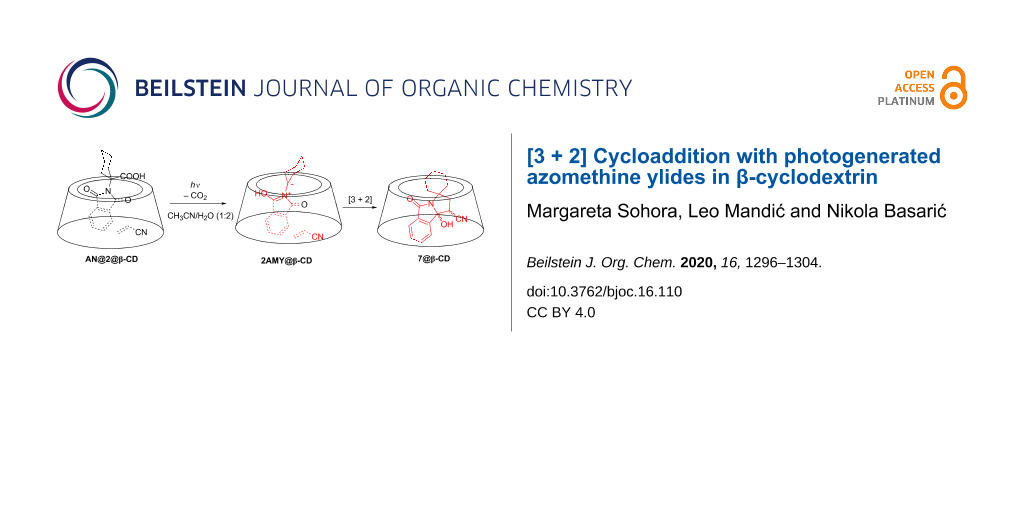
Abstract
Stability constants for the inclusion complexes of cyclohexylphthalimide 2 and adamantylphthalimide 3 with β-cyclodextrin (β-CD) were determined by 1H NMR titration, K = 190 ± 50 M−1, and K = 2600 ± 600 M−1, respectively. Photochemical reactivity of the inclusion complexes 2@β-CD and 3@β-CD was investigated, and we found out that β-CD does not affect the decarboxylation efficiency, while it affects the subsequent photochemical H-abstraction, resulting in different product distribution upon irradiation in the presence of β-CD. The formation of ternary complexes with acrylonitrile (AN) and 2@β-CD or 3@β-CD was also essayed by 1H NMR. Although the formation of such complexes was suggested, stability constants could not be determined. Irradiation of 2@β-CD in the presence of AN in aqueous solution where cycloadduct 7 was formed highly suggests that decarboxylation and [3 + 2] cycloaddition take place in the ternary complex, whereas such a reactivity from bulky adamantane 3 is less likely. This proof of principle that decarboxylation and cycloaddition can be performed in the β-CD cavity has a significant importance for the design of new supramolecular systems for the control of photoreactivity.

Graphical Abstract
Introduction
Cycloadditions are highly useful reactions in organic synthesis providing complex cyclic structures from easily available precursors [1,2]. Among different reactions, [3 + 2] cycloadditions showed applicability in the synthesis of heterocyclic 5-ring compounds [3], as well as in the green synthesis of a number of natural products [4]. One of the useful synthons in [3 + 2] cycloadditions is azomethine ylide [5-7], also used in intramolecular reactions [8]. Azomethine ylides can be formed by several photochemical or thermal catalytic methods [5-7], including photodecarboxylation of phthalimide derivatives of α-amino acids such as N-phthaloylglycine (1) [9,10].
Phthalimide is a versatile chromophore that has been used in the synthesis of complex molecules and natural products [11] since the pioneering work of Kanaoka et al. [12]. Photochemical reactions of phthalimides include H-abstractions, cycloadditions and photoinduced electron transfer (PET)[13]. We became interested in the application of photochemical H-abstraction reactions initiated by phthalimides in organic synthesis [14,15]. Furthermore, H-abstractions were investigated in inclusion complexes, in the cavity of β-cyclodextrins (β-CD) [16]. We found out that H-abstraction reactions were about ten times more efficient in the β-CD complexes than in the isotropic solution, and the macrocyclic host affected the stereochemistry of the reaction. Moreover, we studied photodecarboxylation reactions initiated by the phthalimide chromophore [17-19] and applied them in cyclizations with memory of chirality [20] and diastereoselective peptide cyclizations [21]. Photodecarboxylations were also intensively investigated in a series of nonsteroidal anti-inflammatory drugs [22-24] such as ketoprofen [25-34], due to photoallergic responses initiated by photodecarboxylation of these drugs [35].
Stereoselectivity in photochemical reactions can be achieved by use of supramolecular chemistry [36,37]. For example, stereoselectivity has been reported for photochemical reactions taking place in the inclusion complexes with CD [38-41] or structurally modified CDs [42-47]. Since β-CD is often used in pharmaceutical applications for solubilization of drugs or drug delivery [48], it would be interesting to investigate its effects to the photodecarboxylation reaction. Therefore, we investigated photochemical reactivity of phthalimide derivatives 1–3 (Figure 1) in solution without β-CD and in the β-CD inclusion complexes. Phthalimides 1–3 yield azomethine ylides 1AMY-3AMY that are anticipated to react with acrylonitrile (AN) in [3 + 2] cycloadditions, which should be affected by β-CD.
![[1860-5397-16-110-1]](/bjoc/content/figures/1860-5397-16-110-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Phthalimide derivatives 1–3 and the corresponding azomethine ylides 1AMY-3AMY.
Figure 1: Phthalimide derivatives 1–3 and the corresponding azomethine ylides 1AMY-3AMY.
Results and Discussion
Phthalimide derivatives 1–3 were prepared according to procedures published in precedent literature [17]. The synthesis involves condensation of phthalic anhydride with unprotected amino acid. The synthetic procedures and characterization of compounds are reported in Supporting Information File 1. Irradiation of 1 was conducted first in CH3CN in the presence of AN, with or without addition of H2O (Table 1). Phthalimide 1 most probably undergoes decarboxylation delivering 1AMY from the S1 state [49]. In CH3CN, 1AMY decays with a rate constant of 2.9 × 106 M−1 s−1, and reacts with methyl acrylate in [3 + 2] cycloaddition with the rate constant 2.7 × 107 M−1 s−1 [49]. Protic solvents such as CH3OH or H2O quench azomethyine ylides, giving formal 1,4 H-shifted products. Thus, in the presence of H2O, no cycloaddition products are anticipated. However, it should be probed if the [3 + 2] cycloaddition can compete with the 1,4 H-shift upon irradiation of an inclusion complex.
Under our conditions, photodecarboxylation of 1 was very inefficient in aprotic solvents, and it gave a mixture of simple decarboxylation product 4, formed from 1AMY by 1,4-H shift, in addition to the cycloadducts 5a and 5b (Scheme 1). Only use of a strong irradiation source such as a high pressure Hg lamp (400 W) provided higher yields of the cycloadducts. On the other hand, upon photolysis in the presence of H2O and a base, the photoreaction is about twenty times more efficient, but it delivers simple decarboxylation product 4 only. Attempts to use β-CD to complex both reactants and enhance the efficiency for the cycloaddition failed. In the photolysis of 1, β-CD had no effect (Table 1), which may be ascribed to a small size of 1 that cannot fit well in the large cavity of β-CD and form a stable complex.
