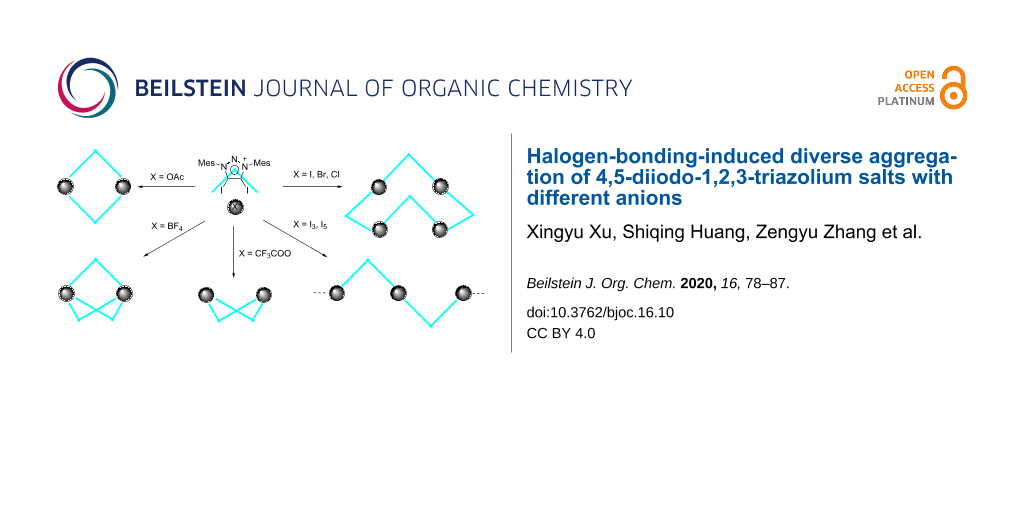
Abstract
The synthesis of 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium salts with different anions have been developed. These triazolium salts show diverse aggregation via halogen bonding between C–I bonds and anions. Triazolium with halide anions exists as a tetramer with saddle conformation. Triazolium tetrafluoroborate exists as a trimer with Chinese lantern shape conformation. Triazolium trifluoroacetate and acetate exist as dimers, respectively, while the former shows boat conformation and the latter forms rectangle conformation. Triazolium salts form a linear polymer with polyiodide.

Graphical Abstract
Introduction
The halogen bond (XB) is a noncovalent interaction between electrophilic halides and Lewis bases or electron-rich regions [1,2]. Computational studies [3-7] and cyrstal architectures including XB-donors (σ-hole) such as perfluorocarbons [8-12], tetraiodoethylene [13], 1,2-diiodo-1,2-difluoroethene [14], diiodoacetylene [15] and iodo/bromoethynyl moieties [16] have revealed that the XB-donors interacting with XB-acceptors (a nucleophilic region) are in approximately linear orientation. Besides, linearity, tunability and hydrophobicity (features of the XB ) are widely applied in crystal engineering, supramolecular chemistry, anion recognition, organocatalysis, materials science and tuning of biomolecular systems [17-27]. 1,2,3-Triazole-based XB-donors, such as 5-iodo-1,2,3-triazoles A [28-33] and 5-iodo-1,2,3-triazolium B [34-37] (Figure 1), are promising candidates for XB donors, which is mainly due to the ease of preparation via a copper-catalyzed click reaction between azide and alkyne [38,39]. 1,2,3-Triazoles and 1,2,3-triazolium-based XB activators have been found applications in catalytic reactions [40,41] and anion recognition [42]. Recently, we reported neutral 4-halo-1,2,3-triazolylidenes C [43], which had a carbene character with σ-donation at the carbon and a σ-hole at the halogen atom. XB is observed by single-crystal X-ray diffraction in their coinage metal complexes. Meanwhile, 4-bromo-1,2,3-triazolylidene can catalyze H/D exchange of aldehydes [44]. Despite a variety of XB donors based on 1,2,3-triazole have been reported, no 4,5-diido-1,2,3-triazolium salts have been reported for an XB interaction. Herein, we report the synthesis and characterization of 4,5-diido-1,2,3-triazolium D with different anions. The crystal structures of these compounds show XB interactions between the triazolium moiety and anions, and different aggregations are formed. Triazolium with halide anions exists as tetramers with saddle conformation. Triazolium tetrafluoroborate exists as trimer with Chinese lantern shape conformation. Triazolium trifluoroacetate and acetate exist as dimers, respectively, while the former shows a boat conformation and the latter forms a rectangle conformation. Triazolium salts form a linear polymer with polyiodide.
![[1860-5397-16-10-1]](/bjoc/content/figures/1860-5397-16-10-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1,2,3-Triazole based XB donors: 1,2,3-triazole A, 1,2,3-triazolium B, 1,2,3-triazolylidene C and diiodotriazolium D.
Figure 1: 1,2,3-Triazole based XB donors: 1,2,3-triazole A, 1,2,3-triazolium B, 1,2,3-triazolylidene C and di...
Results and Discussion
Recently, we found that 4-iodotriazolylidene can be prepared by the treatment of a 4,5-unsubstituted triazolium salt with one equivalent I2 in the presence of two equivalents of potassium tert-butoxide [43]. When 4,5-unsubstituted triazolium salt 1 was treated with two equivalents I2 and two equivalents of potassium tert-butoxide, 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium 2-I was synthesized in a good yield (Scheme 1). The product 2-I was characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry. 2-I has a poor solubility in most organic solvents such as dichloromethane, trichloromethane, tetrahydrofuran, and ethanol. A single crystal of 2-I was obtained by slow diffusion of ether into dimethylformamide solution.
![[1860-5397-16-10-i1]](/bjoc/content/inline/1860-5397-16-10-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium with iodide, Mes: 2,4,6-Me3C6H2.
Scheme 1: Synthesis of 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium with iodide, Mes: 2,4,6-Me3C6H2.
Ion exchange of 2-I with AgBF4 afforded 2-BF4. In contrast, 2-BF4 was soluble in dichloromethane and trichloromethane. A single crystal of 2-BF4 was obtained by slow diffusion of ether into a dichloromethane solution. In a similar manner, 2-OAc and 2-TFA were obtained via removing the iodide anion by AgOAc and CF3COOAg. A single crystal of 2-OAc suitable for X-ray diffraction analysis was obtained by slow diffusion of n-pentane into a dichloromethane solution. A single crystal of 2-TFA suitable for X-ray diffraction analysis was obtained by slow evaporation of dichloromethane solution (Scheme 2).
