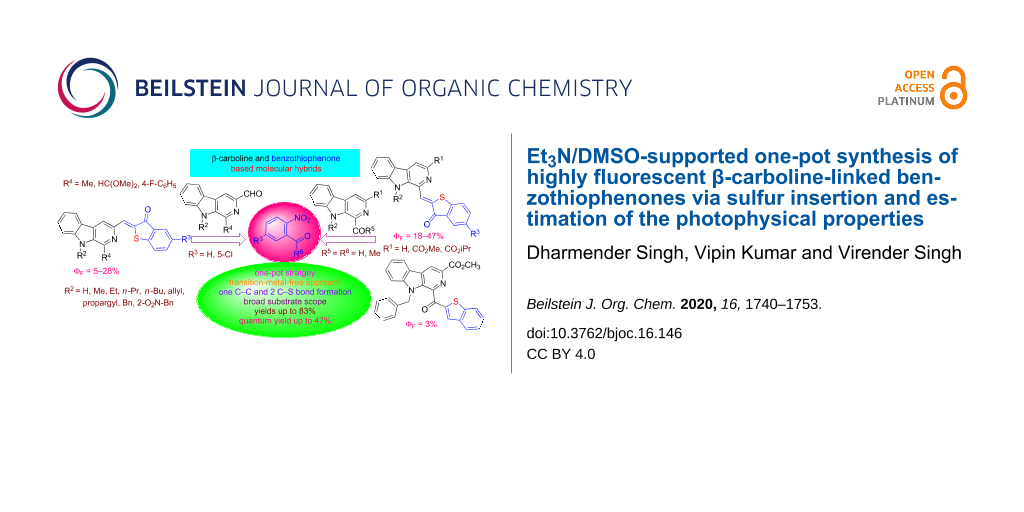
Abstract
A robust transition-metal-free strategy is presented to access novel β-carboline-tethered benzothiophenone derivatives from 1(3)-formyl-β-carbolines using elemental sulfur activated by Et3N/DMSO. This expeditious catalyst-free reaction proceeds through the formation of β-carboline-based 2-nitrochalcones followed by an incorporation of sulfur to generate multifunctional β-carboline-linked benzothiophenones in good to excellent yields. The synthetic strategy could also be extended towards the synthesis of β-carboline-linked benzothiophenes. Moreover, the afforded products emerged as promising fluorophores and displayed excellent light-emitting properties with quantum yields (ΦF) up to 47%.

Graphical Abstract
Introduction
The pyrido[3,4-b]indole moiety, commonly referred as β-carboline, is the core unit of about one quarter of all natural products [1-4] and pharmacologically active compounds endowed with anticancer [5-9], anti-inflammatory, antioxidant, antimalarial, antifungal, and antileishmanial activities (Figure 1) [10-13]. Notably, this privileged scaffold is incorporated in several marketed drugs such as abecarnil, tadalafil, cipargamin, yohimbine, etc. which are used in the treatment of various ailments [14,15]. Apart from their pharmaceutical properties, β-carboline derivatives also found various applications in fields such as organocatalysts, as ligands, and fluorescent probes [16-18]. Importantly, β-carbolines are also used as fluorescence standards. Recently, a novel β-carboline-based fluorescent chemosensor was developed by Batra and co-workers for the quantitative analysis of fluoride ions (F−) at ppb level [19].
![[1860-5397-16-146-1]](/bjoc/content/figures/1860-5397-16-146-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative examples of some commercial drugs and biologically active alkaloids.
Figure 1: Representative examples of some commercial drugs and biologically active alkaloids.