![[1860-5397-16-110-i1]](/bjoc/content/inline/1860-5397-16-110-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Irradiation of 1 in the presence of acrylonitrile (AN).
Scheme 1: Irradiation of 1 in the presence of acrylonitrile (AN).
Table 1: Irradiation conditions, conversions and product ratio for photolysis of 1.a
| irradiation conditions | solvent/ irradiation time | conversion (%) | product ratiob |
|---|---|---|---|
| 300 nm; c(1) = 10 mM, c(AN) = 0.25 M | CH3CN 18 h | 4 | 4/5a/5b = 1:1:1 |
| 300 nm, c(1) = 10 mM, c(AN) = 0.25 M | CH3CN/H2O (1:3) 18 h | 100 | 4/5a/5b = 1:0:0 |
| Hg-HP, c(1) = 10 mM, c(AN) = 0.10 M | CH3CN 18 h | 42 | 4/5a/5b = 2:1:1 |
| 300 nm, c(1) = 0.8 mM, c(AN) = 0.64 M | CH3CN/H2O (1:3) 1 h | 97 | 4/5a/5b = 1:0:0 |
| 300 nm, c(1) = 0.8 mM, c(AN) = 0.25 M, c(β-CD) = 0.08 mM | CH3CN/H2O (1:3) 1 h | 96 | 4/5a/5b = 1:0:0 |
aIrradiations were conducted in CH3CN, or CH3CN-H2O (3:1 v/v) in the presence of a base K2CO3 to deprotonate the acid. Irradiated in a Rayonet reactor using 12 lamps (1 lamp – 8 W) with the output at 300 nm, or by use of a high pressure mercury lamp (400 W Hg-HP). bThe product ratio determined by NMR.
Molecules 2 and 3 are larger, and anticipated to form more stable inclusion complexes with β-CD. Therefore, we performed 1H NMR titrations to determine stability constants for the inclusion complexes 2@β-CD and 3@β-CD. The NMR titration for 2 with β-CD was carried out in CD3CN/D2O (3:7 v/v). The addition of β-CD to the solution of 2 induced downfield shifts of the H-signals corresponding to the cyclohexane 2 and 6 positions, as well as H-signals of the phthalimide, which changed from a singlet to a multiplet (Figure S1 in Supporting Information File 1). The spectral changes are in accordance with the formation of an inclusion complex 2@β-CD, with the dynamics for the complexation faster than the NMR time-scale (millisecond). Nonlinear regression analysis of the chemical shifts depending on the β-CD concentration, to a complexation model with 1:1 stoichiometry, showed good correlation (Figure 2 and Figure S2 in Supporting Information File 1), with the stability constant for 2@β-CD K = 190 ± 50 M−1. Formation of the inclusion complex was also confirmed by a NOESY spectrum where an interaction between the phthalimide H-atoms and the β-CD was observed (Figure S3 in Supporting Information File 1). Note that a compound similar to 2, but with an amino instead of the carboxylic functional group, forms an almost 35 times more stable complex with β-CD (4-amino-N-cyclohexylphthalimide, K = 6200 M−1) [50].
![[1860-5397-16-110-2]](/bjoc/content/figures/1860-5397-16-110-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Dependence of the chemical shift of the H-atom at the cyclohexane 2 position in compound 2 on the β-CD concentration. Dots are experimental values and the red line corresponds to the calculated values by the WINEQNMR program [51] according to the model for the formation of 1:1 stoichiometry of the inclusion complex 2@β-CD.
Figure 2: Dependence of the chemical shift of the H-atom at the cyclohexane 2 position in compound 2 on the β...
An analogous titration in CD3CN/D2O (3:7 v/v) was also conducted for 3 with β-CD. The addition of β-CD to the solution of 3 induced downfield shifts of the signal corresponding to the adamantane 6 position and the phthalimide signals, which changed from singlet to a multiplet (Figure S4 in Supporting Information File 1). Nonlinear regression analysis of the chemical shifts to the β-CD concentration did not provide a good quality of the fit to the model involving 1:1 complex formation (Figure S5 in Supporting Information File 1). However, the approximated association constant for 3@β-CD, K = 2600 ± 600 M−1, is similar to the known association constants for different adamantane derivatives with β-CD (K = 103–105 M−1) [52], in agreement with the anticipated good fit of the adamantane moiety in 3 to the β-CD cavity.
After demonstrating the formation of inclusion complexes 2@β-CD and 3@β-CD, we investigated the possibility for the formation of ternary complexes with AN. Therefore, we titrated solutions of 2 or 3 with AN. The solutions of 2 or 3 contained a high concentration of β-CD to assure that the phthalimide derivative was in the inclusion complex 2@β-CD or 3@β-CD, respectively. The addition of AN to the CD3CN/D2O (3:7 v/v) solution of 2@β-CD induced changes in the spectra, opposite to those observed upon formation of 2@β-CD (compare Figures S1 and S6 in Supporting Information File 1). The spectral changes are in accordance with the formation of a ternary complex AN@2@β-CD (Scheme 2). However, they may also indicate that excess of AN added to the solution competitively binds to β-CD forming a complex AN@β-CD and inducing dissociation of the 2@β-CD. If we assume a model for the complex formation in the stoichiometry 1:1:1, the nonlinear regression analysis of the chemical shift changes depending on the AN concentration and provided a poor fit with the estimated K2 value of 0–6 M−1 (Figure S7 in Supporting Information File 1).
![[1860-5397-16-110-i2]](/bjoc/content/inline/1860-5397-16-110-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Complexation of 2 with β-CD, and formation of a ternary complex AN@2@β-CD.
Scheme 2: Complexation of 2 with β-CD, and formation of a ternary complex AN@2@β-CD.
We investigated also the possibility for the formation of the ternary complex AN@3@β-CD. Therefore, we titrated the solution of 3@β-CD in CD3CN/D2O (3:7 v/v) with AN, whereupon spectral changes were observed (Figure S8 in Supporting Information File 1). The signal of the adamantane H-atom at the adamantane position 6 experienced a downfield shift, whereas the phthalimide signals experienced an upfield shifts. Although the changes were small, we tried to process them using nonlinear regression analysis and model for the complex formation with 1:1:1 stoichiometry. The fit was of poor quality, but it provided an estimation of the constant with the value of K2 = 0–7 M−1 (Figure S9 in Supporting Information File 1).
The NMR titrations did not provide a clear evidence that ternary complexes were formed. However, formation of ternary complexes should affect the photochemical reactivity of 2 and 3 and cycloadditions of the corresponding azomethine ylides with AN. Therefore, we performed irradiation of solutions containing 2 and AN, 3 and AN, or the corresponding complexes 2@β-CD and 3@β-CD with AN. Scheme 3 and Scheme 4 show products formed in the photochemical reactions, whereas ratio of photoproducts obtained is given in Table 2.
![[1860-5397-16-110-i3]](/bjoc/content/inline/1860-5397-16-110-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photochemistry of 2 in the presence of AN, with or without β-CD.
