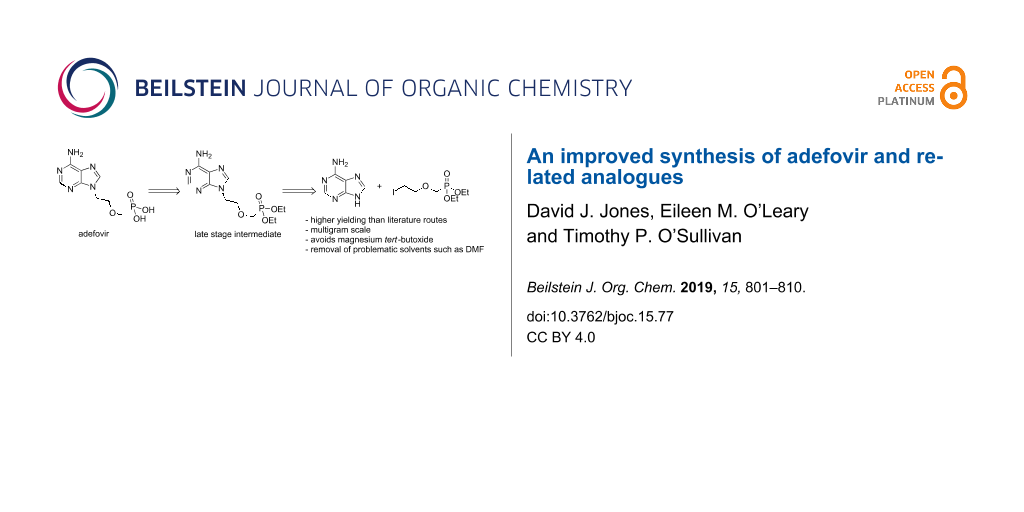
Abstract
An improved synthesis of the antiviral drug adefovir is presented. Problems associated with current routes to adefovir include capricious yields and a reliance on problematic reagents and solvents, such as magnesium tert-butoxide and DMF, to achieve high conversions to the target. A systematic study within our laboratory led to the identification of an iodide reagent which affords higher yields than previous approaches and allows for reactions to be conducted up to 10 g in scale under milder conditions. The use of a novel tetrabutylammonium salt of adenine facilitates alkylations in solvents other than DMF. Additionally, we have investigated how regioselectivity is affected by the substitution pattern of the nucleobase. Finally, this chemistry was successfully applied to the synthesis of several new adefovir analogues, highlighting the versatility of our approach.

Graphical Abstract
Introduction
The acyclic nucleoside phosphonate adefovir (1) [1], administered as its dipivoxil prodrug form (2) [2], is used clinically for the treatment of infections caused by the hepatitis B virus (HBV) [3-5] and the herpes simplex virus (HSV, Figure 1) [5,6]. While 1 possesses inhibitory activity against the human immunodeficiency virus (HIV) [1,5], it has not been approved by the FDA for the treatment of HIV as the dose required to elicit inhibition can cause nephrotoxicity [7].
![[1860-5397-15-77-1]](/bjoc/content/figures/1860-5397-15-77-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Adefovir (1) and its prodrug 2.
Figure 1: Adefovir (1) and its prodrug 2.
The mode of action of adefovir has been widely studied and involves the inhibition of viral replication by the termination of DNA synthesis [8-10]. Although adefovir was described in 1986 for the first time by Holý, De Clercq and co-workers, it is still actively employed in research today [11-13]. Adefovir is often used as a benchmark against which the relative activity of other antiretroviral drugs is measured [14-17]. In addition, the development of new prodrugs of current antiretrovirals remains an important field of study [18]. The efficacy of investigational prodrug strategies is typically measured by derivatising proven therapeutics such as adefovir and comparing the properties of the resulting compounds to the parent substrate [15,19-30]. Additionally, the solid state and spectroscopic properties of adefovir have been the subject of several recent studies [31-34].
It is clear, therefore, that there is still demand for a robust synthetic route to adefovir. The most widely employed method for accessing adefovir is shown in Scheme 1 [17,35,36]. Alkylation of commercially available adenine (3) to form alcohol 4 and further base-mediated alkylation with tosylate 5 affords phosphonate ester intermediate 6. Subsequent dealkylation of 6 using trimethylsilyl bromide (TMSBr) gives adefovir (1). The related analogue tenofovir, developed as an anti-HIV agent, may be prepared in a similar manner [37,38].
![[1860-5397-15-77-i1]](/bjoc/content/inline/1860-5397-15-77-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The poor solubility of adenine and its derivatives in most organic solvents restricts the choice of solvent for this and subsequent reactions to polar aprotic solvents such as DMF, NMP and DMSO [38].
The choice of base for the alkylation of alcohol 4 with tosylate 5 has been the subject of recent studies [35,37-39]. While it was demonstrated that magnesium tert-butoxide (MTB) is the optimum base for this transformation (Scheme 1), it is not without drawbacks. MTB is expensive and hydrolyses on storage when exposed to moisture. The yield for the reaction is often inconsistent, being highly dependent on the quality of the MTB employed. The reaction does not proceed to completion when a stoichiometric amount of the base is used and up to 3.0 equivalents may be required in order to ensure complete consumption of the starting alcohol. The resultant magnesium salts are highly deliquescent and form a sticky resin on exposure to atmospheric moisture. This resin hinders the isolation of phosphonate 6 and stymies scale-up. Finally, several byproducts, including the ethyl ether of 4 and the hemi-dealkylated ester of 6, are produced during this reaction, further reducing the atom economy of this approach [35,38]. In an attempt to overcome these difficulties, some groups have explored telescoping the MTB-mediated alkylation of 4 with the subsequent dealkylation of 6 [38,40]. However, as trimethylsilyl bromide (TMSBr) is moisture sensitive, telescoping the two reactions is difficult due to the hygroscopic nature of both the polar aprotic solvents employed and MTB itself. An improved synthetic route to adefovir (1), which avoids at least some of these difficulties and especially the use of MTB, would be highly desirable.
Results and Discussion
We initially investigated the use of phosphonate reagents bearing alternative leaving groups in an effort to improve the in situ conversion from 4 to 6, thereby improving the yield of this reaction. We selected iodide 7 and triflate 8 for this study and compared the conversion of 4 to 6 by 1H NMR (Table 1, entries 1–3). Unfortunately, we found that tosylate 5 remained the superior alkylating agent under these conditions. The reaction of 4 with iodide 7 afforded only a trace amount of the phosphonate, with mostly unreacted starting material evident in the 1H and 31P NMR spectra of the crude reaction mixture. The reaction with triflate 8 resulted in a significantly lower conversion to 6 than when tosylate 5 was employed. Optimisation of other aspects of this reaction (e.g., choice of base, solvent, temperature and the role of additives) has already been investigated by Brown Ripin [37,40] and Riley [38] which suggested that further investigation had limited potential.