Sulfur-containing organic compounds are broadly associated with numerous bioactive natural products and pharmaceutical drugs [20-22]. Thioaurones (2-benzylidene benzo[b]thiophen-3(2H)-one) are sulfur-containing heterocyclic compounds, an important subclass of flavonoids which were first introduced by O’Sullivan in 1977 [23]. Specifically, thioaurones and their analogs show a variety of biological activities such as anticancer [24], inhibition of tyrosine phosphatase 1B, antioxidant properties, etc. [25-27]. Due to their numerous applications, they have found diverse uses such as thioindigo-like dyes, photoresponsive devices, and photoswitchable biomolecules [28-31]. Moreover, these compounds were also used as synthetic intermediates for various sulfur-containing bioactive molecules (Figure 1) [32-34].
In organic synthesis, aromatic compounds having nitro groups play a vital role as building blocks for the synthesis of nitrogen-containing functional groups and aza-heterocyclic frameworks. However, organic transformations in which aromatic nitro groups act as leaving groups are less reported and require the use of transition-metal catalysts such as Cu, Rh, Pd, etc. [35-37]. Though, several elegant methods have been developed for the synthesis of benzothiophenes, however, these methods rely on the use of organosulfur-based substrates [38-41]. Moreover, these methods are associated with some limitations such as using costly metal catalysts, air-sensitive starting materials, malodorous sulfides or thiols, low yields, and multistep syntheses. To overcome these drawbacks, elemental sulfur has emerged as a surrogate approach, where it can be inserted in situ. In this context, several research groups are actively involved in the development of novel and efficient approaches for the synthesis of sulfur-containing heterocycles [42-46].
In our research endeavors, we have been involved in the exploration of the synthetic potential of 1-formyl-9H-β-carboline (an alkaloid, kumujian C) [47] for preparing chemical libraries of β-carboline-substituted [48-54] and N-fused heterocycles [55,56] which were attributed to the presence of an electrophilic as well as a nucleophilic functionality in this natural product [1]. Encouraged by the applications of β-carboline and benzothiophene motifs in medicinal and materials chemistry, it was envisaged to construct a β-carboline-based novel molecular hybrid containing the benzothiophene moiety (Figure 1) [57,58]. The present study was inspired by the recent findings of Nguyen and co-workers [59-61]. As a part of our ongoing project [62,63], we devised a simple and efficient one-pot practical approach for the construction of β-carboline-tethered benzothiophenone derivatives via incorporation of sulfur. To the best of our knowledge, this is the first report of one-pot synthesis of novel β-carboline-tethered benzothiophenones and evaluation of their light-emitting properties. In this regard, detailed studies are presented and discussed herein.
Results and Discussion
The present study commenced with the synthesis of the β-carboline-based 2-nitrochalcone 1bA which was prepared via a Claisen–Schmidt condensation of 1-formyl-β-carboline (1b) with 2-nitroacetophenone (A) in the presence of KOH (1.05 equiv) in dry MeOH at room temperature (Scheme 1). The analytically pure product was obtained in 86% yield by simple filtration of the precipitate followed by washing with dry MeOH.
![[1860-5397-16-146-i1]](/bjoc/content/inline/1860-5397-16-146-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of β-carboline-linked 2-nitrochalcones.
Scheme 1: Synthesis of β-carboline-linked 2-nitrochalcones.