Scheme 3: Photochemistry of 2 in the presence of AN, with or without β-CD.
![[1860-5397-16-110-i4]](/bjoc/content/inline/1860-5397-16-110-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Photochemistry of 3 in the presence of AN, with or without β-CD.
Scheme 4: Photochemistry of 3 in the presence of AN, with or without β-CD.
Table 2: Irradiation conditions, conversions and product ratio after photolysis of 2 or 3.a
| irradiation conditions | solvent | conversion (%) /unidentified products (%) | product ratiob |
|---|---|---|---|
| 2 | CH3CN | 99/46 | 6/7/8/9 = 25:0:1:0 |
| 2 | CH3CN/H2O (1:1) | 99/70 | 6/7/8/9 = 2.6:0:1:0 |
| 2, c(AN) = 0.78 M | CH3CN | 45/40 | 6/7/8/9 = 2:1:2:0 |
| 2, c(AN) = 0.78 M | CH3CN/H2O (1:1) | 99/61 | 6/7/8/9 = 1.5:0:1:0 |
| 2, c(β-CD) = 22 mM | CH3CN/H2O (1:1) | 99/69 | 6/7/8/9 = 4:0:1:0 |
| 2, c(AN) = 0.78 M, c(β-CD) = 22 mM | CH3CN/H2O (1:1) | 58/36 | 6/7/8/9 = 4.5:1:3:2.5 |
| 3 | CH3CN | 10/7 | 10/11/12/13/14 = 1:0:0:0:0 |
| 3 | CH3CN/H2O (1:1) | 52/50 | 10/11/12/13/14 = 1:0:0:0:0 |
| 3, c(K2CO3) = 0.7 mM | CH3CN/H2O (1:1) | 55/5 | 10/11/12/13/14 = 1:0:0:0:0 |
| 3, c(AN) = 0.74 M, | CH3CN | 33/2 | 10/11/12/13/14 = 1:5:9:8:3 |
| 3, c(AN) = 0.74 M, | CH3CN/H2O (1:1) | 75/56 | 10/11/12/13/14 = 1:5:5:2:1 |
| 3, c(β-CD) = 15 mM | CH3CN/H2O (1:1) | 58/40 | 10/11/12/13/14 = 1:0:5:1:0 |
| 3, c(AN) = 0.74 M, c(β-CD) = 15 mM | CH3CN/H2O (1:1) | 45/21 | 10/11/12/13/14 = 1:1:5:2:1 |
aIrradiations of 2 (c = 2.0 mM) or 3 (c = 1.4 mM) were conducted in CH3CN, or CH3CN/H2O (1:1 v/v), with or without acrylonitrile (AN) and β-CD. All samples were irradiated in a Luzchem reactor using 8 lamps (1 lamp – 8 W) with the output at 300 nm for 30 min (compound 2) or 35 min (compound 3). The detailed procedure can be found in the experimental part. bThe product ratio was determined by HPLC–MS and NMR.
The addition of H2O to the solution of 2 or 3 in CH3CN generally increase the efficiency of the decarboxylation reaction, as well as the efficiency of the secondary photochemical H-abstraction from primarily formed products 6 or 10, giving 8, or 12–14, respectively. Upon addition of AN to the solution of 2 or 3 in CH3CN, in aprotic conditions, 2AMY and 3AMY should be formed and intercepted with AN to yield cycloadducts 7 or 11, respectively. However, the formation of cycloadducts is very inefficient, which may be ascribed to a smaller rate constant for the quenching due to steric hindrance imposed by the bulky cyclohexane or adamantine moiety. Thus, irradiation of 2 gave cycloadducts 7 in ≈9% yield, and 3 gave cycloadducts 11 in ≈6% yield. Furthermore, it is anticipated that the addition of H2O quenches the cycloaddition reaction due to a faster reaction of AMY with H2O then with AN, giving formal 1,4-H-shifted products. Indeed, H2O quenched the cycloaddition of 2AMY with AN, but it did not quench the cycloaddition of 3AMY with AN. Thus, cycloadduct 11 was detected after irradiation of 3 with AN in CH3CN/H2O, but not when a base was added to the solution.
Addition of β-CD did not affect the decarboxylation reaction, since the same conversion to photoproducts was observed in the presence and absence of β-CD. However, for the adamantane derivative in the presence of β-CD, the secondary photochemical H-abstraction became more efficient, resulting in a different product distribution. More efficient H-abstraction reaction in the presence of β-CD have been reported [16].
β-CD affected the cycloaddition reaction of photogenerated AMY with AN. Upon irradiation of 2 with AN in the presence of β-CD, the cycloadduct was formed in ≈5% yield, even though the irradiation was conducted in aqueous solution. The finding suggests that formation of a ternary complex AN@2@β-CD is possible and that photolysis of 2 in such a complex yields 2AMY, which is then readily intercepted with AN in the same complex. Note that cycloadduct 11 was also detected (≈4%), upon irradiation of 3 with AN in the presence of β-CD, suggesting that the photodecarboxylation reaction and subsequent [3 + 2] cycloaddition take place in the ternary complex AN@3@β-CD. However, cycloadduct 11 was formed in a higher yield (≈20%) when 3 was photolyzed with AN in the aqueous solution without β-CD. Although a reason for the different effect of β-CD is not clear, it may be due to a lower stability of the ternary complex AN@3@β-CD, compared to AN@2@β-CD. Namely, the adamantane is a bulky moiety that occupies most of the space in the inclusion complex 3@β-CD, making formation of the ternary complex less likely. Furthermore, if AN@3@β-CD was formed, photogenerated 3AMY@β-CD may not be in the right orientation for the cycloaddition to take place, leading predominantly to the reaction of 3AMY with H2O.
Conclusion
Herein we have demonstrated a proof of principle that β-CD can be used as a molecular container in which two molecules can be complexed, a phthalimide derivative and acrylonitrile, forming a ternary complex. Irradiation of such a complex leads to decarboxylation and formation of the reactive intermediate, azomethine ylide, within the supramolecular host. The subsequent [3 + 2] cycloaddition within the inclusion complex gives heterocyclic cycloadducts, even though it is conducted in aqueous solvent in which ylides have short lifetimes. The reaction needs to be optimized for different substrates with the right choice of host size. However, the proof of principle provides a new idea for the development of supramolecular systems for the tuning of photochemical reactivity.
Experimental
General
1H and 13C NMR spectroscopic data were recorded at room temperature on a Bruker Avance 300 MHz or Bruker Avance 600 MHz spectrometer. CD3OD or CD3CN-D2O were used as deuterated solvent. TMS (1H NMR) or deuterated solvent itself (13C NMR) was used as internal reference. Chemical shifts were reported in ppm. Irradiation experiments were performed in a Rayonet RPR-100 photoreactor equipped with 12 lamps or a Luzchem reactor equipped with 8 lamps with the maximum output at ≈300 nm (1 lamp – 8 W). During the irradiations in the Rayonet reactor, the irradiated solutions were continuously purged with Ar and cooled by a tap water finger condenser. Solvents for the irradiations were of HPLC purity. Chemicals were purchased from the usual commercial sources and used as received. Solvents for chromatographic separations were used from the supplier (p.a. or HPLC grade) as is or were purified by distillation (CH2Cl2). Semipreparative HPLC separations were performed on a Varian Pro Star instrument equipped with a Phenomenex Jupiter C18 5μ 300A column, using CH3OH/H2O + TFA as eluent. HPLC–MS analyses were conducted on an Agilent 1200 Series machine equipped with a DAD detector and a mass spectrometer with a triple quadrupole Agilent 6420 device.