![[1860-5397-16-10-i2]](/bjoc/content/inline/1860-5397-16-10-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium with different anion.
Scheme 2: Synthesis of 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium with different anion.
2-Br and 2-Cl were synthesized by ion exchange between 2-BF4 and the respective potassium halide in acetonitrile. A single crystal of 2-Br was obtained by slow diffusion of n-pentane into a dichloromethane solution. While the single crystal of 2-Cl was obtained by slow evaporation of dichloromethane solution. The treatment of 2-I with 2 equivalents iodine or 4 equivalents iodine afforded triazolium polyiodide 2-I.1.5I2 and 2-I.3.5I2, respectively. The single crystals were obtained by slow diffusion of ether into a dichloromethane solution.
The crystal X-ray analyses of 2-I, 2-Br and 2-Cl show tetrameric aggregation of the 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium moiety with four anion halides that are bridged together to form a saddle shape through XB (Figure 2). 2-I crystallizes in the tetragonal space group ![[Graphic 1]](/bjoc/content/inline/1860-5397-16-10-i3.svg?max-width=637&scale=1.18182) . The distances of I···I are 3.306(1) Å and 3.300(1) Å which are due to the XB. The C–I···I angles are 176.9(3)° and 176.2(3)° (Table 1). The I···I distance is short: the reduction ratio RXB [45], defined as the ratio of the actual distance over the sum of van der Waals radii, amounts to 0.81. The 2-Br crystal package has a similar package diagram. 2-Br crystallizes in the tetragonal space group
. The distances of I···I are 3.306(1) Å and 3.300(1) Å which are due to the XB. The C–I···I angles are 176.9(3)° and 176.2(3)° (Table 1). The I···I distance is short: the reduction ratio RXB [45], defined as the ratio of the actual distance over the sum of van der Waals radii, amounts to 0.81. The 2-Br crystal package has a similar package diagram. 2-Br crystallizes in the tetragonal space group ![[Graphic 2]](/bjoc/content/inline/1860-5397-16-10-i4.svg?max-width=637&scale=1.18182) . The distances of I···Br are 3.107(2) Å, 3.123(2) Å, the C–I···Br angles are 177.1(4)°, 173.4(6)°, and the RXB values are 0.80. The crystal 2-Cl has a monoclinic crystal system and the space group is C2. The distances of I···Cl are 2.963(9) Å, 2.989(6) Å, 2.934(7) Å and 2.98(1) Å. The C–I···Cl angles are 176.8(6)°, 173.4(6)°, 176.2(6)° and 175.9(6)°. The RXB values are 0.77. These crystal package diagrams display that a bent arrangement of the XB donors are around the central halide anion. The measured bent angles of 2-I, 2-Br and 2-Cl by mercury [46] are 146.38(3)°, 144.12(7)° and 145.6(3)°.
. The distances of I···Br are 3.107(2) Å, 3.123(2) Å, the C–I···Br angles are 177.1(4)°, 173.4(6)°, and the RXB values are 0.80. The crystal 2-Cl has a monoclinic crystal system and the space group is C2. The distances of I···Cl are 2.963(9) Å, 2.989(6) Å, 2.934(7) Å and 2.98(1) Å. The C–I···Cl angles are 176.8(6)°, 173.4(6)°, 176.2(6)° and 175.9(6)°. The RXB values are 0.77. These crystal package diagrams display that a bent arrangement of the XB donors are around the central halide anion. The measured bent angles of 2-I, 2-Br and 2-Cl by mercury [46] are 146.38(3)°, 144.12(7)° and 145.6(3)°.
![[1860-5397-16-10-2]](/bjoc/content/figures/1860-5397-16-10-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Packing structure of 2-I (top), 2-Br (middle) and 2-Cl (bottom). Hydrogen atoms have been omitted for clarity. Side view (left) and top view (right).
Figure 2: Packing structure of 2-I (top), 2-Br (middle) and 2-Cl (bottom). Hydrogen atoms have been omitted f...
The crystal X-ray analyses of 2-BF4 shows that the diiodotriazolium moiety has formed with the tetrafluoroborate anion a triangle in which only two anions and three cations are assembled and one tetrafluoroborate is independent (Figure 3). 2-BF4 crystallizes in the triclinic space group ![[Graphic 3]](/bjoc/content/inline/1860-5397-16-10-i5.svg?max-width=637&scale=1.18182) .
.
![[1860-5397-16-10-3]](/bjoc/content/figures/1860-5397-16-10-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Packing structure of 2-BF4. Hydrogen atoms have been omitted for clarity.
Figure 3: Packing structure of 2-BF4. Hydrogen atoms have been omitted for clarity.
The single crystal of 2-OAc crystallizes in the monoclinic space group P21/c. The package diagram shows a dimer which is almost a rectangle (Figure 4). As shown in Table 1, The C–I···O distances are 2.547(2) Å and 2.582(2) Å. The C–I···O angles are 174.60(7)° and 170.38(7)°. The RXB values are 0.72 and 0.73.
![[1860-5397-16-10-4]](/bjoc/content/figures/1860-5397-16-10-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Packing structure of 2-OAc. Hydrogen atoms and solvent molecules have been omitted for clarity.
Figure 4: Packing structure of 2-OAc. Hydrogen atoms and solvent molecules have been omitted for clarity.
The single crystal of 2-TFA crystallizes in the monoclinic space group P21/n, but the packing structure of 2-TFA is different. The package diagram shows that two cations and two acetates form a boat shape (Figure 5). The C–I···O distances are 2.631(8) Å, 2.739(8) Å, 2.666(6) Å and 2.68(1) Å. The C–I···O angles are 175.7(3)°, 172.7(3)°, 176.7(3)° and 176.1(3)°. The RXB values are 0.77, 0.74 and 0.75 (Table 1).
![[1860-5397-16-10-5]](/bjoc/content/figures/1860-5397-16-10-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Packing structure of 2-TFA. Hydrogen atoms and disorder of fluorine atoms have been omitted for clarity. Side view (left) and top view (right).
Figure 5: Packing structure of 2-TFA. Hydrogen atoms and disorder of fluorine atoms have been omitted for cla...