With the objective to synthesize β-carboline-linked benzothiophenone frameworks, we set up screening of conditions for the reaction between the β-carboline-based 2-nitrochalcone 1bA and elemental sulfur by testing different activators (Table 1). Recent findings revealed that a combination of an aliphatic amine with DMSO activated elemental sulfur for an electrophilic addition to generate thioaurones and benzothiophenes [59]. At 70 °C, the use of DIPEA in combination with DMF as the solvent was found to be an excellent sulfur activator, leading to the formation of the desired product 2bA in 50% yield after a short silica gel column chromatographic separation (Table 1, entry 1). The structure of 2bA was confirmed on the basis of spectroscopic data. The 1H NMR spectrum displayed a singlet for one methine proton at δ 8.69 ppm and the presence of additional nine aromatic protons for the β-carboline and benzothiophenone frameworks indicated the formation of the desired product. When elemental sulfur and DIPEA were used in DMSO, a significant increase in the yield (73%) was observed (Table 1, entry 2). At this stage, we realized that DMSO was a better choice for this transformation as the reaction required a shorter time and afforded the product 2bA in a better yield. Then, other amines such as NMP, Et3N, DBU, and DABCO were also investigated. The use of NMP as an activator in DMSO yielded the desired product in only 65% yield (Table 1, entry 3). Interestingly, Et3N in combination with DMSO at 70 °C afforded the anticipated product 2bA in 78% yield within a short span of 20 min (Table 1, entry 4). The reaction at 90 °C gave product 2bA in 76% yield, however, the same reaction performed at 30 °C was found to be sluggish and complete conversion could not be achieved even after 12 h (Table 1, entries 5 and 6). We also observed that the reaction in the absence of Et3N failed to generate the anticipated product 2bA (Table 1, entry 7), which supported the importance of an amine/base with DMSO as an activator. Potassium ethylxanthate [64,65] and sodium sulfide as a sulfur sources in the presence of Et3N in DMSO (Table 1, entries 8 and 9) also did not furnish the desired product 2bA. A decomposition of the product was observed in the case of Na2S. Interestingly, the combination of DABCO and DMSO also afforded the desired product via a clean reaction within 20 min, although only 60% yield of the product was obtained (Table 1, entry 10). This promising result using DABCO encouraged us to explore other amines like DBU, ʟ-proline, pyridine, DMAP, and pyrrolidine as an activator but encouraging results were not obtained (Table 1, entries 11–15). Similarly, the use of KI as an activator also failed to promote the reaction (Table 1, entry 16) [62]. Eventually, we came to the conclusion that a combination of Et3N and DMSO excellently activated elemental sulfur at 70 °C and therefore chose these conditions for the construction of other β-carboline-linked benzothiophenone derivatives.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-146-i9.svg?max-width=637&scale=1.0)
|
||||||
| entry | baseb | sulfur source | solventc | temperature (°C) | time | yieldd (%) of 2bA |
| 1 | DIPEA | S8 | DMF | 70 | 1 h | 50 |
| 2 | DIPEA | S8 | DMSO | 70 | 45 min | 73 |
| 3 | NMP | S8 | DMSO | 80 | 1 h | 65 |
| 4 | Et3N | S8 | DMSO | 70 | 20 min | 78 |
| 5 | Et3N | S8 | DMSO | 90 | 18 min | 76 |
| 6 | Et3N | S8 | DMSO | 30 | 12 h | 45 |
| 7e | – | S8 | DMSO | 90 | 12 h | NR |
| 8e | Et3N | C2H5OCS2K | DMSO | 60 | 10 h | NR |
| 9f | Et3N | Na2S | DMSO | 60 | 20 h | – |
| 10 | DABCO | S8 | DMSO | 60 | 20 min | 60 |
| 11 | DBU | S8 | DMSO | 70 | 40 min | 30 + impurity |
| 12 | L-proline | S8 | DMSO | 70 | 3.5 h | 30 + 1bA |
| 13 | pyridine | S8 | DMSO | 70 | 12 h | NR |
| 14 | DMAP | S8 | DMSO | 70 | 3 h | 35 + 1bA |
| 15 | pyrrolidine | S8 | DMSO | 60 | 3 h | 40 |
| 16e | KI | S8 | DMSO | 70 | 2.5 h | NR |
aAll reactions were performed with 0.12 mmol of 1bA, 0.60 mmol (5.0 equiv) of sulfur powder, and 0.60 mmol (5.0 equiv) of amine/base in 0.5 mL of solvent; b5.0 equiv of amine/base were used except KI (3.0 equiv), L-proline (4.0 equiv), and DMAP (1.5 equiv); cdry solvents were used; disolated yields of the purified product; eNR = no reaction was observed; fdecomposition of starting material was observed.