Photochemistry of 2 and 3 under different conditions
A solution of 2 (30 mg, 0.11 mmol) in CH3CN (27.5 mL) was prepared and transferred to 7 quartz cuvettes (3.9 mL into each). Then, CH3CN (3.9 mL) or H2O (3.9 mL) was added to each of the cuvettes, followed by the addition of β-CD (200 mg, 0.176 mmol), acrylonitrile (AN, 0.4 mL, 6.1 mmol) or nothing (see Table 2). Solutions were purged with N2 for 15 min, sealed and irradiated at the same time in a Luzchem reactor at 300 nm (8 lamps) for 30 min. After the irradiation, solutions were extracted with EtOAc (3 × 3 mL), the extracts were dried over MgSO4, filtered and the solvent was removed on a rotary evaporator. The crude reaction mixtures were filtered through a plug of silica gel by use of CH2Cl2/EtOAc as eluent and were analyzed by 1H NMR and HPLC–MS (Table 2).
Alternatively, a solution of 3 (44 mg, 0.135 mmol) in CH3CN (50 mL) was prepared and transferred to 7 quartz cuvettes (7.0 mL into each). Then, CH3CN (7.0 mL) or H2O (7.0 mL) was added to each of the cuvettes, followed by the addition of β-CD (240 mg, 0.21 mmol), AN (0.7 mL, 10.7 mmol), K2CO3 (1.3 mg, 0.009 mmol), or nothing (see Table 2). Solutions were purged with N2 for 15 min, sealed and irradiated at the same time in a Luzchem reactor at 300 nm (8 lamps) for 35 min. After the above described work up, the composition of the irradiated solutions was analyzed by 1H NMR and HPLC–MS (Table 2).
Preparative irradiation of 2 with AN and with β-CD
Phthalimide 2 (110 mg, 0.403 mmol) was dissolved in CH3CN (50 mL) and this solution was added slowly to the solution of β-CD (4.0 g, 3.52 mmol) in H2O (250 mL). The solution was sonicated for 15 min and then AN (10 mL, 152.6 mmol) was added. After sonicating for additional 15 min, the solution was transferred to fifteen quartz test tubes (each containing 20 mL), purged with N2 for 20 min and sealed. The solutions were irradiated for 1 h in a Luzchem reactor using 8 lamps with the output at 300 nm. When the irradiation was completed, the irradiated solutions were combined and extracted with pentane (3 × 50 mL), and then with CH2Cl2 (2 × 50 mL) and EtOAc (2 × 50 mL). The organic extracts were dried over anhydrous Na2SO4, and CH2Cl2 and EtOAc were combined. The solutions were filtered and the solvent was removed on a rotary evaporator. The photoproducts were separated by chromatography on semipreparative HPLC followed by preparative TLC using 5% MeOH/10% Et2O/85% CH2Cl2 and 40% EtOAc/CH2Cl2 as eluent. Compound 6 (11 mg, 10%) was identified by comparison of the spectra with those from precedent literature [53].
HPLC method: 0–10 min (25% H2O/MeOH), 10–30 min (25–0% H2O/MeOH), 30–40 min (MeOH), 40-45 min (0–25% H2O/MeOH).
9b'-Hydroxy-5'-oxo-1',2',5',9b'-tetrahydrospiro[cyclohexane-1,3'-pyrrolo[2,1-a]isoindole]-1'-carbonitrile (7): 2 mg (2%), oily crystals; 1H NMR (CD3OD, 600 MHz) δ 7.81 (dd, J = 1.0, 7.6 Hz, 1H), 7.70 (dt, J = 1.3, 7.6 Hz, 1H), 7.65 (dt, J = 1.3, 7.6 Hz, 1H), 7.48 (dd, J = 1.0, 7.6 Hz, 1H), 4.55 (br s, 3H), 3.48–3.42 (m, 1H), 2.65–2.59 (m, 1H), 2.06–2.00 (m, 2H), 1.86–1.78 (m, 2H), 1.53–1.27 (m, 7H); MS m/z (% relative intensity): 282 (100), 283 (18.4), 284 (1.6).
1,3,4,4a,5,11a-Hexahydro-6H-dibenzo[b,e]azepine-6,11(2H)-dione (8): 4 mg (4%), oily crystals; 1H NMR (CD3OD, 300 MHz) δ 7.83–7.77 (m, 1H), 7.71–7.61 (m, 2H), 7.55–7.49 (m, 1H), 4.18 (d, J =2.4 Hz, 1H), 2.74–2.64 (m, 1H), 2.31–2.20 (m, 1H), 1.97–1.84 (m, 5H), 1.47–1.34 (m, 1H); 13C NMR (CD3OD, 75 MHz) δ 207.1 (s, 1C), 172.2 (s, 1C), 139.1 (s, 1C), 133.3 (s, 1C), 133.2 (d, 1C), 133.1 (d, 1C), 130.0 (d, 1C), 128.8 (d, 1C), 57.3 (d, 1C), 49.7 (d, 1C), 30.1 (t, 1C), 26.2 (t, 1C), 23.7 (t, 1C), 21.3 (t, 1C); MS m/z (% relative intensity): 229 (100), 230 (15.1), 231 (1.1).
1,6-Dioxo-1,4,5,6-tetrahydro-2H-spiro[benzo[c]azocine-3,1'-cyclohexane]-5-carbonitrile (9): 3 mg (3%), oily crystals; 1H NMR (CD3OD, 300 MHz) δ 8.19-8.15 (m, 1H), 8.04–8.00 (m, 1H), 7.80–7.66 (m, 2H), 2.84 (dd, J = 3.3, 12.0 Hz, 1H), 2.50–2.30 (m, 2H), 2.10–1.70 (m, 8H), 1.55–1.35 (m, 2H); 13C NMR (CD3OD, 75 MHz) δ 202.5 (s, 1C), 134.9 (s, 1C), 134.7 (d, 1C), 133.4 (d, 1C), 132.9 (d, 1C), 130.6 (d, 1C), 120.7 (s, 1C, CN), 62.0 (d, 1C), 36.1 (t, 1C), 35.7 (t, 1C), 30.7 (s, 1C), 28.0 (t, 1C), 26.5 (t, 1C), 21.7 (t, 1C), 11.7 (t, 1C), signals for 2 quaternary C-atoms were not observed; MS m/z (% relative intensity): 282 (100), 283 (18.4), 284 (1.6).