Table 1: Impact of the leaving group on in situ conversion of 4 to 6.
![[Graphic 1]](/bjoc/content/inline/1860-5397-15-77-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | Electrophilea | Leaving group (X) | Conversionb | Yieldc |
| 1 | 5 | OTs | 85% | 43% |
| 2 | 7 | I | 56% | 24% |
| 3 | 8 | OTf | trace | – |
aReactions were carried out on a 3 mmol scale, 0.1 M concentration using 1.50 equiv of the electrophile. bMeasured by analysis of the 1H NMR spectrum of the crude reaction mixture after removal of the solvent in vacuo, comparing the NCH2 signals of 4 and 6 at 4.19 ppm and 4.33 ppm, respectively. cYield following purification by column chromatography.
Given the issues encountered in employing MTB, we next investigated the introduction of the phosphonate ester as the nucleophile rather than as the electrophile (Figure 2).
![[1860-5397-15-77-2]](/bjoc/content/figures/1860-5397-15-77-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Retrosynthetic analysis of 6 to synthons 9 and 10.
Figure 2: Retrosynthetic analysis of 6 to synthons 9 and 10.
Commercially available alcohol 10 was prepared by adapting a previously reported literature procedure where diethyl phosphonate (11) was condensed with paraformaldehyde in the presence of catalytic triethylamine (Scheme 2) [41]. Iodide 9 was accessed in two steps from adenine (3). The highly regioselective alkylation of adenine (3) with an excess (4.0 equiv) of 1,2-dibromoethane furnished bromide 12 in 78% yield. Despite the poor solubility of 12 in acetonitrile, the compound was successfully converted to iodide 9 via a Finkelstein reaction. Unfortunately, subsequent alkylation of 10 with iodide 9 in the presence of sodium hydride furnished 6 in a poor yield of 21%. Formation of self-alkylation product 13 constituted the major reaction pathway when the reaction was stirred for prolonged periods of time or heated to higher temperatures. Consequently, the overall yield of this process, at 14%, is lower than that of the process outlined in Scheme 1.
![[1860-5397-15-77-i2]](/bjoc/content/inline/1860-5397-15-77-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Forward synthesis of 6 from 9 and 10.
Scheme 2: Forward synthesis of 6 from 9 and 10.
Given the ease of alkylation of 3 with 1,2-dibromoethane, we wondered if alkylation of adenine with advanced intermediate 14 might represent a facile route to 6 (Figure 3).
![[1860-5397-15-77-3]](/bjoc/content/figures/1860-5397-15-77-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Retrosynthesis of 6 to synthons 14 and 3.
Figure 3: Retrosynthesis of 6 to synthons 14 and 3.
Holý and co-workers have previously utilised a similar alkyl chloride reagent to access adefovir analogues, but found that recourse to an isopropyl ester intermediate was necessary as the corresponding ethyl ester was prone to unwanted dealkylation under their harsh reaction conditions [13]. We reasoned that iodide 14 would facilitate alkylation under much milder conditions, and that the ethyl esters would, therefore, remain intact. Other comparable alkylating agents have also been employed in the synthesis of acyclic nucleoside phosphonates including mesylates [42], tosylates [16,43,44] and alkyl chlorides [45-51]. Alkylation reactions conducted with these electrophiles generally require higher temperatures. Furthermore, these reagents typically afford products in low to moderate yields as the reactions fail to reach completion or else furnish multiple side-products. The successful application of such iodide-based electrophiles is precedented, as demonstrated by the work of Ubasawa et al. in their preparation of purine analogue 17 from 15 (Scheme 3) [52]. A similar approach is outlined in a 1998 patent by the same group [53].
![[1860-5397-15-77-i3]](/bjoc/content/inline/1860-5397-15-77-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Application of related alkyl iodide 15 [52].
Scheme 3: Application of related alkyl iodide 15 [52].
Iodide 14 was prepared over two steps from commercially available 2-chloroethyl chloromethyl ether (18, Scheme 4). Firstly, 18 was subjected to an Arbuzov rearrangement to furnish phosphonate 19 in quantitative yield. A subsequent Finkelstein reaction afforded iodide 14. During the optimisation of this transformation, we observed that 2.0 equivalents of sodium iodide were required in order to achieve complete consumption of 19. Iodide 14 was stored under a nitrogen atmosphere over copper wire in an amber container and found to be stable for over one year. Gratifyingly, alkylation of adenine with iodide 14 proceeded smoothly under mild conditions to furnish phosphonate 6 in 70% yield. The overall yield of 6 from 18 was 58% which compared very favourably with that reported in Scheme 1. Interestingly, the unnatural N7-regioisomer 20 was also isolated in 16% yield, which was readily separable from 6 by column chromatography. The synthesis of 20 has been reported only once before and the resulting phosphonic acid also possesses antiviral activity [54]. The authors report that 20 was prepared in four steps while our three step route affords 20 in a comparable overall yield of 13%.
![[1860-5397-15-77-i4]](/bjoc/content/inline/1860-5397-15-77-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 6 and 20 via iodide 14.
Scheme 4: Synthesis of 6 and 20 via iodide 14.
We conducted a temperature, solvent and base screen to determine what impact these parameters have on the regioselectivity of the alkylation (Table 2). The overall regioselectivity appeared insensitive to the reaction conditions with only a slight decrease in selectivity for 6 observed when hydroxylic bases were employed in the reaction (Table 2, entries 1 and 2). Temperature was observed to have no effect on the regioselectivity (Table 2, entries 7, 9 and 10). Similarly, no improvement was observed when other aprotic solvents were used in place of DMF (Table 2, entries 8, 12, 13, 15 and 16). The same regioselectivity was observed when the reaction was performed in either NMP or DMF, even though adenine (3) was more soluble in NMP. No reaction was observed in acetonitrile, acetone or ethyl acetate, presumably due to the insolubility of adenine (3) at room temperature. When wet DMF was employed (Table 2, entry 11), no change in the regioselectivity occurred, although the reaction was found to proceed more slowly, having not reached completion after 30 hours. Conducting the reaction in ethanol (Table 2, entry 17) resulted in degradation of the iodide starting material and the formation of only trace amounts of product. No reaction was observed in cyrene (Table 2, entry 18), often employed as a green alternative to DMF [55].
Table 2: Impact of base, solvent and temperature on reaction performance and regioselectivity.