With the standardized conditions identified, the scope of this domino approach was investigated with diversely substituted β-carboline-based 2-nitrochalcones 1aA-bA, 1dA, and 1hA prepared from aldehydes 1a-b, 1d and 1h in 76–91% yields (Scheme 1). The methodology was found to be general in nature and produced the fluorescent β-carboline-linked benzothiophenone derivatives 2aA-bA, 2dA and 2hA within 15–45 min in DMSO at 70 °C as depicted in Scheme 2. The analytically pure products were obtained in 42–86% yields after a short silica gel column chromatographic purification. It was observed that N-alkyl-β-carboline-based 2-nitrochalcones 1bA, 1dA, and 1hA reacted faster and delivered the products 2bA, 2dA, and 2hA in better yields (78-86%). Conversely, the substrate 1aA bearing a free NH was found to be slow reacting and produced 2aA in a lower yield (42%).
![[1860-5397-16-146-i2]](/bjoc/content/inline/1860-5397-16-146-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of β-carboline-linked benzothiophenone frameworks.
Scheme 2: Synthesis of β-carboline-linked benzothiophenone frameworks.
From the perspective of green chemistry, one-pot reactions are preferred as less waste is generated due to the avoidance of work-up, isolation, and purification of intermediates [66]. Accordingly, the feasibility of a one-pot synthesis of the targeted products was attempted. Therefore, after the formation of the 2-nitrochalcone 1bA, excess of MeOH was decanted, and the crude product was redissolved in 1 mL of DMSO followed by the sequential addition of Et3N (5 equiv) and elemental sulfur (5 equiv). To our pleasure, the reaction at 70 °C smoothly afforded the corresponding β-carboline-linked benzothiophenone derivative 2bA in less than 30 min. More importantly, a clean reaction was observed during the one-pot strategy which surely avoided the isolation of the intermediate (2-nitrochalcone derivative 1bA), and a significant increment in the overall yield of 2bA (from 67% to 74%) was also noted. Similarly, a remarkable improvement in the yields of 2aA (from 32% to 35%), 2dA (from 78% to 83%), and 2hA (from 69% to 78%) was also observed during the one-pot approach. The comparison of the product yields obtained through both approaches are summarized in Scheme 3.
![[1860-5397-16-146-i3]](/bjoc/content/inline/1860-5397-16-146-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Comparison of outcome of one-pot vs two-pot approach.
Scheme 3: Comparison of outcome of one-pot vs two-pot approach.
Having successfully established a one-pot strategy, we next turned our attention to the generality and scope of the method. Interestingly, diversely substituted 1-formyl-β-carbolines 1a–m (except 1k) reacted efficiently with nitroacetophenones A,B in one pot furnishing the anticipated products 2aA–nA, 2bB, and 2hB as depicted in Scheme 4. The synthesized products were purified through silica gel column chromatography and further washed with anhydrous methanol to yield the analytically pure products in 35–83% yields (two-step yield), except for 2kA, which was obtained in trace amounts only. We observed that N-alkyl derivatives 1bA–jA, 1lA–nA, 1bB, and 1hB reacted faster and led to higher product yields. The substrate B bearing a chloro substituent required longer reaction times and afforded the targeted products 2bB and 2hB in slightly lower yields.
![[1860-5397-16-146-i4]](/bjoc/content/inline/1860-5397-16-146-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: One-pot synthesis of β-carboline C-1-tethered benzothiophenone derivatives.
Scheme 4: One-pot synthesis of β-carboline C-1-tethered benzothiophenone derivatives.
Encouraged by the results obtained from the one-pot synthesis of β-carboline C-1 substituted benzothiophenone derivatives, we were interested if the scope of this one-pot strategy could be extended for the synthesis of β-carboline C-3-tethered benzothiophenones (Scheme 5). Thus, the Claisen–Schmidt condensation of 3-formyl-9H-β-carbolines 3a–g [51] with substituted 2-nitroacetophenones (A and B) in the presence of KOH delivered the corresponding 2-nitrochalcones (3aA–gA and 3eB). The in situ-generated β-carboline-based 2-nitrochalcones were further treated with elemental sulfur in the presence of Et3N in DMSO at 70 °C straightforwardly affording the cyclized products 4aA–gA and 4eB in 48–79% yield as presented in Scheme 5.