Preparative irradiation of phthalimide 3 with AN and with β-CD
Phthalimide 3 (150 mg, 0.461 mmol) was dissolved in CH3CN (100 mL) and this solution was added slowly to the solution of β-CD (5.23 g, 4.61 mmol) in H2O (370 mL). The solution was sonicated for 15 min and then AN (10 mL, 152.6 mmol) was added. After sonicating for additional 15 min, the solution was transferred to twenty quartz test tubes (each containing ≈20 mL), purged with N2 for 20 min and sealed. The solutions were irradiated for 4 h in a Luzchem reactor using 8 lamps with the output at 300 nm. When the irradiation was completed, the irradiated solutions were combined and extracted with pentane (3 × 50 mL), and then with CH2Cl2 (2 × 50 mL) and EtOAc (2 × 50 mL). The organic extracts were dried over anhydrous Na2SO4, and CH2Cl2 and EtOAc were combined. The solutions were filtered and the solvent was removed on a rotary evaporator. The photoproducts were separated by chromatography on a column of silica gel using 2–10% MeOH/CH2Cl2 followed by preparative TLC with 5% MeOH/DCM and 5% MeOH/10% Et2O/DCM. The separation afforded only products 10, 12, 13, and 14, which were identified by comparison of the spectra with literature precedent [54].
Preparative irradiation of phthalimide 3 with AN and without β-CD
A solution of phthalimide 3 (142 mg, 0.450 mmol) and AN (5 mL, 76.33 mmol) in CH3CN (100 mL) was poured to a quartz Erlenmayer flask. The solution was purged with N2 for 30 min and then irradiated for 20 h with continuous stirring. After the irradiation, the solvent was removed on a rotary evaporator and the crude reaction mixture was chromatographed on a column of SiO2 with 0–10% MeOH/CH2Cl2 as eluent, followed by chromatography on a semipreparative HPLC with 0–50% H2O/MeOH, 0.1% TFA as eluent, and finally by preparative TLC with 3% MeOH/CH2Cl2 as eluent.
HPLC method: 0–5 min (35% H2O/MeOH, 0.1% TFA), 5–20 min (35–0% H2O/MeOH, 0.1% TFA), 20–25 min (MeOH), 25–30 min (0−35% H2O/MeOH, 0.1% TFA).
9b'-Methyl-5'-oxo-1',2',5',9b'-tetrahydrospiro[adamantane-2,3'-pyrrolo[2,1-a]isoindole]-1'-carbonitrile (11): 2 mg (2%), oily crystals; 1H NMR (CD3OD, 300 MHz) δ 7.70–7.65 (m, 2H), 7.59 (d, J = 7.6 Hz, 1H), 7.53 (dt, J = 0.6, 7.4 Hz, 1H), 4.03 (d, J = 7.9 Hz, 1H), 2.30–2.26 (m, 1H), 2.04–2.00 (m, 1H), 1.97–1.91 (m, 5H), 1.89–1.85 (m, 3H), 1.80–1.76 (m, 1H); 13C NMR (CD3OD, 75 MHz) δ 208.6 (s, 1C), 135.4 (s, 1C), 134.7 (d, 1C), 132.9 (s, 1C), 130.8 (d, 1C), 124.2 (d, 1C), 123.3 (d, 1C), 98.1 (s, 1C), 71.6 (s, 1C), 39.0 (t, 1C), 37.9 (t, 1C), 35.5 (t, 1C), 35.3 (d, 1C), 34.3 (d, 1C), 33.5 (t, 1C), 33.2 (t, 1C), 30.7 (d, 1C), 28.6 (d, 1C), 28.3 (d, 1C), 27.3 (t, 1C), the singlet corresponding to the CN was not observed; MS m/z (% relative intensity): 334 (100), 335 (22.7), 336 (2.5).
NMR titrations with β-CD
A solution of 2 (c = 7.21 mM), or 3 (c = 2.83 mM) in CD3CN/D2O (3:7 v/v, 1.0 or 0.5 mL, respectively) in NMR tube was titrated with a solution of β-CD (c = 20.5 mM). After each addition of β-CD, an 1H NMR spectrum was recorded. The changes of chemical shifts depending on the β-CD concentration were processed by nonlinear regression analysis using WinEQNMR software [51]. The titration was performed at 25 °C.
NMR titrations with AN
A solution of 2@β-CD (prepared by mixing 2 in the concentration of 2.71 mM with β-CD in the concentration of 8.20 mM), or 3@β-CD (prepared by mixing 3 in the concentration of 1.54 mM with β-CD in the concentration of 20.5 mM) in CD3CN/D2O (3:7 v/v, 1.0 mL) was titrated with acrylonitrile (AN). After each addition of AN, an 1H NMR spectrum was recorded. The changes of chemical shifts depending on the AN concentration were processed by nonlinear regression analysis using WinEQNMR software [51]. The titration was performed at 25 °C.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization of the known compounds, NMR spectra from the titration experiments and copies of 1H and 13C NMR spectra of all compounds. | ||
| Format: PDF | Size: 5.2 MB | Download |
References
-
Kobayashi, S.; Jørgensen, K. A., Eds. Cycloaddition Reactions in Organic Synthesis; WILEY-VCH: Weinheim, 2002. doi:10.1002/3527600256
Return to citation in text: [1] -
Chiacchio, U.; Padwa, A.; Romeo, G. Curr. Org. Chem. 2009, 13, 422–447. doi:10.2174/138527209787582268
Return to citation in text: [1] -
Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366–5412. doi:10.1021/cr5007182
Return to citation in text: [1] -
Martina, K.; Tagliapietra, S.; Veselov, V. V.; Cravotto, G. Front. Chem. (Lausanne, Switz.) 2019, 7, No. 95. doi:10.3389/fchem.2019.00095
Return to citation in text: [1] -
Nájera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594
Return to citation in text: [1] [2] -
Hladíková, V.; Váňa, J.; Hanusek, J. Beilstein J. Org. Chem. 2018, 14, 1317–1348. doi:10.3762/bjoc.14.113
Return to citation in text: [1] [2] -
Ryan, J. H. ARKIVOC 2015, No. i, 160–183. doi:10.3998/ark.5550190.p008.928
Return to citation in text: [1] [2] -
Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765–2809. doi:10.1021/cr040004c
Return to citation in text: [1] -
Yoon, U. C.; Kim, D. U.; Lee, C. W.; Choi, Y. S.; Lee, Y.-J.; Ammon, H. L.; Mariano, P. S. J. Am. Chem. Soc. 1995, 117, 2698–2710. doi:10.1021/ja00115a004
Return to citation in text: [1] -
Yoon, U. C.; Cho, S. J.; Lee, Y.-J.; Mancheno, M. J.; Mariano, P. S. J. Org. Chem. 1995, 60, 2353–2360. doi:10.1021/jo00113a012
Return to citation in text: [1] -
Griesbeck, A. G.; Hoffmann, N.; Warzecha, K.-D. Acc. Chem. Res. 2007, 40, 128–140. doi:10.1021/ar068148w
Return to citation in text: [1] -
Kanaoka, Y. Acc. Chem. Res. 1978, 11, 407–413. doi:10.1021/ar50131a002
Return to citation in text: [1] -
Horvat, M.; Mlinarić-Majerski, K.; Basarić, N. Croat. Chem. Acta 2010, 83, 179–188.