![[Graphic 2]](/bjoc/content/inline/1860-5397-15-77-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Solvent | Temperature (°C) | N9:N7a | Conversionb |
| 1 | NaOH | DMF | rt | 3:1 | 100% |
| 2 | KOH | DMF | rt | 3:1 | 100% |
| 3 | NEt3 | DMF | rt | –c | trace |
| 4 | DBU | DMF | rt | 4:1d | 33% |
| 5 | DMAP | DMF | rt | –c | N.P. |
| 6 | Cs2CO3 | DMF | rt | 4:1 | 100% |
| 7 | K2CO3 | DMF | rt | 4:1 | 100% |
| 8 | K2CO3 | NMP | rt | 4:1 | 100% |
| 9 | K2CO3 | DMF | 0 | 4:1 | 100% |
| 10 | K2CO3 | DMF | 50 | 4:1 | 100% |
| 11 | K2CO3 | water:DMF (30% v/v) | rt | 4:1 | 59% |
| 12 | K2CO3 | MeCN | rt | –c | N.P. |
| 13 | DBU | MeCN | rt | –c | N.P. |
| 14 | NaH | DMF | rt | 4:1 | 100% |
| 15 | K2CO3 | EtOAc | rt | –c | N.P. |
| 16 | K2CO3 | acetone | rt | –c | N.P. |
| 17 | K2CO3 | EtOH | rt | –e | trace |
| 18 | K2CO3 | cyrene | rt | –c | N.P. |
aDetermined by integration of the NCH2 signals from 6 and 20 at 4.33 ppm and 4.50 ppm, respectively, in the 1H NMR spectra of the crude reaction mixture after removal of the solvent in vacuo. bDetermined by integration of the C(8)H in 3, 6 and 20 in the 1H NMR spectra of the crude reaction mixture. cNo reaction observed, only starting material evident in the 1H NMR spectra of the crude reaction mixtures. dReaction did not reach completion. eDegradation of iodide 14 evident in 1H NMR and 31P NMR spectra of crude reaction mixture. Only traces of product formed. N.P. = no product formed.
In a bid to replace DMF as the solvent, we turned our attention to more soluble adenine analogues and investigated the formation of 21, a novel tetrabutylammonium salt of adenine (Scheme 5). Preparation of 21 was facile and the salt was isolated quantitatively as a colourless powder following work-up. Gratifyingly, the subsequent alkylation with iodide 14 in acetonitrile proceeded in comparable yields to those in DMF, constituting an attractive alternative to current literature methods for accessing 6.
![[1860-5397-15-77-i5]](/bjoc/content/inline/1860-5397-15-77-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of phosphonate 6 using novel salt 21.
Scheme 5: Synthesis of phosphonate 6 using novel salt 21.
To explore the utility of iodide 14 in the synthesis of novel antivirals, we examined its reactivity towards other 6-substituted purine nucleobase analogues (Scheme 6). Alkylation of both 6-chloropurine (22) and N6-benzyladenine (25) afforded the corresponding N9-regioisomers as the major product as determined by 1H NMR spectroscopy (Scheme 6a,b). The major isomers were then isolated and subsequently converted to phosphonate 6 in order to confirm the degree of regioselectivity. Interestingly, the amidine moiety in 28 favoured alkylation at N7 and 29 was isolated in 79% yield following purification by column chromatography (Scheme 6c).
![[1860-5397-15-77-i6]](/bjoc/content/inline/1860-5397-15-77-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Application of iodide 14 in the synthesis of adefovir analogues.
Scheme 6: Application of iodide 14 in the synthesis of adefovir analogues.
Pal and co-workers previously reported the N7-selective alkylation of 28 using other electrophiles [56]. They confirmed that the reaction had occurred at N7 via HMBC analysis and the identification of a correlation between the methylene protons adjacent to the purine ring with C-5. A similar analysis by us revealed the same correlation, confirming 29 as the major product (Figure 4). While the amidine is itself an important moiety in medicinal chemistry [57], it can also be readily converted to other functional groups including nitrogen-containing heterocycles [58]. Consequently, the use of the amidine moiety to direct the N7-selective alkylation with iodide 14 may likewise facilitate the preparation of other N7-functionalised adefovir analogues for evaluation.
![[1860-5397-15-77-4]](/bjoc/content/figures/1860-5397-15-77-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: HMBC spectrum confirms N7-selectivity for the major product 29.
Figure 4: HMBC spectrum confirms N7-selectivity for the major product 29.
Finally, we investigated whether a similar strategy could be exploited to access adefovir dipivoxil (2) directly. Although we were able to successfully prepare novel phosphonate 33 via phosphonic acid 31, subsequent attempts at converting 33 to the corresponding iodide 34, or alkylation of salt 21 were unsuccessful and hence we were unable to access 2 (Scheme 7). Instead, the major product isolated in both cases was novel heterocycle 35. It is likely that cleavage of one of the pivaloxymethyl groups, followed by intramolecular cyclisation, results in the formation of 35. Microwave heating of 33 in the presence of DBU also furnishes cyclic phosphonate 35 in excellent yields.
![[1860-5397-15-77-i7]](/bjoc/content/inline/1860-5397-15-77-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Attempted synthesis of adefovir dipivoxil (2) exploiting phosphonate 33.
Scheme 7: Attempted synthesis of adefovir dipivoxil (2) exploiting phosphonate 33.
Conclusion
We have described a synthetic route to phosphonate intermediate 6 which has a number of distinct advantages when compared to previous approaches. While the standard approach to adefovir relies heavily on the use of magnesium tert-butoxide, an expensive, troublesome and capricious reagent, our improved synthesis of adefovir avoids this. As adenine is introduced later in our synthetic sequence, fewer steps are carried out in hazardous, environmentally harmful solvents such as DMF or NMP. Furthermore, the introduction of a more reactive electrophile in 14 means that the critical alkylation step is conducted at room temperature. Additionally, the preparation of chloride 19 is a solventless reaction and the subsequent conversion of 19 to iodide 14 takes place in acetone, a green solvent. Our route also produces fewer byproducts and is higher yielding than the standard synthesis of adefovir, making it a highly attractive alternative for those interested in the study of this molecule and its analogues, particularly on a laboratory scale. In addition, through the use of salt 21, the preparation of 6 can be achieved without the use of DMF as a solvent. Furthermore, this approach gives synthetically useful quantities of N7-substituted analogue 20, via a more concise route than the current literature procedure. The preparation of a number of novel adefovir analogues using iodide 14 highlights the utility of this reagent. Finally, strategic incorporation of an amidine moiety allows for regioselective alkylations with 14 and facilitates the synthesis of novel N7-substituted adefovir analogues.
Supporting Information
| Supporting Information File 1: Experimental part and NMR spectra. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Holý, A.; Votruba, I.; Merta, A.; Černý, J.; Veselý, J.; Vlach, J.; Šedivá, K.; Rosenberg, I.; Otmar, M.; Hřebabecký, H.; Trávníĉek, M.; Vonka, V.; Snoeck, R.; De Clercq, E. Antiviral Res. 1990, 13, 295–311. doi:10.1016/0166-3542(90)90014-x
Return to citation in text: [1] [2] -
Starrett, J. E., Jr.; Tortolani, D. R.; Hitchcock, M. J. M.; Martin, J. C.; Mansuri, M. M. Antiviral Res. 1992, 19, 267–273. doi:10.1016/0166-3542(92)90084-i
Return to citation in text: [1] -
Yuen, M.-F.; Lai, C.-L. Expert Opin. Pharmacother. 2004, 5, 2361–2367. doi:10.1517/14656566.5.11.2361
Return to citation in text: [1] -
Dando, T. M.; Plosker, G. L. Drugs 2003, 63, 2215–2234. doi:10.2165/00003495-200363200-00007
Return to citation in text: [1] -
Balzarini, J. Adv. Exp. Med. Biol. 1994, 370, 459–464.