![[1860-5397-16-146-i5]](/bjoc/content/inline/1860-5397-16-146-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: One-pot synthesis of β-carboline C-3-linked benzothiophenone derivatives.
Scheme 5: One-pot synthesis of β-carboline C-3-linked benzothiophenone derivatives.
It was observed that the reaction time and yields were affected by the nature of the substituent at the C1 (R4) and N-9 (R2) position of β-carboline ring. Substrates bearing a dimethoxymethyl group (3c and 3d) reacted smoothly and within shorter reaction time. Similarly, N-alkyl (R2) 3-formyl-β-carbolines (3b, 3d, 3f, and 3g) also reacted faster and delivered higher yields as compared to free NH derivatives (3a, 3c, and 3e). In case of the dihalogenated product 4eB, a slow reaction accompanied with a low yield was detected due to presence of electron-withdrawing substituents in starting compound B. The slightly lower yields obtained for 2bB, 2hB, and 4eB were possibly due to the low reactivity of substrate B during the condensation process (step 1), as in the cyclization process, the presence of the electron-withdrawing substituents in B seemed to favor the anticipated SNAr mechanism by stabilizing the negatively charged intermediate 10 (Figure 2). Overall, it was noted that the 1-formyl-β-carbolines 1a–m reacted faster and afforded the corresponding products in higher yields as compared to 3-formyl-β-carbolines 3a–g which may be attributed to the higher electrophilicity of the formyl group at C1 position of the β-carboline ring.
Inspired by the results of the above study, it was envisaged to check the generality of this strategy for the synthesis of β-carboline linked benzothiophene derivatives. Accordingly, we employed 1-acetyl-β-carboline 5 [67,68] for Claisen–Schimdt condensation with 2-nitrobenzaldehyde (C) in the presence of Cs2CO3 and anhydrous THF at room temperature. Product 5C was obtained as a major product along with some unidentified impurities. The evaporation of excess solvent (THF) followed by the treatment of the resultant crude, 2-nitrochalcone 5C with Et3N and elemental sulfur in DMSO at 70 °C furnished the expected product 6C, albeit in a low yield (39%) as shown in Scheme 6. It is important to mention that the Claisen–Schmidt condensation of 5 with 2-nitrobenzaldehyde (C) could not be achieved with KOH in MeOH or Cs2CO3 in DMSO.
![[1860-5397-16-146-i6]](/bjoc/content/inline/1860-5397-16-146-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: One-pot synthesis of β-carboline-linked benzothiophene derivative 6C.
Scheme 6: One-pot synthesis of β-carboline-linked benzothiophene derivative 6C.
To probe the reaction mechanism, a control experiment was conducted with model substrate 1bA in the presence of a radical scavenger (TEMPO) to check the possibility of a radical mechanism vs an electrophilic addition of sulfur [69] (Scheme 7). It was observed that the reaction could not be completed even after 24 h in the presence of TEMPO whereas only 20 min were required for completion under standard conditions. Thus, it is assumed that the reaction proceeds through a radical pathway.
![[1860-5397-16-146-i7]](/bjoc/content/inline/1860-5397-16-146-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Control experiment in the presence of a radical scavenger.
Scheme 7: Control experiment in the presence of a radical scavenger.