Triazolium polyiodide 2-I·1.5I2 was made by 2-I with iodine and crystallizes in the monoclinic space group P21/c. The crystal package diagram displays that a bent arrangement of XB donors are around the central iodine atom of the I3− anion (Figure 6). 2-I·3.5I2 crystallizes in the monoclinic space group P21/c (Figure 7). There are two I3− and one I2 molecule in 2-I·1.5I2, while two I5− and three I2 molucules can be found in 2-I·3.5I2.
![[1860-5397-16-10-6]](/bjoc/content/figures/1860-5397-16-10-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Packing structure of 2-I.1.5I2. Hydrogen atoms have been omitted for clarity.
Figure 6: Packing structure of 2-I.1.5I2. Hydrogen atoms have been omitted for clarity.
![[1860-5397-16-10-7]](/bjoc/content/figures/1860-5397-16-10-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Packing structure of 2-I.3.5I2. Hydrogen atoms have been omitted for clarity.
Figure 7: Packing structure of 2-I.3.5I2. Hydrogen atoms have been omitted for clarity.
Table 1: XB interactions of the crystals.
| compound | interaction | I···X distance (Å) | RXB | angle C–I···X (deg) | C–I bond length (Å) |
| 2-I | I1···I3 | 3.306(1) | 0.81 | C1–I1···I3 176.9(3) | C1–I1 2.101(9) |
| I2···I3 | 3.300(1) | 0.81 | C2–I2···I3 176.2(3) | C2–I2 2.089(10) | |
| 2-Br | I1···Br1 | 3.107(2) | 0.80 | C1–I1···Br1 173.4(4) | C1–I1 2.10(1) |
| I2···Br1 | 3.123(2) | 0.80 | C2–I2···Br1 177.1(4) | C2–I2 2.10(1) | |
| 2-Cl | I1···Cl1 | 2.960(8) | 0.77 | C1–I1···Cl1 176.8(6) | C1–I1 2.09(2) |
| I2···Cl2 | 2.973(5) | 0.77 | C2–I2···Cl2 173.4(6) | C2–I2 2.05(2) | |
| I3···Cl2 | 2.975(7) | 0.77 | C3–I3···Cl2 176.2(6) | C3–I3 2.08(2) | |
| I4···Cl1 | 2.97(1) | 0.77 | C4–I4···Cl1 175.9(6) | C4–I4 2.09(3) | |
| 2-OAc | I1···O1 | 2.582(2) | 0.73 | C1–I1···O1 170.38(7) | C1–I1 2.085(2) |
| I2···O2 | 2.547(2) | 0.72 | C2–I2···O2 174.60(7) | C2–I2 2.105(2) | |
| 2-TFA | I1···O3 | 2.644(8) | 0.74 | C1–I1···O3 175.3(3) | C1–I1 2.082(7) |
| I2···O1 | 2.710(8) | 0.77 | C2–I2···O1 173.0(3) | C2–I2 2.059(7) | |
| I3···O2 | 2.677(6) | 0.75 | C3–I3···O2 177.0(3) | C3–I3 2.087(8) | |
| I4···O4 | 2.676(8) | 0.75 | C4–I4···O4 176.3(3) | C4–I4 2.072(7) | |
| 2-I.1.5 I2 | I1···I5 | 3.482(2) | 0.85 | C1–I1···I5 167.48(3) | C1–I1 2.061(4) |
| I2···I8 | 3.538(1) | 0.87 | C2–I2···I8 155.79(3) | C2–I2 2.062(4) | |
| I3···I5 | 3.394(1) | 0.83 | C3–I3···I5 176.97(2) | C3–I3 2.070(4) | |
| 2-I.3.5 I2 | I1···I5 | 3.5454(7) | 0.87 | C1–I1···I5 175.8(2) | C1–I1 2.047(7) |
| I2···I9 | 3.5829(7) | 0.88 | C2–I2···I9 174.2(2) | C2–I2 2.068(7) | |
| I4···I15 | 3.7356(7) | 0.92 | C4–I4···I15 177.8(2) | C4–I4 2.063(7) | |
| I3···I12 | 3.7237(8) | 0.92 | C3–I3···I12 149.2(2) | C3–I3 2.048(7) | |
The XB interaction with neural halogen acceptors was also investigated. Diffusion of ether into the mixture of 4,4'-bipyridine (bpy) and 2-BF4 in dichloromethane leads to the crystallization of 2-BF4·0.5bpy (Figure 8). It crystallizes in the monoclinic space group P21/c. The acceptor 4,4'-bipyridine provides a complementary link for 1D chain formation. The C–I···N angles are 168.2(3)° and 173.4(4)°, close to linear, which is consistent with the high directionality of the interaction. The C–I···N distances are 2.599(9) Å and 2.580(9) Å. The RXB value of C–I···N is 0.70. The C–I···F distances are 3.137(10) Å and 2.932(20) Å.
![[1860-5397-16-10-8]](/bjoc/content/figures/1860-5397-16-10-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Packing structure of 2-BF4.0.5bpy. Hydrogen atoms and dichloromethane have been omitted for clarity.
Figure 8: Packing structure of 2-BF4.0.5bpy. Hydrogen atoms and dichloromethane have been omitted for clarity....
To better understand the 1,2,3-triazole based XB donors, model 1,2,3-triazole A, 1,2,3-triazolium B, 1,2,3-triazolylidene complex C-CuI and diiodotriazolium D were calculated by DFT calculations (Figure 9). The calculation results show that σ holes in diiodotriazolium D are mainly located in the elongation of two C–I bonds. The DFT calculation also shows that σ hole of in diiodotriazolium D and 1,2,3-triazolium B are comparable, and much larger than the 1,2,3-triazole A and 1,2,3-triazolylidene complex C-CuI due to positive charge effect.
![[1860-5397-16-10-9]](/bjoc/content/figures/1860-5397-16-10-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: 1,2,3-Triazole-based halogen model calculation: electrostatic potential surfaces mapped on total density (iso value 0.01). C was calculated using 1,2,3-triazolylidene copper iodide complex C-CuI.
Figure 9: 1,2,3-Triazole-based halogen model calculation: electrostatic potential surfaces mapped on total de...