Return to citation in text: [1] [2] [3] -
De Clercq, E. Med. Res. Rev. 2015, 35, 698–719. doi:10.1002/med.21340
Return to citation in text: [1] -
Law, S.-t.; Li, K. K.; Ho, Y. Y. J. Clin. Pharm. Ther. 2012, 37, 128–131. doi:10.1111/j.1365-2710.2011.01278.x
Return to citation in text: [1] -
Naesens, L.; Balzarini, J.; Bischofberger, N.; De Clercq, E. Antimicrob. Agents Chemother. 1996, 40, 22–28. doi:10.1128/aac.40.1.22
Return to citation in text: [1] -
Rossi, L.; Dominici, S.; Serafini, S.; Casabianca, A.; Cerasi, A.; Chiarantini, L.; Celeste, A. G.; Cappellacci, L.; Franchetti, P.; Grifantini, M.; Magnani, M. J. Antimicrob. Chemother. 2002, 50, 365–374. doi:10.1093/jac/dkf125
Return to citation in text: [1] -
Neyts, J.; De Clercq, E. Biochem. Pharmacol. 1994, 47, 39–41. doi:10.1016/0006-2952(94)90435-9
Return to citation in text: [1] -
De Clercq, E.; Holý, A.; Rosenberg, I.; Sakuma, T.; Balzarini, J.; Maudgal, P. C. Nature 1986, 323, 464–467. doi:10.1038/323464a0
Return to citation in text: [1] -
De Clercq, E.; Sakuma, T.; Baba, M.; Pauwels, R.; Balzarini, J.; Rosenberg, I.; Holý, A. Antiviral Res. 1987, 8, 261–272. doi:10.1016/s0166-3542(87)80004-9
Return to citation in text: [1] -
Holý, A.; Günter, J.; Dvořáková, H.; Masojídková, M.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E. J. Med. Chem. 1999, 42, 2064–2086. doi:10.1021/jm9811256
Return to citation in text: [1] [2] -
De Clercq, E.; Holý, A. Nat. Rev. Drug Discovery 2005, 4, 928–940. doi:10.1038/nrd1877
Return to citation in text: [1] -
Meier, C.; Görbig, U.; Müller, C.; Balzarini, J. J. Med. Chem. 2005, 48, 8079–8086. doi:10.1021/jm050641a
Return to citation in text: [1] [2] -
Hocková, D.; Holý, A.; Masojídková, M.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. J. Med. Chem. 2003, 46, 5064–5073. doi:10.1021/jm030932o
Return to citation in text: [1] [2] -
Barral, K.; Priet, S.; Sire, J.; Neyts, J.; Balzarini, J.; Canard, B.; Alvarez, K. J. Med. Chem. 2006, 49, 7799–7806. doi:10.1021/jm060030y
Return to citation in text: [1] [2] -
Pradere, U.; Garnier-Amblard, E. C.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2014, 114, 9154–9218. doi:10.1021/cr5002035
Return to citation in text: [1] -
Tichý, T.; Andrei, G.; Dračínský, M.; Holý, A.; Balzarini, J.; Snoeck, R.; Krečmerová, M. Bioorg. Med. Chem. 2011, 19, 3527–3539. doi:10.1016/j.bmc.2011.04.016
Return to citation in text: [1] -
Mehellou, Y.; Rattan, H. S.; Balzarini, J. J. Med. Chem. 2018, 61, 2211–2226. doi:10.1021/acs.jmedchem.7b00734
Return to citation in text: [1] -
Holý, A.; Dvořáková, H.; Jindřich, J.; Masojídková, M.; Buděšínský, M.; Balzarini, J.; Andrei, G.; De Clercq, E. J. Med. Chem. 1996, 39, 4073–4088. doi:10.1021/jm960314q
Return to citation in text: [1] -
Tang, Y.-b.; Peng, Z.-g.; Liu, Z.-y.; Li, Y.-p.; Jiang, J.-d.; Li, Z.-r. Bioorg. Med. Chem. Lett. 2007, 17, 6350–6353. doi:10.1016/j.bmcl.2007.08.065
Return to citation in text: [1] -
Benzaria, S.; Pélicano, H.; Johnson, R.; Maury, G.; Imbach, J.-L.; Aubertin, A.-M.; Obert, G.; Gosselin, G. J. Med. Chem. 1996, 39, 4958–4965. doi:10.1021/jm960289o
Return to citation in text: [1] -
Starrett, J. E., Jr.; Tortolani, D. R.; Russell, J.; Hitchcock, M. J.; Whiterock, V.; Martin, J. C.; Mansuri, M. M. J. Med. Chem. 1994, 37, 1857–1864. doi:10.1021/jm00038a015
Return to citation in text: [1] -
Fu, X.; Jiang, S.; Li, C.; Xin, J.; Yang, Y.; Ji, R. Bioorg. Med. Chem. Lett. 2007, 17, 465–470. doi:10.1016/j.bmcl.2006.10.021
Return to citation in text: [1] -
Erion, M. D.; Reddy, K. R.; Boyer, S. H.; Matelich, M. C.; Gomez-Galeno, J.; Lemus, R. H.; Ugarkar, B. G.; Colby, T. J.; Schanzer, J.; van Poelje, P. D. J. Am. Chem. Soc. 2004, 126, 5154–5163. doi:10.1021/ja031818y
Return to citation in text: [1] -
Ballatore, C.; McGuigan, C.; De Clercq, E.; Balzarini, J. Bioorg. Med. Chem. Lett. 2001, 11, 1053–1056. doi:10.1016/s0960-894x(01)00128-7
Return to citation in text: [1] -
Adelfinskaya, O.; Terrazas, M.; Froeyen, M.; Marliere, P.; Nauwelaerts, K.; Herdewijn, P. Nucleic Acids Res. 2007, 35, 5060–5072. doi:10.1093/nar/gkm498
Return to citation in text: [1] -
Serafinowska, H. T.; Ashton, R. J.; Bailey, S.; Harnden, M. R.; Jackson, S. M.; Sutton, D. J. Med. Chem. 1995, 38, 1372–1379. doi:10.1021/jm00008a015
Return to citation in text: [1] -
Dang, Q.; Kasibhatla, S. R.; Jiang, T.; Fan, K.; Liu, Y.; Taplin, F.; Schulz, W.; Cashion, D. K.; Reddy, K. R.; van Poelje, P. D.; Fujitaki, J. M.; Potter, S. C.; Erion, M. D. J. Med. Chem. 2008, 51, 4331–4339. doi:10.1021/jm8001235