Based on our observations during the present study and previous reports [69], a plausible mechanism for the formation of the benzothiophenone ring is depicted in Figure 2. It is anticipated that an initial formation of trisulfur radical anion (S3·−) occurs via the reaction of elemental sulfur with triethylamine in DMSO. The addition of the trisulfur radical anion to the double bond in 2-nitrochalcone (1bA) may yield the intermediate 7. The further abstraction of hydrogen in intermediate 7 may result in formation of intermediate 8. The cleavage of the S–S bond in 8 under basic conditions may generate sulfur anion 9. The nucleophilic substitution reaction (SNAr) by transit the sulfur anion in 9 followed by dismissal of the nitrite ion may result in the formation of the β-carboline-tethered benzothiophenone derivative 2bA. It is anticipated that the role of DMSO is to stabilize the ionic intermediates, specifically 10 and to accelerate the transformation.
![[1860-5397-16-146-2]](/bjoc/content/figures/1860-5397-16-146-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Photophysical studies
Fluorescence, offering a nondestructive exceptional technique to monitor a system of interest at the molecular level [70-72], has found wide-ranging applications in several research areas such as medicine, pharmaceutics, biology, environment, and food science [73,74]. Therefore, the light-emitting properties of the novel β-carboline C1 as well as C3-substituted benzothiophenone derivatives 2aA–nA, 2bB, 2hB, 4aA–gA, 4eB, and 6C were evaluated to stimulate their further exploration for possible applications in the field of materials science as chemosensors, ligands, and fluorescent probes. In order to investigate the fluorescence properties, compound 2bA was chosen as the model substrate for optimization of various parameters like time, concentration, and solvent. In order to obtain the maximum emission, various solvents were screened. The synthesized compounds showed best solubility and displayed a maximum intensity in CHCl3 (Supporting Information File 1) as compared to other solvents (DMSO, DMF, and MeOH). No considerable change in the fluorescence intensity of was observed even after 5 h of sample preparation. Next, after careful analysis of concentrations, a 4 µM concentration in CHCl3 was found to be optimal for the photophysical studies of the synthesized derivatives.
The fluorescence quantum efficiency (ΦF) was measured relative to quinine sulfate (ΦR = 0.546 in 0.1 M H2SO4 under 350 nm excitation) as a reference compound. For the measurement of UV–vis absorption and fluorescence emission of the samples, stock solutions of 1.0 mM concentration were prepared using analytical grade CHCl3 as the solvent, and diluted to the final concentration of 4.0 μM. Next, we carefully measured the photophysical properties at room temperature including absorption, excitation, emission, Stokes shift, fluorescence quantum efficiency, molar extinction coefficient, and brightness. The photophysical data of the β-carboline C1 or C3-tethered benzothiophenone derivatives are summarized in Table 2, and their graphical data are depicted in Figure 3 and Figure 4. The quantum yields were calculated based on Equation 1.
![[1860-5397-16-146-3]](/bjoc/content/figures/1860-5397-16-146-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Fluorescence spectra of 2aA–nA, 2bB, 2hB, and 6C.
Figure 3: Fluorescence spectra of 2aA–nA, 2bB, 2hB, and 6C.
![[1860-5397-16-146-4]](/bjoc/content/figures/1860-5397-16-146-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Fluorescence spectra of 4aA–gA, and 4eB.
Figure 4: Fluorescence spectra of 4aA–gA, and 4eB.
![[1860-5397-16-146-i8]](/bjoc/content/inline/1860-5397-16-146-i8.svg?max-width=590&scale=1.18182)
where R means reference and S sample, respectively.
Generally weak fluorescence emissions were observed for the thiophene-based chromophores due to a remarkable spin–orbit coupling which is originating from the heavy atom effect of sulfur [73,74]. It is worth mentioning that the majority of the β-carboline-substituted benzothiophenone derivatives showed good fluorescence. The emission maxima of the fluorophores showed a wide region for fluorescent emissions (λem, 490–582 nm in CHCl3) along with large Stokes shifts (up to 293 nm), excellent quantum yields (up to 47%), and high brightness (up to 11196). The brightness of the fluorophores was calculated by multiplication of the quantum yield (Φ) with its molar extinction coefficient (ε).