Conclusion
In summary, we synthesized 4,5-diiodo-1,3-dimesityl-1,2,3-triazolium salts with different anions. When the anion is chloride, bromide or iodide, the crystal is a tetramer. Strong XB was observed in these forms. When the anion is changed to tetrafluoroborate, it takes Chinese lantern shape as a trimer. Triazolium trifluoroacetate and acetate exist as a dimer, while the former shows a boat conformation and the latter forms a rectangle conformation. Triazolium salts form a linear polymer with polyiodide. 2-BF4 forms co-crystals with 4,4'-bipyridine via halogen bonding. DFT calculation results show that the σ holes of 4,5-diiodo-1,2,3-triazolium is similar to the σ hole of 5-iodo-1,2,3-triazoliums salts.
Experimental
General
Unless otherwise noted, all reagents were obtained from commercial sources and used without further purification. All solvents were dried then stored over 4 Å molecular sieves prior to use. All syntheses were carried out under an atmosphere of dry nitrogen or in a glovebox. At the same time, the syntheses were performed under a standard Schlenk vacuum line. 1H NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. High-resolution mass spectra (HRMS) were acquired with a Thermo Scientific (Q-Exactive) instrument using electrospray ionization mode (ESI). Elemental analyses (C, H, N) were performed on Flash EA 1112 Analyzer.
Synthesis
2-I: 4,5-Unsubstituted triazolium salt 1 (450 mg, 1 mmol), potassium tert-butoxide (250 mg, 2.2 mmol) and I2 (510 mg, 2 mmol) were added in a Schlenk tube under nitrogen, then THF (20 mL) was added at −78 °C. The mixture was stirred for 12 hours. After the evaporation of THF, dichloromethane (100 mL) was added and inorganic salts were removed by filtration. The pure product (521 mg) was obtained by evaporation of dichloromethane and washed with ether. The yield was 76%. A single crystal of 2-I was obtained by slow diffusion of ether into a dimethylformamide solution due to the poor solubility. 1H NMR (400 MHz, CDCl3) δ 7.07 (s, 4H), 2.41 (s, 6H), 2.00 (s, 12H); 13C NMR (100 MHz, DMSO-d6) 143.2, 134.9, 131.9, 130.2, 111.8, 21.2, 17.1; HRMS (m/z): [M – I−]+ calcd for C20H22I2N3+, 557.9898; found, 557.9891; anal. calcd. for C20H22I3N3 (685.13): C, 35.06, H, 3.24, N, 6.13%; found: C, 35.41, H, 3.32, N, 6.06%.
2-BF4: AgBF4 (60 mg, 0.30 mmol) and 2-I (205 mg, 0.30 mmol) were added in a Schlenk tube, then dichloromethane (5 mL) was added and the solution stirred under nitrogen in the dark for 6 h. After AgI was removed by filtration, the solution was washed with water. The pure product was obtained by evaporation of the dichloromethane phase (178 mg, 92% yield). A single crystal of 2-BF4 was obtained by slow diffusion of ether into a dichloromethane solution. 1H NMR (400 MHz, CDCl3) δ 7.12 (s, 4H), 2.40 (s, 6H), 2.02 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 134.8, 134.2, 131.5, 130.3, 104.4, 12.4, 17.3; anal. calcd. for C20H22BF4I2N3 (645.03): C, 37.24, H, 3.44, N, 6.51%; found: C, 37.51, H, 3.52, N, 6.42%.
2-Cl: Potassium chloride (740 mg, 10 mmol) and 2-BF4 (120 mg, 0.19 mmol) were mixed in acetonitrile (30 mL) in a round bottom flask, then the mixture was stirred for 24 h under air. Then the acetonitrile was removed under reduced pressure. Dichloromethane (50 mL) was added and the excess potassium chloride was removed by filtration. Then the dichloromethane was removed by evaporation to give the final white product (2-Cl) (99 mg, 87% yield). 1H NMR (400 MHz, CDCl3) δ 7.05 (s, 4H), 2.39 (s, 6H), 1.98 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 142.9, 134.2, 132.1, 130.1, 113.2, 21.4, 17.4; anal. calcd. for C20H22ClI2N3 (593.68): C, 40.46, H, 3.74, N, 7.08%; found: C, 40.81, H, 3.79, N, 6.96%. A single crystal of 2-Cl was obtained by slow evaporation of a dichloromethane solution.
2-Br: Potassium bromide (1190 mg, 10 mmol) and 2-BF4 (128 mg, 0.2 mmol) were mixed in acetonitrile (30 mL) in a round bottom flask, then the mixture was stirred under air for 24 h. Then the acetonitrile was removed under reduced pressure, dichloromethane (70 mL) was added and the excess potassium bromide was removed by filtration. Then the dichloromethane was removed by evaporation to give the white product (2-Br) (112 mg, 88% yield). 1H NMR (400 MHz, CDCl3) δ 7.05 (s, 4H), 2.39 (s, 6H), 1.99 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 143.0, 134.2, 132.1, 130.1, 113.8, 21.4, 17.4; anal. calcd. for C20H22BrI2N3 (638.13): C, 37.64, H, 3.48, N, 6.59%; found: C, 37.91, H, 3.65, N, 6.41%.
2-OAc: AgOAc (52 mg, 0.30 mmol) and 2-I (205 mg, 0.30 mmol) were added in a Schlenk tube, then dichloromethane (6 mL) was added and the solution stirred in a glove box in the dark for 6 h. Then AgI was removed by filtration and the solution was washed with water (10 mL) to remove the excess of silver. The pure product was obtained by evaporation of dichloromethane solution (176 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 7.00 (s, 4H), 2.32 (s, 6H), 1.92 (s, 12H), 1.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.6, 142.6, 133.9, 131.8, 129.8, 110.8, 23.6, 21.2, 17.2; anal. calcd. for C22H25I2N3O2 (617.27): C, 42.81, H, 4.08, N, 6.81%; found: C, 42.96, H, 4.26, N, 6.72%. The single crystal of 2-OAc was obtained by slow diffusion of n-pentane into a dichloromethane solution.
2-TFA: CF3COOAg (77 mg, 0.35 mmol) and 2-I (205 mg, 0.3 mmol) were added in a Schlenk tube, then dichloromethane (6 mL) was added and the solution stirred in the glovebox for 6 h in the dark. Then AgI was removed by filtration and the solution was washed with water (5 mL) to remove the excess of silver. The pure product was obtained by evaporation of the dichloromethane solution (125 mg, 53%). 1H NMR (400 MHz, CDCl3) δ 7.08 (s, 4H), 2.40 (s, 6H), 2.00 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 161.0 (q, J = 35.0 Hz), 143.3, 134.2, 131.9, 130.2, 116.5 (q, J = 297.2 Hz), 110.0, 21.4, 17.4; anal. calcd. for C22H22F3I2N3O2 (671.24): C, 39.37, H, 3.30, N, 6.26%; found: C, 39.51, H, 3.49, N, 6.18%.