Return to citation in text: [1] -
An, J.-H.; Jin, F.; Kim, H. S.; Ryu, H. C.; Kim, J. S.; Kim, H. M.; Kiyonga, A. N.; Min, D. S.; Youn, W.; Kim, K. H.; Jung, K. Arch. Pharmacal Res. 2016, 39, 646–659. doi:10.1007/s12272-016-0721-0
Return to citation in text: [1] -
Hou, G.; Yin, Q.; Yang, Y.; Hu, Y.; Zhang, M.; Wang, J. J. Chem. Eng. Data 2008, 53, 1021–1023. doi:10.1021/je700664g
Return to citation in text: [1] -
Prohens, R.; Barbas, R.; Portell, A.; Font-Bardia, M.; Alcobé, X.; Puigjaner, C. Cryst. Growth Des. 2015, 15, 475–484. doi:10.1021/cg501542h
Return to citation in text: [1] -
Yim, J.-H.; Kim, W.-S.; Lim, J. S. Ind. Eng. Chem. Res. 2014, 53, 1663–1671. doi:10.1021/ie402543d
Return to citation in text: [1] -
Roux, L.; Priet, S.; Payrot, N.; Weck, C.; Fournier, M.; Zoulim, F.; Balzarini, J.; Canard, B.; Alvarez, K. Eur. J. Med. Chem. 2013, 63, 869–881. doi:10.1016/j.ejmech.2013.02.039
Return to citation in text: [1] [2] [3] -
Schultze, L. M.; Chapman, H. H.; Dubree, N. J. P.; Jones, R. J.; Kent, K. M.; Lee, T. T.; Louie, M. S.; Postich, M. J.; Prisbe, E. J.; Rohloff, J. C.; Yu, R. H. Tetrahedron Lett. 1998, 39, 1853–1856. doi:10.1016/s0040-4039(98)00131-2
Return to citation in text: [1] -
Brown Ripin, D. H.; Teager, D. S.; Fortunak, J.; Basha, S. M.; Bivins, N.; Boddy, C. N.; Byrn, S.; Catlin, K. K.; Houghton, S. R.; Jagadeesh, S. T.; Kumar, K. A.; Melton, J.; Muneer, S.; Rao, L. N.; Rao, R. V.; Ray, P. C.; Reddy, N. G.; Reddy, R. M.; Shekar, K. C.; Silverton, T.; Smith, D. T.; Stringham, R. W.; Subbaraju, G. V.; Talley, F.; Williams, A. Org. Process Res. Dev. 2010, 14, 1194–1201. doi:10.1021/op1001337
Return to citation in text: [1] [2] [3] -
Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Yu, R. H.; Schultze, L. M.; Rohloff, J. C.; Dudzinski, P. W.; Kelly, D. E. Org. Process Res. Dev. 1999, 3, 53–55. doi:10.1021/op980067v
Return to citation in text: [1] -
Houghton, S. R.; Melton, J.; Fortunak, J.; Brown Ripin, D. H.; Boddy, C. N. Tetrahedron 2010, 66, 8137–8144. doi:10.1016/j.tet.2010.08.037
Return to citation in text: [1] [2] -
Phillion, D. P.; Andrew, S. S. Tetrahedron Lett. 1986, 27, 1477–1480. doi:10.1016/s0040-4039(00)84289-6
Return to citation in text: [1] -
Choi, J.-R.; Cho, D.-G.; Roh, K. Y.; Hwang, J.-T.; Ahn, S.; Jang, H. S.; Cho, W.-Y.; Kim, K. W.; Cho, Y.-G.; Kim, J.; Kim, Y.-Z. J. Med. Chem. 2004, 47, 2864–2869. doi:10.1021/jm0305265
Return to citation in text: [1] -
Pomeisl, K.; Beier, P.; Pohl, R.; Krečmerová, M. ChemistrySelect 2016, 1, 2102–2106. doi:10.1002/slct.201600445
Return to citation in text: [1] -
Baszczyňski, O.; Jansa, P.; Dračínský, M.; Klepetářová, B.; Holý, A.; Votruba, I.; Clercq, E. d.; Balzarini, J.; Janeba, Z. Bioorg. Med. Chem. 2011, 19, 2114–2124. doi:10.1016/j.bmc.2011.02.050
Return to citation in text: [1] -
Česnek, M.; Holý, A.; Masojídková, M. Tetrahedron 2002, 58, 2985–2996. doi:10.1016/s0040-4020(02)00186-2
Return to citation in text: [1] -
Krečmerová, M.; Dračínský, M.; Snoeck, R.; Balzarini, J.; Pomeisl, K.; Andrei, G. Bioorg. Med. Chem. 2017, 25, 4637–4648. doi:10.1016/j.bmc.2017.06.046
Return to citation in text: [1] -
Fu, X.-Z.; Jiang, F.-J.; Ou, Y.; Fu, S.; Cha, Y.-F.; Zhang, S.; Liu, Z.-Y.; Zhou, W.; Wang, A.-M.; Wang, Y.-L. Chin. Chem. Lett. 2014, 25, 115–118. doi:10.1016/j.cclet.2013.09.013
Return to citation in text: [1] -
Doláková, P.; Dračínský, M.; Masojídková, M.; Šolínová, V.; Kašička, V.; Holý, A. Eur. J. Med. Chem. 2009, 44, 2408–2424. doi:10.1016/j.ejmech.2008.09.031
Return to citation in text: [1] -
Semaine, W.; Johar, M.; Tyrrell, D. L. J.; Kumar, R.; Agrawal, B. J. Med. Chem. 2006, 49, 2049–2054. doi:10.1021/jm058271d
Return to citation in text: [1] -
Valiaeva, N.; Beadle, J. R.; Aldern, K. A.; Trahan, J.; Hostetler, K. Y. Antiviral Res. 2006, 72, 10–19. doi:10.1016/j.antiviral.2006.03.007
Return to citation in text: [1] -
Pomeisl, K.; Holý, A.; Pohl, R. Tetrahedron Lett. 2007, 48, 3065–3067. doi:10.1016/j.tetlet.2007.02.107
Return to citation in text: [1] -
Sekiya, K.; Takashima, H.; Ueda, N.; Kamiya, N.; Yuasa, S.; Fujimura, Y.; Ubasawa, M. J. Med. Chem. 2002, 45, 3138–3142. doi:10.1021/jm020036x
Return to citation in text: [1] [2] -
Ubasawa, M.; Sekiya, K.; Takashima, H.; Ueda, N.; Yuasa, S.; Kamiya, N. Phosphonate nucleotide compounds. WO Patent WO98006726A1, Feb 13, 1998.