In case of the β-carboline C1-substituted benzothiophenones 2aA–nA, 2bB, and 2hB, the substituents R1 and R2 significantly affected the fluorescence of the compounds. It was noted that the fluorescence increased with lengthening of the alkyl chain at N-9 (R2) and followed the order n-Bu > n-Pr > Et > Me > H. The β-carboline derivative with free N–H (N-9), 2aA, showed a low fluorescence quantum yield (ΦF = 18%) in this series (Table 2 and Figure 3). The presence of a benzyl group at R2 (2hA), improved the photophysical properties including a higher quantum yield (ΦF = 47%). With variation of the substituents at the R1 position, a regular pattern of fluorescence was observed, i.e., CO2iPr > CO2Me > H which may be attributed to the electron-withdrawing nature of the substituents (ester group) at C3 position of the β-carboline ring.
Table 2: Photophysical data of β-carboline-tethered benzothiophenone derivatives.
| compound | UV–visa | fluorescence |
Stokes shift
(nm) |
molar extinction coefficient (ε)
(M−1·cm−1) |
brightness | ||
| λEx (nm) | λEm (nm) | intensity (a.u.) | quantum yield (ΦF)b | ||||
| 2aA |
300.50
481.00 |
539.95
540.88 |
47.61
38.78 |
0.186
0.295 |
239.45
59.88 |
13750
6750 |
2557
1991 |
| 2bA |
305.20
487.78 |
531.94
536.86 |
213.81
178.31 |
0.229
0.342 |
226.74
49.08 |
45750
25000 |
10477
8550 |
| 2cA |
305.40
488.18 |
533.89
532.83 |
233.89
193.36 |
0.265
0.347 |
234.49
44.65 |
42250
26250 |
11196
9109 |
| 2dA |
306.13
487.50 |
534.02
534.02 |
182.87
143.78 |
0.274
0.383 |
227.72
47.42 |
31000
17250 |
8494
6607 |
| 2eA |
304.93
487.49 |
531.94
534.02 |
197.94
160.50 |
0.279
0.400 |
227.01
46.53 |
33250
18250 |
9277
7300 |
| 2fA |
303.41
483.69 |
530.89
531.94 |
144.88
121.01 |
0.256
0.374 |
227.48
48.25 |
27750
15500 |
7104
5797 |
| 2gA |
303.61
480.49 |
534.02
535.07 |
121.25
100.11 |
0.221
0.328 |
230.41
54.58 |
28500
15500 |
6298
5084 |
| 2hA |
303.21
482.90 |
532.98
532.83 |
39.57
31.74 |
0.302
0.473 |
229.77
49.93 |
6750
3250 |
2038
1537 |
| 2iA |
306.21
480.08 |
525.07
524.91 |
83.13
67.85 |
0.243
0.337 |
218.86
44.83 |
17000
9500 |
4131
3201 |
| 2jA |
305.89
487.51 |
534.02
534.98 |
181.82
148.43 |
0.304
0.378 |
228.13
47.47 |
29250
18750 |
8892
7087 |
| 2lA |
289.98
492.53 |
542.05
541.08 |
72.89
99.43 |
0.189
0.253 |
252.07
48.55 |
19500
18500 |
3685
4680 |
| 2mA |
311.59
491.81 |
537.01
538.95 |
49.49
66.73 |
0.223
0.366 |
225.42
47.14 |
10250
8000 |
2286
2928 |
| 2nA |
309.56
487.74 |
531.94
531.79 |
104.13
126.29 |
0.213
0.349 |
222.38
44.05 |
22750
16250 |
4846
5671 |
| 2bB |
309.67
486.62 |
531.94
535.97 |
162.21
127.26 |
0.301
0.378 |
222.27
49.35 |
25500
15500 |
7675
5859 |
| 2hB |
308.54
482.71 |
515.78
521.54 |
140.57
116.95 |
0.330
0.258 |
207.24
38.83 |
18750
8000 |
6187
2064 |
| 4aA |
289.82
463.50 |
522.94
512.83 |
37.76
39.76 |
0.052
0.095 |
293.12
49.33 |
60750
24250 |
3159
2304 |
| 4bA |
289.62
380.09 470.77 |
514.64
518.04 517.02 |
69.79
40.58 89.14 |
0.167
0.179 0.219 |
225.02
137.95 46.25 |
22500
10750 17750 |
3757
1924 3887 |
| 4cA |
289.17
465.41 |
514.91
518.05 |
38.82
36.06 |
0.079
0.179 |
225.74
52.64 |
25000