Return to citation in text: [1] -
Basarić, N.; Horvat, M.; Mlinarić-Majerski, K.; Zimmermann, E.; Neudörfl, J.; Griesbeck, A. G. Org. Lett. 2008, 10, 3965–3968. doi:10.1021/ol801362x
Return to citation in text: [1] -
Horvat, M.; Görner, H.; Warzecha, K.-D.; Neudörfl, J.; Griesbeck, A. G.; Mlinarić-Majerski, K.; Basarić, N. J. Org. Chem. 2009, 74, 8219–8231. doi:10.1021/jo901753z
Return to citation in text: [1] -
Cindro, N.; Halasz, I.; Mlinarić-Majerski, K.; Basarić, N. Eur. J. Org. Chem. 2013, 929–938. doi:10.1002/ejoc.201201332
Return to citation in text: [1] [2] -
Mandić, L.; Mlinarić-Majerski, K.; Griesbeck, A. G.; Basarić, N. Eur. J. Org. Chem. 2016, 4404–4414. doi:10.1002/ejoc.201600491
Return to citation in text: [1] [2] -
Sohora, M.; Šumanovac Ramljak, T.; Mlinarić-Majerski, K.; Basarić, N. Croat. Chem. Acta 2014, 87, 431–446. doi:10.5562/cca2482
Return to citation in text: [1] -
Horvat, M.; Mlinarić-Majerski, K.; Griesbeck, A. G.; Basarić, N. Photochem. Photobiol. Sci. 2011, 10, 610–617. doi:10.1039/c0pp00357c
Return to citation in text: [1] -
Šumanovac Ramljak, T.; Sohora, M.; Antol, I.; Kontrec, D.; Basarić, N.; Mlinarić-Majerski, K. Tetrahedron Lett. 2014, 55, 4078–4081. doi:10.1016/j.tetlet.2014.05.118
Return to citation in text: [1] -
Sohora, M.; Vazdar, M.; Sović, I.; Mlinarić-Majerski, K.; Basarić, N. J. Org. Chem. 2018, 83, 14905–14922. doi:10.1021/acs.joc.8b01785
Return to citation in text: [1] -
Boscá, F.; Miranda, M. A. J. Photochem. Photobiol., B 1998, 43, 1–26. doi:10.1016/s1011-1344(98)00062-1
Return to citation in text: [1] -
Boscá, F.; Marín, M.-L.; Miranda, M. A. Photochem. Photobiol. 2001, 74, 637–655. doi:10.1562/0031-8655(2001)074<0637:potnai>2.0.co;2
Return to citation in text: [1] -
Boscá, F.; Marín, M.-L.; Miranda, M. A. In Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W.; Lenci, F., Eds.; CRC Press: Boca Raton, 2004.
Return to citation in text: [1] -
Monti, S.; Sortino, S.; De Guidi, G.; Marconi, G. J. Chem. Soc., Faraday Trans. 1997, 93, 2269–2275. doi:10.1039/a700367f
Return to citation in text: [1] -
Martínez, L. J.; Scaiano, J. C. J. Am. Chem. Soc. 1997, 119, 11066–11070. doi:10.1021/ja970818t
Return to citation in text: [1] -
Cosa, G.; Martínez, L. J.; Scaiano, J. C. Phys. Chem. Chem. Phys. 1999, 1, 3533–3537. doi:10.1039/a903268a
Return to citation in text: [1] -
Laferrière, M.; Sanramé, C. N.; Scaiano, J. C. Org. Lett. 2004, 6, 873–875. doi:10.1021/ol036313r
Return to citation in text: [1] -
Blake, J. A.; Gagnon, E.; Lukeman, M.; Scaiano, J. C. Org. Lett. 2006, 8, 1057–1060. doi:10.1021/ol052953d
Return to citation in text: [1] -
Chuang, Y. P.; Xue, J.; Du, Y.; Li, M.; An, H.-Y.; Phillips, D. L. J. Phys. Chem. B 2009, 113, 10530–10539. doi:10.1021/jp903234m
Return to citation in text: [1] -
Li, M.-D.; Du, Y.; Chuang, Y. P.; Xue, J.; Phillips, D. L. Phys. Chem. Chem. Phys. 2010, 12, 4800–4808. doi:10.1039/b919330h
Return to citation in text: [1] -
Xu, Y.; Chen, X.; Fang, W.-H.; Phillips, D. L. Org. Lett. 2011, 13, 5472–5475. doi:10.1021/ol202182k
Return to citation in text: [1] -
Li, M.-D.; Yeung, C. S.; Guan, X.; Ma, J.; Li, W.; Ma, C.; Phillips, D. L. Chem. – Eur. J. 2011, 17, 10935–10950. doi:10.1002/chem.201003297
Return to citation in text: [1] -
Li, M.-D.; Su, T.; Ma, J.; Liu, M.; Liu, H.; Li, X.; Phillips, D. L. Chem. – Eur. J. 2013, 19, 11241–11250. doi:10.1002/chem.201300285
Return to citation in text: [1] -
Boscá, F.; Miranda, M. A.; Carganico, G.; Mauleón, D. Photochem. Photobiol. 1994, 60, 96–101. doi:10.1111/j.1751-1097.1994.tb05073.x
Return to citation in text: [1] -
Ramamurthy, V.; Inoue,, Y., Eds. Supramolecular Photochemistry; Wiley: Hoboken, 2011. doi:10.1002/9781118095300
Return to citation in text: [1] -
Ramamurthy, V.; Sivaguru, J. Chem. Rev. 2016, 116, 9914–9993. doi:10.1021/acs.chemrev.6b00040
Return to citation in text: [1] -
Aoyama, H.; Miyazaki, K.-I.; Sakamoto, M.; Omote, Y. Tetrahedron 1987, 43, 1513–1518. doi:10.1016/s0040-4020(01)90267-4
Return to citation in text: [1] -
Koodanjeri, S.; Joy, A.; Ramamurthy, V. Tetrahedron 2000, 56, 7003–7009. doi:10.1016/s0040-4020(00)00523-8
Return to citation in text: [1] -
Shailaja, J.; Karthikeyan, S.; Ramamurthy, V. Tetrahedron Lett. 2002, 43, 9335–9339. doi:10.1016/s0040-4039(02)02338-9
Return to citation in text: [1] -
Vízvárdi, K.; Desmet, K.; Luyten, I.; Sandra, P.; Hoornaert, G.; Van der Eycken, E. Org. Lett. 2001, 3, 1173–1175. doi:10.1021/ol0156345
Return to citation in text: [1] -
Inoue, Y.; Dong, F.; Yamamoto, K.; Tong, L.-H.; Tsuneishi, H.; Hakushi, T.; Tai, A. J. Am. Chem. Soc. 1995, 117, 11033–11034. doi:10.1021/ja00149a037
Return to citation in text: [1] -