2-I.1.5I2: 2-I (35 mg, 0.05 mol) and I2 (25 mg, 0.1 mmol) were mixed in dichloromethane (6 mL) in a round bottom flask. The single crystals were obtained by slow diffusion of ether into a dichloromethane solution. Brown solid, 21 mg, 39% yield. 1H NMR (400 MHz, CDCl3) δ 7.20 (s, 4H), 2.47 (s, 6H), 2.09 (s, 12H); anal. calcd. for C22H22I6N3 (1065.84): C, 22.54, H, 2.08, N, 3.94%; found: 22.63, H, 2.19, N, 3.82%.
2-I.3.5I2: 2-I (35 mg, 0.05 mol) and I2 (50 mg, 0.2 mmol) were mixed in dichloromethane (10 mL) in a round bottom flask. The single crystals were obtained by slow diffusion of ether into a dichloromethane solution, Brown solid, 63 mg, 81% yield. 1H NMR (400 MHz, CDCl3) δ 7.21 (s, 4H),2.48 (s, 6H), 2.10 (s, 12H); anal. calcd. for C22H22I10N3 (1073.46): C, 15.72, H, 1.41, N, 2.67%; found: C, 15.93, H, 1.66, N, 2.41%.
2-BF4.0.5bpy: 2-BF4 (65 mg, 0.1 mmol) and 4,4'-bipyridine (16 mg, 0.1 mmol) was mixed in dichloromethane (4 mL) in a round bottom flask. The single crystals were obtained by slow diffusion of ether into a dichloromethane solution. Colourless solid, 28 mg, 31% yield. 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 6.1 Hz, 2H), 7.57 (d, J = 6.2 Hz, 2H), 7.11 (s, 4H), 2.39 (s, 6H), 2.02 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 150.0, 145.7, 143.5, 134.0, 131.6, 130.2, 121.9, 107.6, 21.4, 17.2.
X-ray diffraction measurements
Single crystal diffraction data for 2-I, 2-Br and 2-Cl were collected at 200 K using an IμS micro-focus sealed X-ray tube with Mo Kα radiation (λ = 0.71073 Å) on a Bruker D8 venture Kappa Duo diffractometer equipped with a PHOTON 100 detector. Low-temperature holding was achieved by a Cryostream Cooler (Oxford Cryosystems). Single crystal diffraction data for 2-OAc, 2-TFA, 2-BF4.0.5bpy, 2-I.1.5I2 and 2-I.3.5I2 were collected at 150 K while 2-BF4 was collected at 298 K. All the data were collected 0.5 degree per step and using the ω scan mode. Frames were integrated using the Bruker SAINT [47] software. Semi-empirical absorption correction was applied with the SADABS program [48].
All the structures were solved by SHELXT [49] and refined by SHELXL [50] programs against |F|2 using all data following established refinement strategies [51] through olex2 [52]. Their packing diagrams were prepared by using Mercury [46].
Computational details
All calculations were performed with the Gaussian 16 (G16) program package [53]. The DFT method using the M06-2X functional [54] relying on relativistic pseudo-potentials was used, namely the small core ECP46MWB [55] for I atoms. The C, H and N atoms were treated with a basis set of 6-311G** [56]. The Cu atom was treated with a basis set of LanL2DZ [57]. Geometry optimizations were performed without any constraints, and the frequency analysis confirmed that there were no image frequencies for these structures. Visualization of the electrostatic potential was performed using the Gauss View 6.0 package [58].
References
-
Desiraju, G. R.; Ho, P. S.; Kloo, L.; Legon, A. C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Pure Appl. Chem. 2013, 85, 1711–1713. doi:10.1351/pac-rec-12-05-10
Return to citation in text: [1] -
Hassel, O. Science 1970, 170, 497–502. doi:10.1126/science.170.3957.497
Return to citation in text: [1] -
Valerio, G.; Raos, G.; Meille, S. V.; Metrangolo, P.; Resnati, G. J. Phys. Chem. A 2000, 104, 1617–1620. doi:10.1021/jp993415j
Return to citation in text: [1] -
Politzer, P.; Murray, J. S.; Clark, T. Phys. Chem. Chem. Phys. 2010, 12, 7748. doi:10.1039/c004189k
Return to citation in text: [1] -
Lu, Y.; Li, H.; Zhu, X.; Zhu, W.; Liu, H. J. Phys. Chem. A 2011, 115, 4467–4475. doi:10.1021/jp111616x
Return to citation in text: [1] -
Tsuzuki, S.; Wakisaka, A.; Ono, T.; Sonoda, T. Chem. – Eur. J. 2012, 18, 951–960. doi:10.1002/chem.201102562
Return to citation in text: [1] -
Huber, S. M.; Scanlon, J. D.; Jimenez-Izal, E.; Ugalde, J. M.; Infante, I. Phys. Chem. Chem. Phys. 2013, 15, 10350. doi:10.1039/c3cp50892g
Return to citation in text: [1] -
Liantonio, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Stevenazzi, A. Cryst. Growth Des. 2003, 3, 799–803. doi:10.1021/cg034098f
Return to citation in text: [1] -
Lucassen, A. C. B.; Vartanian, M.; Leitus, G.; van der Boom, M. E. Cryst. Growth Des. 2005, 5, 1671–1673. doi:10.1021/cg0501433
Return to citation in text: [1] -
Cardillo, P.; Corradi, E.; Lunghi, A.; Meille, S. V.; Messina, M. T.; Metrangolo, P.; Resnati, G. Tetrahedron 2000, 56, 5535–5550. doi:10.1016/s0040-4020(00)00476-2
Return to citation in text: [1] -
Saccone, M.; Cavallo, G.; Metrangolo, P.; Pace, A.; Pibiri, I.; Pilati, T.; Resnati, G.; Terraneo, G. CrystEngComm 2013, 15, 3102. doi:10.1039/c3ce40268a
Return to citation in text: [1] -
Walsh, R. B.; Padgett, C. W.; Metrangolo, P.; Resnati, G.; Hanks, T. W.; Pennington, W. T. Cryst. Growth Des. 2001, 1, 165–175. doi:10.1021/cg005540m
Return to citation in text: [1] -
Bailey, R. D.; Hook, L. L.; Watson, R. P.; Hanks, T. W.; Pennington, W. T. Cryst. Eng. 2000, 3, 155–171. doi:10.1016/s1463-0184(00)00039-3
Return to citation in text: [1] -