Return to citation in text: [1] -
Hakimelahi, G. H.; Ly, T. W.; Moosavi-Movahedi, A. A.; Jain, M. L.; Zakerinia, M.; Davari, H.; Mei, H.-C.; Sambaiah, T.; Moshfegh, A. A.; Hakimelahi, S. J. Med. Chem. 2001, 44, 3710–3720. doi:10.1021/jm010216r
Return to citation in text: [1] -
Sherwood, J.; De bruyn, M.; Constantinou, A.; Moity, L.; McElroy, C. R.; Farmer, T. J.; Duncan, T.; Raverty, W.; Hunt, A. J.; Clark, J. H. Chem. Commun. 2014, 50, 9650–9652. doi:10.1039/c4cc04133j
Return to citation in text: [1] -
Pal, A.; Salandria, K. J.; Arico, J. W.; Schlegel, M. K.; McLaughlin, L. W. Chem. Commun. 2013, 49, 2936–2938. doi:10.1039/c3cc37265k
Return to citation in text: [1] -
Greenhill, J. V.; Lue, P. 5 Amidines and Guanidines in Medicinal Chemistry. In Progress in Medicinal Chemistry; Ellis, G. P.; Luscombe, D. K., Eds.; Elsevier: Amsterdam, Netherlands, 1993; pp 203–326. doi:10.1016/s0079-6468(08)70378-3
Return to citation in text: [1] -
Boyd, G. V. Amidines and Imidates; John Wiley & Sons Ltd.: New York, 1991.
Return to citation in text: [1]
| 52. | Sekiya, K.; Takashima, H.; Ueda, N.; Kamiya, N.; Yuasa, S.; Fujimura, Y.; Ubasawa, M. J. Med. Chem. 2002, 45, 3138–3142. doi:10.1021/jm020036x |
| 54. | Hakimelahi, G. H.; Ly, T. W.; Moosavi-Movahedi, A. A.; Jain, M. L.; Zakerinia, M.; Davari, H.; Mei, H.-C.; Sambaiah, T.; Moshfegh, A. A.; Hakimelahi, S. J. Med. Chem. 2001, 44, 3710–3720. doi:10.1021/jm010216r |
| 55. | Sherwood, J.; De bruyn, M.; Constantinou, A.; Moity, L.; McElroy, C. R.; Farmer, T. J.; Duncan, T.; Raverty, W.; Hunt, A. J.; Clark, J. H. Chem. Commun. 2014, 50, 9650–9652. doi:10.1039/c4cc04133j |
| 1. | Holý, A.; Votruba, I.; Merta, A.; Černý, J.; Veselý, J.; Vlach, J.; Šedivá, K.; Rosenberg, I.; Otmar, M.; Hřebabecký, H.; Trávníĉek, M.; Vonka, V.; Snoeck, R.; De Clercq, E. Antiviral Res. 1990, 13, 295–311. doi:10.1016/0166-3542(90)90014-x |
| 1. | Holý, A.; Votruba, I.; Merta, A.; Černý, J.; Veselý, J.; Vlach, J.; Šedivá, K.; Rosenberg, I.; Otmar, M.; Hřebabecký, H.; Trávníĉek, M.; Vonka, V.; Snoeck, R.; De Clercq, E. Antiviral Res. 1990, 13, 295–311. doi:10.1016/0166-3542(90)90014-x |
| 5. | Balzarini, J. Adv. Exp. Med. Biol. 1994, 370, 459–464. |
| 38. | Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364 |
| 5. | Balzarini, J. Adv. Exp. Med. Biol. 1994, 370, 459–464. |
| 6. | De Clercq, E. Med. Res. Rev. 2015, 35, 698–719. doi:10.1002/med.21340 |
| 35. | Roux, L.; Priet, S.; Payrot, N.; Weck, C.; Fournier, M.; Zoulim, F.; Balzarini, J.; Canard, B.; Alvarez, K. Eur. J. Med. Chem. 2013, 63, 869–881. doi:10.1016/j.ejmech.2013.02.039 |
| 37. | Brown Ripin, D. H.; Teager, D. S.; Fortunak, J.; Basha, S. M.; Bivins, N.; Boddy, C. N.; Byrn, S.; Catlin, K. K.; Houghton, S. R.; Jagadeesh, S. T.; Kumar, K. A.; Melton, J.; Muneer, S.; Rao, L. N.; Rao, R. V.; Ray, P. C.; Reddy, N. G.; Reddy, R. M.; Shekar, K. C.; Silverton, T.; Smith, D. T.; Stringham, R. W.; Subbaraju, G. V.; Talley, F.; Williams, A. Org. Process Res. Dev. 2010, 14, 1194–1201. doi:10.1021/op1001337 |
| 38. | Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364 |
| 39. | Yu, R. H.; Schultze, L. M.; Rohloff, J. C.; Dudzinski, P. W.; Kelly, D. E. Org. Process Res. Dev. 1999, 3, 53–55. doi:10.1021/op980067v |
| 3. | Yuen, M.-F.; Lai, C.-L. Expert Opin. Pharmacother. 2004, 5, 2361–2367. doi:10.1517/14656566.5.11.2361 |
| 4. | Dando, T. M.; Plosker, G. L. Drugs 2003, 63, 2215–2234. doi:10.2165/00003495-200363200-00007 |
| 5. | Balzarini, J. Adv. Exp. Med. Biol. 1994, 370, 459–464. |
| 17. | Barral, K.; Priet, S.; Sire, J.; Neyts, J.; Balzarini, J.; Canard, B.; Alvarez, K. J. Med. Chem. 2006, 49, 7799–7806. doi:10.1021/jm060030y |
| 35. | Roux, L.; Priet, S.; Payrot, N.; Weck, C.; Fournier, M.; Zoulim, F.; Balzarini, J.; Canard, B.; Alvarez, K. Eur. J. Med. Chem. 2013, 63, 869–881. doi:10.1016/j.ejmech.2013.02.039 |
| 36. | Schultze, L. M.; Chapman, H. H.; Dubree, N. J. P.; Jones, R. J.; Kent, K. M.; Lee, T. T.; Louie, M. S.; Postich, M. J.; Prisbe, E. J.; Rohloff, J. C.; Yu, R. H. Tetrahedron Lett. 1998, 39, 1853–1856. doi:10.1016/s0040-4039(98)00131-2 |
| 2. | Starrett, J. E., Jr.; Tortolani, D. R.; Hitchcock, M. J. M.; Martin, J. C.; Mansuri, M. M. Antiviral Res. 1992, 19, 267–273. doi:10.1016/0166-3542(92)90084-i |
