, J.-P.; Wang, H.-X.; Wang, H.-X.; Xie, M.-S.; Qu, G.-R.; Niu, H.-Y.; Guo, H.-M. Eur. J. Org. Chem. 2014, 2225–2230. doi:10.1002/ejoc.201301897 |
| 35. | Pratibha; Singh, S.; Sivakumar, S.; Verma, S. Eur. J. Inorg. Chem. 2017, 4202–4209. doi:10.1002/ejic.201700806 |
| 39. | Bucevicius, J.; Turks, M.; Tumkevicius, S. Synlett 2018, 29, 525–529. doi:10.1055/s-0036-1590942 |
| 25. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 50. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2013, 54, 6557–6561. doi:10.1016/j.tetlet.2013.09.095 |
| 51. | Novosjolova, I.; Bizdēna, Ē.; Turks, M. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 1236–1241. doi:10.1080/10426507.2014.989435 |
| 39. | Bucevicius, J.; Turks, M.; Tumkevicius, S. Synlett 2018, 29, 525–529. doi:10.1055/s-0036-1590942 |
| 49. | Pignatello, R.; Puleo, A.; Acquaviva, R.; Di Giacomo, C.; Vanella, A.; Puglisi, G. J. Drug Delivery Sci. Technol. 2004, 14, 181–186. doi:10.1016/s1773-2247(04)50098-9 |
| 42. | Lu, W.; Sengupta, S.; Petersen, J. L.; Akhmedov, N. G.; Shi, X. J. Org. Chem. 2007, 72, 5012–5015. doi:10.1021/jo070515+ |
| 43. | Zatloukal, M.; Jorda, R.; Gucký, T.; Řezníčková, E.; Voller, J.; Pospíšil, T.; Malínková, V.; Adamcová, H.; Kryštof, V.; Strnad, M. Eur. J. Med. Chem. 2013, 61, 61–72. doi:10.1016/j.ejmech.2012.06.036 |
| 44. | Gucký, T.; Jorda, R.; Zatloukal, M.; Bazgier, V.; Berka, K.; Řezníčková, E.; Béres, T.; Strnad, M.; Kryštof, V. J. Med. Chem. 2013, 56, 6234–6247. doi:10.1021/jm4006884 |
| 45. | Morales, F.; Ramírez, A.; Conejo-García, A.; Morata, C.; Marchal, J. A.; Campos, J. M. Eur. J. Med. Chem. 2014, 76, 118–124. doi:10.1016/j.ejmech.2014.02.012 |
| 46. | Calderón-Arancibia, J.; Espinosa-Bustos, C.; Cañete-Molina, Á.; Tapia, R. A.; Faúndez, M.; Torres, M. J.; Aguirre, A.; Paulino, M.; Salas, C. O. Molecules 2015, 20, 6808–6826. doi:10.3390/molecules20046808 |
| 47. | Malínková, V.; Řezníčková, E.; Jorda, R.; Gucký, T.; Kryštof, V. Bioorg. Med. Chem. 2017, 25, 6523–6535. doi:10.1016/j.bmc.2017.10.032 |
| 48. | Komodziński, K.; Lepczyńska, J.; Gdaniec, Z.; Bartolotti, L.; Delley, B.; Franzen, S.; Skalski, B. Photochem. Photobiol. Sci. 2014, 13, 563–573. doi:10.1039/c3pp50385b |
| 39. | Bucevicius, J.; Turks, M.; Tumkevicius, S. Synlett 2018, 29, 525–529. doi:10.1055/s-0036-1590942 |
| 25. | Kovaļovs, A.; Novosjolova, I.; Bizdēna, Ē.; Bižāne, I.; Skardziute, L.; Kazlauskas, K.; Jursenas, S.; Turks, M. Tetrahedron Lett. 2013, 54, 850–853. doi:10.1016/j.tetlet.2012.11.095 |
| 29. | Bucevicius, J.; Skardziute, L.; Dodonova, J.; Kazlauskas, K.; Bagdziunas, G.; Jursenas, S.; Tumkevicius, S. RSC Adv. 2015, 5, 38610–38622. doi:10.1039/c5ra05482f |
| 40. | Cīrule, D.; Ozols, K.; Platnieks, O.; Bizdēna, Ē.; Māliņa, I.; Turks, M. Tetrahedron 2016, 72, 4177–4185. doi:10.1016/j.tet.2016.05.043 |
| 41. | Ozols, K.; Cīrule, D.; Novosjolova, I.; Stepanovs, D.; Liepinsh, E.; Bizdēna, Ē.; Turks, M. Tetrahedron Lett. 2016, 57, 1174–1178. doi:10.1016/j.tetlet.2016.02.003 |
© 2019 Šišuļins et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