Burton, D. D.; Fontana, F.; Metrangolo, P.; Pilati, T.; Resnati, G. Tetrahedron Lett. 2003, 44, 645–648. doi:10.1016/s0040-4039(02)02710-7
Return to citation in text: [1] -
Perkins, C.; Libri, S.; Adams, H.; Brammer, L. CrystEngComm 2012, 14, 3033–3038. doi:10.1039/c2ce00029f
Return to citation in text: [1] -
Aakeröy, C. B.; Baldrighi, M.; Desper, J.; Metrangolo, P.; Resnati, G. Chem. – Eur. J. 2013, 19, 16240–16247. doi:10.1002/chem.201302162
Return to citation in text: [1] -
Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484
Return to citation in text: [1] -
Brown, A.; Beer, P. D. Chem. Commun. 2016, 52, 8645–8658. doi:10.1039/c6cc03638d
Return to citation in text: [1] -
Beale, T. M.; Chudzinski, M. G.; Sarwar, M. G.; Taylor, M. S. Chem. Soc. Rev. 2013, 42, 1667–1680. doi:10.1039/c2cs35213c
Return to citation in text: [1] -
Erdélyi, M. Chem. Soc. Rev. 2012, 41, 3547. doi:10.1039/c2cs15292d
Return to citation in text: [1] -
Jentzsch, A. V.; Matile, S. Top. Curr. Chem. 2014, 358, 205–239. doi:10.1007/128_2014_541
Return to citation in text: [1] -
Gilday, L. C.; Robinson, S. W.; Barendt, T. A.; Langton, M. J.; Mullaney, B. R.; Beer, P. D. Chem. Rev. 2015, 115, 7118–7195. doi:10.1021/cr500674c
Return to citation in text: [1] -
Scholfield, M. R.; Ford, M. C.; Carlsson, A.-C. C.; Butta, H.; Mehl, R. A.; Ho, P. S. Biochemistry 2017, 56, 2794–2802. doi:10.1021/acs.biochem.7b00022
Return to citation in text: [1] -
Bulfield, D.; Huber, S. M. Chem. – Eur. J. 2016, 22, 14434–14450. doi:10.1002/chem.201601844
Return to citation in text: [1] -
Schulze, B.; Schubert, U. S. Chem. Soc. Rev. 2014, 43, 2522. doi:10.1039/c3cs60386e
Return to citation in text: [1] -
Molina, P.; Zapata, F.; Caballero, A. Chem. Rev. 2017, 117, 9907–9972. doi:10.1021/acs.chemrev.6b00814
Return to citation in text: [1] -
Gilday, L. C.; White, N. G.; Beer, P. D. Dalton Trans. 2013, 42, 15766. doi:10.1039/c3dt52093e
Return to citation in text: [1] -
Robinson, S. W.; Mustoe, C. L.; White, N. G.; Brown, A.; Thompson, A. L.; Kennepohl, P.; Beer, P. D. J. Am. Chem. Soc. 2015, 137, 499–507. doi:10.1021/ja511648d
Return to citation in text: [1] -
Maugeri, L.; Jamieson, E. M. G.; Cordes, D. B.; Slawin, A. M. Z.; Philp, D. Chem. Sci. 2017, 8, 938–945. doi:10.1039/c6sc03696a
Return to citation in text: [1] -
Mungalpara, D.; Stegmüller, S.; Kubik, S. Chem. Commun. 2017, 53, 5095–5098. doi:10.1039/c7cc02424j
Return to citation in text: [1] -
Tepper, R.; Bode, S.; Geitner, R.; Jäger, M.; Görls, H.; Vitz, J.; Dietzek, B.; Schmitt, M.; Popp, J.; Hager, M. D.; Schubert, U. S. Angew. Chem., Int. Ed. 2017, 56, 4047–4051. doi:10.1002/anie.201610406
Return to citation in text: [1] -
Kaasik, M.; Kaabel, S.; Kriis, K.; Järving, I.; Aav, R.; Rissanen, K.; Kanger, T. Chem. – Eur. J. 2017, 23, 7337–7344. doi:10.1002/chem.201700618
Return to citation in text: [1] -
Dreger, A.; Engelage, E.; Mallick, B.; Beer, P. D.; Huber, S. M. Chem. Commun. 2018, 54, 4013–4016. doi:10.1039/c8cc00527c
Return to citation in text: [1] -
Kilah, N. L.; Wise, M. D.; Serpell, C. J.; Thompson, A. L.; White, N. G.; Christensen, K. E.; Beer, P. D. J. Am. Chem. Soc. 2010, 132, 11893–11895. doi:10.1021/ja105263q
Return to citation in text: [1] -
Kilah, N. L.; Wise, M. D.; Beer, P. D. Cryst. Growth Des. 2011, 11, 4565–4571. doi:10.1021/cg200811a
Return to citation in text: [1] -
Kniep, F.; Rout, L.; Walter, S. M.; Bensch, H. K. V.; Jungbauer, S. H.; Herdtweck, E.; Huber, S. M. Chem. Commun. 2012, 48, 9299. doi:10.1039/c2cc34392d
Return to citation in text: [1] -
Tepper, R.; Schulze, B.; Jäger, M.; Friebe, C.; Scharf, D. H.; Görls, H.; Schubert, U. S. J. Org. Chem. 2015, 80, 3139–3150. doi:10.1021/acs.joc.5b00028
Return to citation in text: [1] -
Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 8018–8021. doi:10.1002/anie.200903558
Return to citation in text: [1] -
Chen, Z.; Liu, Z.; Cao, G.; Li, H.; Ren, H. Adv. Synth. Catal. 2017, 359, 202–224. doi:10.1002/adsc.201600918
Return to citation in text: [1] -
Kaasik, M.; Metsala, A.; Kaabel, S.; Kriis, K.; Järving, I.; Kanger, T. J. Org. Chem. 2019, 84, 4294–4303. doi:10.1021/acs.joc.9b00248
Return to citation in text: [1] -
Haraguchi, R.; Hoshino, S.; Sakai, M.; Tanazawa, S.-g.; Morita, Y.; Komatsu, T.; Fukuzawa, S.-i. Chem. Commun. 2018, 54, 10320–10323. doi:10.1039/c8cc05309j
Return to citation in text: [1] -
Borissov, A.; Marques, I.; Lim, J. Y. C.; Félix, V.; Smith, M. D.; Beer, P. D. J. Am. Chem. Soc. 2019, 141, 4119–4129. doi:10.1021/jacs.9b00148
Return to citation in text: [1] -
Xu, X.; Zhang, Z.; Huang, S.; Cao, L.; Liu, W.; Yan, X. Dalton Trans. 2019, 48, 6931–6941. doi:10.1039/c9dt01018a
Return to citation in text: [1] [2] -
Liu, W.; Zhao, L.-L.; Melaimi, M.; Cao, L.; Xu, X.; Bouffard, J.; Bertrand, G.; Yan, X. Chem 2019, 5, 2484–2494. doi:10.1016/j.chempr.2019.08.011
Return to citation in text: [1] -
Alvarez, S. Dalton Trans. 2013, 42, 8617. doi:10.1039/c3dt50599e
Return to citation in text: [1] -
Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466–470. doi:10.1107/s0021889807067908
Return to citation in text: [1] [2] -
Bruker SAINT, v8.34A; Bruker AXS: Madison, WI, 2013.