| 37. | Brown Ripin, D. H.; Teager, D. S.; Fortunak, J.; Basha, S. M.; Bivins, N.; Boddy, C. N.; Byrn, S.; Catlin, K. K.; Houghton, S. R.; Jagadeesh, S. T.; Kumar, K. A.; Melton, J.; Muneer, S.; Rao, L. N.; Rao, R. V.; Ray, P. C.; Reddy, N. G.; Reddy, R. M.; Shekar, K. C.; Silverton, T.; Smith, D. T.; Stringham, R. W.; Subbaraju, G. V.; Talley, F.; Williams, A. Org. Process Res. Dev. 2010, 14, 1194–1201. doi:10.1021/op1001337 |
| 38. | Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364 |
| 14. | De Clercq, E.; Holý, A. Nat. Rev. Drug Discovery 2005, 4, 928–940. doi:10.1038/nrd1877 |
| 15. | Meier, C.; Görbig, U.; Müller, C.; Balzarini, J. J. Med. Chem. 2005, 48, 8079–8086. doi:10.1021/jm050641a |
| 16. | Hocková, D.; Holý, A.; Masojídková, M.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. J. Med. Chem. 2003, 46, 5064–5073. doi:10.1021/jm030932o |
| 17. | Barral, K.; Priet, S.; Sire, J.; Neyts, J.; Balzarini, J.; Canard, B.; Alvarez, K. J. Med. Chem. 2006, 49, 7799–7806. doi:10.1021/jm060030y |
| 15. | Meier, C.; Görbig, U.; Müller, C.; Balzarini, J. J. Med. Chem. 2005, 48, 8079–8086. doi:10.1021/jm050641a |
| 19. | Tichý, T.; Andrei, G.; Dračínský, M.; Holý, A.; Balzarini, J.; Snoeck, R.; Krečmerová, M. Bioorg. Med. Chem. 2011, 19, 3527–3539. doi:10.1016/j.bmc.2011.04.016 |
| 20. | Mehellou, Y.; Rattan, H. S.; Balzarini, J. J. Med. Chem. 2018, 61, 2211–2226. doi:10.1021/acs.jmedchem.7b00734 |
| 21. | Holý, A.; Dvořáková, H.; Jindřich, J.; Masojídková, M.; Buděšínský, M.; Balzarini, J.; Andrei, G.; De Clercq, E. J. Med. Chem. 1996, 39, 4073–4088. doi:10.1021/jm960314q |
| 22. | Tang, Y.-b.; Peng, Z.-g.; Liu, Z.-y.; Li, Y.-p.; Jiang, J.-d.; Li, Z.-r. Bioorg. Med. Chem. Lett. 2007, 17, 6350–6353. doi:10.1016/j.bmcl.2007.08.065 |
| 23. | Benzaria, S.; Pélicano, H.; Johnson, R.; Maury, G.; Imbach, J.-L.; Aubertin, A.-M.; Obert, G.; Gosselin, G. J. Med. Chem. 1996, 39, 4958–4965. doi:10.1021/jm960289o |
| 24. | Starrett, J. E., Jr.; Tortolani, D. R.; Russell, J.; Hitchcock, M. J.; Whiterock, V.; Martin, J. C.; Mansuri, M. M. J. Med. Chem. 1994, 37, 1857–1864. doi:10.1021/jm00038a015 |
| 25. | Fu, X.; Jiang, S.; Li, C.; Xin, J.; Yang, Y.; Ji, R. Bioorg. Med. Chem. Lett. 2007, 17, 465–470. doi:10.1016/j.bmcl.2006.10.021 |
| 26. | Erion, M. D.; Reddy, K. R.; Boyer, S. H.; Matelich, M. C.; Gomez-Galeno, J.; Lemus, R. H.; Ugarkar, B. G.; Colby, T. J.; Schanzer, J.; van Poelje, P. D. J. Am. Chem. Soc. 2004, 126, 5154–5163. doi:10.1021/ja031818y |
| 27. | Ballatore, C.; McGuigan, C.; De Clercq, E.; Balzarini, J. Bioorg. Med. Chem. Lett. 2001, 11, 1053–1056. doi:10.1016/s0960-894x(01)00128-7 |
| 28. | Adelfinskaya, O.; Terrazas, M.; Froeyen, M.; Marliere, P.; Nauwelaerts, K.; Herdewijn, P. Nucleic Acids Res. 2007, 35, 5060–5072. doi:10.1093/nar/gkm498 |
| 29. | Serafinowska, H. T.; Ashton, R. J.; Bailey, S.; Harnden, M. R.; Jackson, S. M.; Sutton, D. J. Med. Chem. 1995, 38, 1372–1379. doi:10.1021/jm00008a015 |
| 30. | Dang, Q.; Kasibhatla, S. R.; Jiang, T.; Fan, K.; Liu, Y.; Taplin, F.; Schulz, W.; Cashion, D. K.; Reddy, K. R.; van Poelje, P. D.; Fujitaki, J. M.; Potter, S. C.; Erion, M. D. J. Med. Chem. 2008, 51, 4331–4339. doi:10.1021/jm8001235 |
| 11. | De Clercq, E.; Holý, A.; Rosenberg, I.; Sakuma, T.; Balzarini, J.; Maudgal, P. C. Nature 1986, 323, 464–467. doi:10.1038/323464a0 |
| 12. | De Clercq, E.; Sakuma, T.; Baba, M.; Pauwels, R.; Balzarini, J.; Rosenberg, I.; Holý, A. Antiviral Res. 1987, 8, 261–272. doi:10.1016/s0166-3542(87)80004-9 |
| 13. | Holý, A.; Günter, J.; Dvořáková, H.; Masojídková, M.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E. J. Med. Chem. 1999, 42, 2064–2086. doi:10.1021/jm9811256 |
| 31. | An, J.-H.; Jin, F.; Kim, H. S.; Ryu, H. C.; Kim, J. S.; Kim, H. M.; Kiyonga, A. N.; Min, D. S.; Youn, W.; Kim, K. H.; Jung, K. Arch. Pharmacal Res. 2016, 39, 646–659. doi:10.1007/s12272-016-0721-0 |
| 32. | Hou, G.; Yin, Q.; Yang, Y.; Hu, Y.; Zhang, M.; Wang, J. J. Chem. Eng. Data 2008, 53, 1021–1023. doi:10.1021/je700664g |
| 33. | Prohens, R.; Barbas, R.; Portell, A.; Font-Bardia, M.; Alcobé, X.; Puigjaner, C. Cryst. Growth Des. 2015, 15, 475–484. doi:10.1021/cg501542h |
| 34. | Yim, J.-H.; Kim, W.-S.; Lim, J. S. Ind. Eng. Chem. Res. 2014, 53, 1663–1671. doi:10.1021/ie402543d |
| 8. | Naesens, L.; Balzarini, J.; Bischofberger, N.; De Clercq, E. Antimicrob. Agents Chemother. 1996, 40, 22–28. doi:10.1128/aac.40.1.22 |
| 9. | Rossi, L.; Dominici, S.; Serafini, S.; Casabianca, A.; Cerasi, A.; Chiarantini, L.; Celeste, A. G.; Cappellacci, L.; Franchetti, P.; Grifantini, M.; Magnani, M. J. Antimicrob. Chemother. 2002, 50, 365–374. doi:10.1093/jac/dkf125 |