Inoue, Y.; Wada, T.; Sugahara, N.; Yamamoto, K.; Kimura, K.; Tong, L.-H.; Gao, X.-M.; Hou, Z.-J.; Liu, Y. J. Org. Chem. 2000, 65, 8041–8050. doi:10.1021/jo001262m
Return to citation in text: [1] -
Fukuhara, G.; Mori, T.; Wada, T.; Inoue, Y. Chem. Commun. 2005, 4199–4201. doi:10.1039/b504948b
Return to citation in text: [1] -
Fukuhara, G.; Mori, T.; Wada, T.; Inoue, Y. J. Org. Chem. 2006, 71, 8233–8243. doi:10.1021/jo061389x
Return to citation in text: [1] -
Lu, R.; Yang, C.; Cao, Y.; Wang, Z.; Wada, T.; Jiao, W.; Mori, T.; Inoue, Y. Chem. Commun. 2008, 374–376. doi:10.1039/b714300a
Return to citation in text: [1] -
Lu, R.; Yang, C.; Cao, Y.; Tong, L.; Jiao, W.; Wada, T.; Wang, Z.; Mori, T.; Inoue, Y. J. Org. Chem. 2008, 73, 7695–7701. doi:10.1021/jo801439n
Return to citation in text: [1] -
Uekama, K.; Hirayama, F.; Irie, T. Chem. Rev. 1998, 98, 2045–2076. doi:10.1021/cr970025p
Return to citation in text: [1] -
Takahashi, Y.; Miyashi, T.; Yoon, U. C.; Oh, S. W.; Mancheno, M.; Su, Z.; Falvey, D. F.; Mariano, P. S. J. Am. Chem. Soc. 1999, 121, 3926–3932. doi:10.1021/ja9841862
Return to citation in text: [1] [2] -
Wintgens, V.; Amiel, C. J. Photochem. Photobiol., A 2004, 168, 217–226. doi:10.1016/j.jphotochem.2004.06.002
Return to citation in text: [1] -
Hynes, M. J. J. Chem. Soc., Dalton Trans. 1993, 311–312. doi:10.1039/dt9930000311
Return to citation in text: [1] [2] [3] -
Rekharsky, M. V.; Inoue, Y. Chem. Rev. 1998, 98, 1875–1918. doi:10.1021/cr970015o
Return to citation in text: [1] -
Worlikar, S. A.; Larock, R. C. J. Org. Chem. 2008, 73, 7175–7180. doi:10.1021/jo800936h
Return to citation in text: [1] -
Basarić, N.; Horvat, M.; Franković, O.; Mlinarić-Majerski, K.; Neudörfl, J.; Griesbeck, A. G. Tetrahedron 2009, 65, 1438–1443. doi:10.1016/j.tet.2008.12.010
Return to citation in text: [1]
| 52. | Rekharsky, M. V.; Inoue, Y. Chem. Rev. 1998, 98, 1875–1918. doi:10.1021/cr970015o |
| 16. | Cindro, N.; Halasz, I.; Mlinarić-Majerski, K.; Basarić, N. Eur. J. Org. Chem. 2013, 929–938. doi:10.1002/ejoc.201201332 |
| 53. | Worlikar, S. A.; Larock, R. C. J. Org. Chem. 2008, 73, 7175–7180. doi:10.1021/jo800936h |
| 1. | Kobayashi, S.; Jørgensen, K. A., Eds. Cycloaddition Reactions in Organic Synthesis; WILEY-VCH: Weinheim, 2002. doi:10.1002/3527600256 |
| 2. | Chiacchio, U.; Padwa, A.; Romeo, G. Curr. Org. Chem. 2009, 13, 422–447. doi:10.2174/138527209787582268 |
| 21. | Sohora, M.; Vazdar, M.; Sović, I.; Mlinarić-Majerski, K.; Basarić, N. J. Org. Chem. 2018, 83, 14905–14922. doi:10.1021/acs.joc.8b01785 |
| 5. | Nájera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594 |
| 6. | Hladíková, V.; Váňa, J.; Hanusek, J. Beilstein J. Org. Chem. 2018, 14, 1317–1348. doi:10.3762/bjoc.14.113 |
| 7. | Ryan, J. H. ARKIVOC 2015, No. i, 160–183. doi:10.3998/ark.5550190.p008.928 |
| 22. | Boscá, F.; Miranda, M. A. J. Photochem. Photobiol., B 1998, 43, 1–26. doi:10.1016/s1011-1344(98)00062-1 |
| 23. | Boscá, F.; Marín, M.-L.; Miranda, M. A. Photochem. Photobiol. 2001, 74, 637–655. doi:10.1562/0031-8655(2001)074<0637:potnai>2.0.co;2 |
| 24. | Boscá, F.; Marín, M.-L.; Miranda, M. A. In Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W.; Lenci, F., Eds.; CRC Press: Boca Raton, 2004. |
| 4. | Martina, K.; Tagliapietra, S.; Veselov, V. V.; Cravotto, G. Front. Chem. (Lausanne, Switz.) 2019, 7, No. 95. doi:10.3389/fchem.2019.00095 |
| 17. | Mandić, L.; Mlinarić-Majerski, K.; Griesbeck, A. G.; Basarić, N. Eur. J. Org. Chem. 2016, 4404–4414. doi:10.1002/ejoc.201600491 |
| 18. | Sohora, M.; Šumanovac Ramljak, T.; Mlinarić-Majerski, K.; Basarić, N. Croat. Chem. Acta 2014, 87, 431–446. doi:10.5562/cca2482 |
| 19. | Horvat, M.; Mlinarić-Majerski, K.; Griesbeck, A. G.; Basarić, N. Photochem. Photobiol. Sci. 2011, 10, 610–617. doi:10.1039/c0pp00357c |
| 3. | Hashimoto, T.; Maruoka, K. Chem. Rev. 2015, 115, 5366–5412. doi:10.1021/cr5007182 |
| 20. | Šumanovac Ramljak, T.; Sohora, M.; Antol, I.; Kontrec, D.; Basarić, N.; Mlinarić-Majerski, K. Tetrahedron Lett. 2014, 55, 4078–4081. doi:10.1016/j.tetlet.2014.05.118 |
| 14. | Basarić, N.; Horvat, M.; Mlinarić-Majerski, K.; Zimmermann, E.; Neudörfl, J.; Griesbeck, A. G. Org. Lett. 2008, 10, 3965–3968. doi:10.1021/ol801362x |
| 15. | Horvat, M.; Görner, H.; Warzecha, K.-D.; Neudörfl, J.; Griesbeck, A. G.; Mlinarić-Majerski, K.; Basarić, N. J. Org. Chem. 2009, 74, 8219–8231. doi:10.1021/jo901753z |
| 51. | Hynes, M. J. J. Chem. Soc., Dalton Trans. 1993, 311–312. doi:10.1039/dt9930000311 |
| 11. | Griesbeck, A. G.; Hoffmann, N.; Warzecha, K.-D. Acc. Chem. Res. 2007, 40, 128–140. doi:10.1021/ar068148w |
| 16. | Cindro, N.; Halasz, I.; Mlinarić-Majerski, K.; Basarić, N. Eur. J. Org. Chem. 2013, 929–938. doi:10.1002/ejoc.201201332 |