Return to citation in text: [1] -
SADABS, V2008/1; Bruker AXS: Madison, WI, 2008.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273315099842
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Müller, P. Crystallogr. Rev. 2009, 15, 57–83. doi:10.1080/08893110802547240
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Theor. Chim. Acta 1990, 77, 123–141. doi:10.1007/bf01114537
Return to citation in text: [1] -
McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980
Return to citation in text: [1] -
Dunning, T. H., Jr.; Hay, P. J. In Modern Theoretical Chemistry; Schaefer, H. F., III., Ed.; Plenum Press: New York, 1976; Vol. 3, pp 1–28.
Return to citation in text: [1] -
GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, 2016.
Return to citation in text: [1]
| 55. | Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Theor. Chim. Acta 1990, 77, 123–141. doi:10.1007/bf01114537 |
| 56. | McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639–5648. doi:10.1063/1.438980 |
| 57. | Dunning, T. H., Jr.; Hay, P. J. In Modern Theoretical Chemistry; Schaefer, H. F., III., Ed.; Plenum Press: New York, 1976; Vol. 3, pp 1–28. |
| 1. | Desiraju, G. R.; Ho, P. S.; Kloo, L.; Legon, A. C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Pure Appl. Chem. 2013, 85, 1711–1713. doi:10.1351/pac-rec-12-05-10 |
| 2. | Hassel, O. Science 1970, 170, 497–502. doi:10.1126/science.170.3957.497 |
| 14. | Burton, D. D.; Fontana, F.; Metrangolo, P.; Pilati, T.; Resnati, G. Tetrahedron Lett. 2003, 44, 645–648. doi:10.1016/s0040-4039(02)02710-7 |
| 44. | Liu, W.; Zhao, L.-L.; Melaimi, M.; Cao, L.; Xu, X.; Bouffard, J.; Bertrand, G.; Yan, X. Chem 2019, 5, 2484–2494. doi:10.1016/j.chempr.2019.08.011 |
| 13. | Bailey, R. D.; Hook, L. L.; Watson, R. P.; Hanks, T. W.; Pennington, W. T. Cryst. Eng. 2000, 3, 155–171. doi:10.1016/s1463-0184(00)00039-3 |
| 43. | Xu, X.; Zhang, Z.; Huang, S.; Cao, L.; Liu, W.; Yan, X. Dalton Trans. 2019, 48, 6931–6941. doi:10.1039/c9dt01018a |
| 8. | Liantonio, R.; Metrangolo, P.; Pilati, T.; Resnati, G.; Stevenazzi, A. Cryst. Growth Des. 2003, 3, 799–803. doi:10.1021/cg034098f |
| 9. | Lucassen, A. C. B.; Vartanian, M.; Leitus, G.; van der Boom, M. E. Cryst. Growth Des. 2005, 5, 1671–1673. doi:10.1021/cg0501433 |
| 10. | Cardillo, P.; Corradi, E.; Lunghi, A.; Meille, S. V.; Messina, M. T.; Metrangolo, P.; Resnati, G. Tetrahedron 2000, 56, 5535–5550. doi:10.1016/s0040-4020(00)00476-2 |
| 11. | Saccone, M.; Cavallo, G.; Metrangolo, P.; Pace, A.; Pibiri, I.; Pilati, T.; Resnati, G.; Terraneo, G. CrystEngComm 2013, 15, 3102. doi:10.1039/c3ce40268a |
| 12. | Walsh, R. B.; Padgett, C. W.; Metrangolo, P.; Resnati, G.; Hanks, T. W.; Pennington, W. T. Cryst. Growth Des. 2001, 1, 165–175. doi:10.1021/cg005540m |
| 42. | Borissov, A.; Marques, I.; Lim, J. Y. C.; Félix, V.; Smith, M. D.; Beer, P. D. J. Am. Chem. Soc. 2019, 141, 4119–4129. doi:10.1021/jacs.9b00148 |
| 3. | Valerio, G.; Raos, G.; Meille, S. V.; Metrangolo, P.; Resnati, G. J. Phys. Chem. A 2000, 104, 1617–1620. doi:10.1021/jp993415j |
| 4. | Politzer, P.; Murray, J. S.; Clark, T. Phys. Chem. Chem. Phys. 2010, 12, 7748. doi:10.1039/c004189k |
| 5. | Lu, Y.; Li, H.; Zhu, X.; Zhu, W.; Liu, H. J. Phys. Chem. A 2011, 115, 4467–4475. doi:10.1021/jp111616x |
| 6. | Tsuzuki, S.; Wakisaka, A.; Ono, T.; Sonoda, T. Chem. – Eur. J. 2012, 18, 951–960. doi:10.1002/chem.201102562 |
| 7. | Huber, S. M.; Scanlon, J. D.; Jimenez-Izal, E.; Ugalde, J. M.; Infante, I. Phys. Chem. Chem. Phys. 2013, 15, 10350. doi:10.1039/c3cp50892g |
| 43. | Xu, X.; Zhang, Z.; Huang, S.; Cao, L.; Liu, W.; Yan, X. Dalton Trans. 2019, 48, 6931–6941. doi:10.1039/c9dt01018a |
| 28. | Robinson, S. W.; Mustoe, C. L.; White, N. G.; Brown, A.; Thompson, A. L.; Kennepohl, P.; Beer, P. D. J. Am. Chem. Soc. 2015, 137, 499–507. doi:10.1021/ja511648d |
| 29. | Maugeri, L.; Jamieson, E. M. G.; Cordes, D. B.; Slawin, A. M. Z.; Philp, D. Chem. Sci. 2017, 8, 938–945. doi:10.1039/c6sc03696a |