| 10. | Neyts, J.; De Clercq, E. Biochem. Pharmacol. 1994, 47, 39–41. doi:10.1016/0006-2952(94)90435-9 |
| 56. | Pal, A.; Salandria, K. J.; Arico, J. W.; Schlegel, M. K.; McLaughlin, L. W. Chem. Commun. 2013, 49, 2936–2938. doi:10.1039/c3cc37265k |
| 7. | Law, S.-t.; Li, K. K.; Ho, Y. Y. J. Clin. Pharm. Ther. 2012, 37, 128–131. doi:10.1111/j.1365-2710.2011.01278.x |
| 18. | Pradere, U.; Garnier-Amblard, E. C.; Coats, S. J.; Amblard, F.; Schinazi, R. F. Chem. Rev. 2014, 114, 9154–9218. doi:10.1021/cr5002035 |
| 57. | Greenhill, J. V.; Lue, P. 5 Amidines and Guanidines in Medicinal Chemistry. In Progress in Medicinal Chemistry; Ellis, G. P.; Luscombe, D. K., Eds.; Elsevier: Amsterdam, Netherlands, 1993; pp 203–326. doi:10.1016/s0079-6468(08)70378-3 |
| 37. | Brown Ripin, D. H.; Teager, D. S.; Fortunak, J.; Basha, S. M.; Bivins, N.; Boddy, C. N.; Byrn, S.; Catlin, K. K.; Houghton, S. R.; Jagadeesh, S. T.; Kumar, K. A.; Melton, J.; Muneer, S.; Rao, L. N.; Rao, R. V.; Ray, P. C.; Reddy, N. G.; Reddy, R. M.; Shekar, K. C.; Silverton, T.; Smith, D. T.; Stringham, R. W.; Subbaraju, G. V.; Talley, F.; Williams, A. Org. Process Res. Dev. 2010, 14, 1194–1201. doi:10.1021/op1001337 |
| 40. | Houghton, S. R.; Melton, J.; Fortunak, J.; Brown Ripin, D. H.; Boddy, C. N. Tetrahedron 2010, 66, 8137–8144. doi:10.1016/j.tet.2010.08.037 |
| 35. | Roux, L.; Priet, S.; Payrot, N.; Weck, C.; Fournier, M.; Zoulim, F.; Balzarini, J.; Canard, B.; Alvarez, K. Eur. J. Med. Chem. 2013, 63, 869–881. doi:10.1016/j.ejmech.2013.02.039 |
| 38. | Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364 |
| 38. | Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364 |
| 40. | Houghton, S. R.; Melton, J.; Fortunak, J.; Brown Ripin, D. H.; Boddy, C. N. Tetrahedron 2010, 66, 8137–8144. doi:10.1016/j.tet.2010.08.037 |
| 52. | Sekiya, K.; Takashima, H.; Ueda, N.; Kamiya, N.; Yuasa, S.; Fujimura, Y.; Ubasawa, M. J. Med. Chem. 2002, 45, 3138–3142. doi:10.1021/jm020036x |
| 53. | Ubasawa, M.; Sekiya, K.; Takashima, H.; Ueda, N.; Yuasa, S.; Kamiya, N. Phosphonate nucleotide compounds. WO Patent WO98006726A1, Feb 13, 1998. |
| 16. | Hocková, D.; Holý, A.; Masojídková, M.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. J. Med. Chem. 2003, 46, 5064–5073. doi:10.1021/jm030932o |
| 43. | Pomeisl, K.; Beier, P.; Pohl, R.; Krečmerová, M. ChemistrySelect 2016, 1, 2102–2106. doi:10.1002/slct.201600445 |
| 44. | Baszczyňski, O.; Jansa, P.; Dračínský, M.; Klepetářová, B.; Holý, A.; Votruba, I.; Clercq, E. d.; Balzarini, J.; Janeba, Z. Bioorg. Med. Chem. 2011, 19, 2114–2124. doi:10.1016/j.bmc.2011.02.050 |
| 45. | Česnek, M.; Holý, A.; Masojídková, M. Tetrahedron 2002, 58, 2985–2996. doi:10.1016/s0040-4020(02)00186-2 |
| 46. | Krečmerová, M.; Dračínský, M.; Snoeck, R.; Balzarini, J.; Pomeisl, K.; Andrei, G. Bioorg. Med. Chem. 2017, 25, 4637–4648. doi:10.1016/j.bmc.2017.06.046 |
| 47. | Fu, X.-Z.; Jiang, F.-J.; Ou, Y.; Fu, S.; Cha, Y.-F.; Zhang, S.; Liu, Z.-Y.; Zhou, W.; Wang, A.-M.; Wang, Y.-L. Chin. Chem. Lett. 2014, 25, 115–118. doi:10.1016/j.cclet.2013.09.013 |
| 48. | Doláková, P.; Dračínský, M.; Masojídková, M.; Šolínová, V.; Kašička, V.; Holý, A. Eur. J. Med. Chem. 2009, 44, 2408–2424. doi:10.1016/j.ejmech.2008.09.031 |
| 49. | Semaine, W.; Johar, M.; Tyrrell, D. L. J.; Kumar, R.; Agrawal, B. J. Med. Chem. 2006, 49, 2049–2054. doi:10.1021/jm058271d |
| 50. | Valiaeva, N.; Beadle, J. R.; Aldern, K. A.; Trahan, J.; Hostetler, K. Y. Antiviral Res. 2006, 72, 10–19. doi:10.1016/j.antiviral.2006.03.007 |
| 51. | Pomeisl, K.; Holý, A.; Pohl, R. Tetrahedron Lett. 2007, 48, 3065–3067. doi:10.1016/j.tetlet.2007.02.107 |
| 13. | Holý, A.; Günter, J.; Dvořáková, H.; Masojídková, M.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E. J. Med. Chem. 1999, 42, 2064–2086. doi:10.1021/jm9811256 |
| 42. | Choi, J.-R.; Cho, D.-G.; Roh, K. Y.; Hwang, J.-T.; Ahn, S.; Jang, H. S.; Cho, W.-Y.; Kim, K. W.; Cho, Y.-G.; Kim, J.; Kim, Y.-Z. J. Med. Chem. 2004, 47, 2864–2869. doi:10.1021/jm0305265 |
| 38. | Riley, D. L.; Walwyn, D. R.; Edlin, C. D. Org. Process Res. Dev. 2016, 20, 742–750. doi:10.1021/acs.oprd.5b00364 |
| 41. | Phillion, D. P.; Andrew, S. S. Tetrahedron Lett. 1986, 27, 1477–1480. doi:10.1016/s0040-4039(00)84289-6 |
© 2019 Jones et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)