| 9. | Yoon, U. C.; Kim, D. U.; Lee, C. W.; Choi, Y. S.; Lee, Y.-J.; Ammon, H. L.; Mariano, P. S. J. Am. Chem. Soc. 1995, 117, 2698–2710. doi:10.1021/ja00115a004 |
| 10. | Yoon, U. C.; Cho, S. J.; Lee, Y.-J.; Mancheno, M. J.; Mariano, P. S. J. Org. Chem. 1995, 60, 2353–2360. doi:10.1021/jo00113a012 |
| 54. | Basarić, N.; Horvat, M.; Franković, O.; Mlinarić-Majerski, K.; Neudörfl, J.; Griesbeck, A. G. Tetrahedron 2009, 65, 1438–1443. doi:10.1016/j.tet.2008.12.010 |
| 5. | Nájera, C.; Sansano, J. M. Curr. Org. Chem. 2003, 7, 1105–1150. doi:10.2174/1385272033486594 |
| 6. | Hladíková, V.; Váňa, J.; Hanusek, J. Beilstein J. Org. Chem. 2018, 14, 1317–1348. doi:10.3762/bjoc.14.113 |
| 7. | Ryan, J. H. ARKIVOC 2015, No. i, 160–183. doi:10.3998/ark.5550190.p008.928 |
| 13. | Horvat, M.; Mlinarić-Majerski, K.; Basarić, N. Croat. Chem. Acta 2010, 83, 179–188. |
| 51. | Hynes, M. J. J. Chem. Soc., Dalton Trans. 1993, 311–312. doi:10.1039/dt9930000311 |
| 36. | Ramamurthy, V.; Inoue,, Y., Eds. Supramolecular Photochemistry; Wiley: Hoboken, 2011. doi:10.1002/9781118095300 |
| 37. | Ramamurthy, V.; Sivaguru, J. Chem. Rev. 2016, 116, 9914–9993. doi:10.1021/acs.chemrev.6b00040 |
| 25. | Monti, S.; Sortino, S.; De Guidi, G.; Marconi, G. J. Chem. Soc., Faraday Trans. 1997, 93, 2269–2275. doi:10.1039/a700367f |
| 26. | Martínez, L. J.; Scaiano, J. C. J. Am. Chem. Soc. 1997, 119, 11066–11070. doi:10.1021/ja970818t |
| 27. | Cosa, G.; Martínez, L. J.; Scaiano, J. C. Phys. Chem. Chem. Phys. 1999, 1, 3533–3537. doi:10.1039/a903268a |
| 28. | Laferrière, M.; Sanramé, C. N.; Scaiano, J. C. Org. Lett. 2004, 6, 873–875. doi:10.1021/ol036313r |
| 29. | Blake, J. A.; Gagnon, E.; Lukeman, M.; Scaiano, J. C. Org. Lett. 2006, 8, 1057–1060. doi:10.1021/ol052953d |
| 30. | Chuang, Y. P.; Xue, J.; Du, Y.; Li, M.; An, H.-Y.; Phillips, D. L. J. Phys. Chem. B 2009, 113, 10530–10539. doi:10.1021/jp903234m |
| 31. | Li, M.-D.; Du, Y.; Chuang, Y. P.; Xue, J.; Phillips, D. L. Phys. Chem. Chem. Phys. 2010, 12, 4800–4808. doi:10.1039/b919330h |
| 32. | Xu, Y.; Chen, X.; Fang, W.-H.; Phillips, D. L. Org. Lett. 2011, 13, 5472–5475. doi:10.1021/ol202182k |
| 33. | Li, M.-D.; Yeung, C. S.; Guan, X.; Ma, J.; Li, W.; Ma, C.; Phillips, D. L. Chem. – Eur. J. 2011, 17, 10935–10950. doi:10.1002/chem.201003297 |
| 34. | Li, M.-D.; Su, T.; Ma, J.; Liu, M.; Liu, H.; Li, X.; Phillips, D. L. Chem. – Eur. J. 2013, 19, 11241–11250. doi:10.1002/chem.201300285 |
| 35. | Boscá, F.; Miranda, M. A.; Carganico, G.; Mauleón, D. Photochem. Photobiol. 1994, 60, 96–101. doi:10.1111/j.1751-1097.1994.tb05073.x |
| 50. | Wintgens, V.; Amiel, C. J. Photochem. Photobiol., A 2004, 168, 217–226. doi:10.1016/j.jphotochem.2004.06.002 |
| 51. | Hynes, M. J. J. Chem. Soc., Dalton Trans. 1993, 311–312. doi:10.1039/dt9930000311 |
| 49. | Takahashi, Y.; Miyashi, T.; Yoon, U. C.; Oh, S. W.; Mancheno, M.; Su, Z.; Falvey, D. F.; Mariano, P. S. J. Am. Chem. Soc. 1999, 121, 3926–3932. doi:10.1021/ja9841862 |
| 49. | Takahashi, Y.; Miyashi, T.; Yoon, U. C.; Oh, S. W.; Mancheno, M.; Su, Z.; Falvey, D. F.; Mariano, P. S. J. Am. Chem. Soc. 1999, 121, 3926–3932. doi:10.1021/ja9841862 |
| 48. | Uekama, K.; Hirayama, F.; Irie, T. Chem. Rev. 1998, 98, 2045–2076. doi:10.1021/cr970025p |
| 17. | Mandić, L.; Mlinarić-Majerski, K.; Griesbeck, A. G.; Basarić, N. Eur. J. Org. Chem. 2016, 4404–4414. doi:10.1002/ejoc.201600491 |
| 38. | Aoyama, H.; Miyazaki, K.-I.; Sakamoto, M.; Omote, Y. Tetrahedron 1987, 43, 1513–1518. doi:10.1016/s0040-4020(01)90267-4 |
| 39. | Koodanjeri, S.; Joy, A.; Ramamurthy, V. Tetrahedron 2000, 56, 7003–7009. doi:10.1016/s0040-4020(00)00523-8 |
| 40. | Shailaja, J.; Karthikeyan, S.; Ramamurthy, V. Tetrahedron Lett. 2002, 43, 9335–9339. doi:10.1016/s0040-4039(02)02338-9 |
| 41. | Vízvárdi, K.; Desmet, K.; Luyten, I.; Sandra, P.; Hoornaert, G.; Van der Eycken, E. Org. Lett. 2001, 3, 1173–1175. doi:10.1021/ol0156345 |
| 42. | Inoue, Y.; Dong, F.; Yamamoto, K.; Tong, L.-H.; Tsuneishi, H.; Hakushi, T.; Tai, A. J. Am. Chem. Soc. 1995, 117, 11033–11034. doi:10.1021/ja00149a037 |
| 43. | Inoue, Y.; Wada, T.; Sugahara, N.; Yamamoto, K.; Kimura, K.; Tong, L.-H.; Gao, X.-M.; Hou, Z.-J.; Liu, Y. J. Org. Chem. 2000, 65, 8041–8050. doi:10.1021/jo001262m |
| 44. | Fukuhara, G.; Mori, T.; Wada, T.; Inoue, Y. Chem. Commun. 2005, 4199–4201. doi:10.1039/b504948b |
| 45. | Fukuhara, G.; Mori, T.; Wada, T.; Inoue, Y. J. Org. Chem. 2006, 71, 8233–8243. doi:10.1021/jo061389x |
| 46. | Lu, R.; Yang, C.; Cao, Y.; Wang, Z.; Wada, T.; Jiao, W.; Mori, T.; Inoue, Y. Chem. Commun. 2008, 374–376. doi:10.1039/b714300a |
| 47. | Lu, R.; Yang, C.; Cao, Y.; Tong, L.; Jiao, W.; Wada, T.; Wang, Z.; Mori, T.; Inoue, Y. J. Org. Chem. 2008, 73, 7695–7701. doi:10.1021/jo801439n |
© 2020 Sohora et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)