| 30. | Mungalpara, D.; Stegmüller, S.; Kubik, S. Chem. Commun. 2017, 53, 5095–5098. doi:10.1039/c7cc02424j |
| 31. | Tepper, R.; Bode, S.; Geitner, R.; Jäger, M.; Görls, H.; Vitz, J.; Dietzek, B.; Schmitt, M.; Popp, J.; Hager, M. D.; Schubert, U. S. Angew. Chem., Int. Ed. 2017, 56, 4047–4051. doi:10.1002/anie.201610406 |
| 32. | Kaasik, M.; Kaabel, S.; Kriis, K.; Järving, I.; Aav, R.; Rissanen, K.; Kanger, T. Chem. – Eur. J. 2017, 23, 7337–7344. doi:10.1002/chem.201700618 |
| 33. | Dreger, A.; Engelage, E.; Mallick, B.; Beer, P. D.; Huber, S. M. Chem. Commun. 2018, 54, 4013–4016. doi:10.1039/c8cc00527c |
| 38. | Hein, J. E.; Tripp, J. C.; Krasnova, L. B.; Sharpless, K. B.; Fokin, V. V. Angew. Chem., Int. Ed. 2009, 48, 8018–8021. doi:10.1002/anie.200903558 |
| 39. | Chen, Z.; Liu, Z.; Cao, G.; Li, H.; Ren, H. Adv. Synth. Catal. 2017, 359, 202–224. doi:10.1002/adsc.201600918 |
| 17. | Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. Chem. Rev. 2016, 116, 2478–2601. doi:10.1021/acs.chemrev.5b00484 |
| 18. | Brown, A.; Beer, P. D. Chem. Commun. 2016, 52, 8645–8658. doi:10.1039/c6cc03638d |
| 19. | Beale, T. M.; Chudzinski, M. G.; Sarwar, M. G.; Taylor, M. S. Chem. Soc. Rev. 2013, 42, 1667–1680. doi:10.1039/c2cs35213c |
| 20. | Erdélyi, M. Chem. Soc. Rev. 2012, 41, 3547. doi:10.1039/c2cs15292d |
| 21. | Jentzsch, A. V.; Matile, S. Top. Curr. Chem. 2014, 358, 205–239. doi:10.1007/128_2014_541 |
| 22. | Gilday, L. C.; Robinson, S. W.; Barendt, T. A.; Langton, M. J.; Mullaney, B. R.; Beer, P. D. Chem. Rev. 2015, 115, 7118–7195. doi:10.1021/cr500674c |
| 23. | Scholfield, M. R.; Ford, M. C.; Carlsson, A.-C. C.; Butta, H.; Mehl, R. A.; Ho, P. S. Biochemistry 2017, 56, 2794–2802. doi:10.1021/acs.biochem.7b00022 |
| 24. | Bulfield, D.; Huber, S. M. Chem. – Eur. J. 2016, 22, 14434–14450. doi:10.1002/chem.201601844 |
| 25. | Schulze, B.; Schubert, U. S. Chem. Soc. Rev. 2014, 43, 2522. doi:10.1039/c3cs60386e |
| 26. | Molina, P.; Zapata, F.; Caballero, A. Chem. Rev. 2017, 117, 9907–9972. doi:10.1021/acs.chemrev.6b00814 |
| 27. | Gilday, L. C.; White, N. G.; Beer, P. D. Dalton Trans. 2013, 42, 15766. doi:10.1039/c3dt52093e |
| 40. | Kaasik, M.; Metsala, A.; Kaabel, S.; Kriis, K.; Järving, I.; Kanger, T. J. Org. Chem. 2019, 84, 4294–4303. doi:10.1021/acs.joc.9b00248 |
| 41. | Haraguchi, R.; Hoshino, S.; Sakai, M.; Tanazawa, S.-g.; Morita, Y.; Komatsu, T.; Fukuzawa, S.-i. Chem. Commun. 2018, 54, 10320–10323. doi:10.1039/c8cc05309j |
| 16. | Aakeröy, C. B.; Baldrighi, M.; Desper, J.; Metrangolo, P.; Resnati, G. Chem. – Eur. J. 2013, 19, 16240–16247. doi:10.1002/chem.201302162 |
| 15. | Perkins, C.; Libri, S.; Adams, H.; Brammer, L. CrystEngComm 2012, 14, 3033–3038. doi:10.1039/c2ce00029f |
| 34. | Kilah, N. L.; Wise, M. D.; Serpell, C. J.; Thompson, A. L.; White, N. G.; Christensen, K. E.; Beer, P. D. J. Am. Chem. Soc. 2010, 132, 11893–11895. doi:10.1021/ja105263q |
| 35. | Kilah, N. L.; Wise, M. D.; Beer, P. D. Cryst. Growth Des. 2011, 11, 4565–4571. doi:10.1021/cg200811a |
| 36. | Kniep, F.; Rout, L.; Walter, S. M.; Bensch, H. K. V.; Jungbauer, S. H.; Herdtweck, E.; Huber, S. M. Chem. Commun. 2012, 48, 9299. doi:10.1039/c2cc34392d |
| 37. | Tepper, R.; Schulze, B.; Jäger, M.; Friebe, C.; Scharf, D. H.; Görls, H.; Schubert, U. S. J. Org. Chem. 2015, 80, 3139–3150. doi:10.1021/acs.joc.5b00028 |
| 46. | Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466–470. doi:10.1107/s0021889807067908 |
| 54. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 52. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 46. | Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466–470. doi:10.1107/s0021889807067908 |
| 50. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 49. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273315099842 |
© 2020 Xu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)