9250 |
1975
1656 |
| 4dA |
293.41
470.59 |
518.05
520.01 |
119.83
126.18 |
0.160
0.291 |
224.64
49.42 |
35750
19500 |
5720
5674 |
| 4eA |
282.56
376.41 466.89 |
521.94
519.85 521.04 |
107.64
63.43 99.98 |
0.111
0.141 0.196 |
239.38
143.44 54.15 |
46750
23500 23250 |
5189
3313 4557 |
| 4fA |
280.74
383.74 473.79 |
524.92
520.89 522.98 |
51.79
29.12 48.80 |
0.080
0.141 0.158 |
244.18
137.15 49.19 |
32500
11750 13750 |
2600
1657 2172 |
| 4gA |
288.53
380.11 471.56 |
514.02
517.01 517.90 |
170.41
86.20 173.56 |
0.252
0.284 0.257 |
225.49
136.90 46.34 |
29500
13000 21500 |
7437
3692 5525 |
| 4eB |
291.19
464.27 |
511.94
508.04 |
36.39
29.47 |
0.150
0.225 |
220.75
43.77 |
12250
4250 |
1837
956 |
| 6C |
263.14
320.64 |
525.96
490.41 |
13.60
4.48 |
0.034
0.016 |
262.82
169.77 |
15000
18750 |
510
300 |
aMeasured at 4 µM concentration in CHCl3; bquantum yields (ΦF) were determined with reference to quinine sulfate.
Interestingly, a similar trend was observed in case of β-carboline C3-substituted benzothiophenone derivatives (4aA–gA and 4eB). Compared to 4aA and 4aC bearing aliphatic substituents at C1 (R4), compound 4eA with aromatic substituent exhibited better fluorescence due to extended conjugation. The effect of R2 substituent in these derivatives (4bA, 4dA, 4fA and 4gA) was also investigated and it was found that N-alkylation improved the photophysical properties along with higher quantum yields (Figure 4). With regard to the impact of R3 substituent, thiopheneone derivatives with chloro substitution (2bB and 4eB) displayed a higher quantum yield than unsubstituted derivatives (2bA and 4eA) as evident from Table 2. Overall, it can be concluded that β-carboline C1 substituted benzothiophenone derivatives exhibited better photophysical properties including high quantum yield, brightness and significant bathochromic shift in the emission wavelengths. In short, β-carboline-substituted benzothiophenone derivatives emerged as excellent fluorophores and displayed remarkable photophysical properties with quantum yield (ΦF) up to 47%. It is believed that these compounds may find applications in materials science and biomedical investigations.
Conclusion
In summary, an efficient synthesis of highly fluorescent β-carboline-linked benzothiophenone derivatives was successfully accomplished through a one-pot metal-free approach for the first time. The transformation could be executed from β-carboline-based 2-nitrochalcones via a one-pot, two-step procedure starting from 1(3)-formyl-β-carbolines (a framework represented by alkaloid kumujian C). The combination of Et3N and DMSO played a vital role in the activation of sulfur resulting in the formation of two C–S bonds in a single operation. This strategy offers several advantages, such as one-pot procedure, operational simplicity, easy purification, use of inexpensive reagents, and wide functional group compatibility. Importantly, the presence of two important pharmacophores along with the exocyclic double bond with Michael acceptor properties in the title compounds offers the opportunity to explore their biological potential. Moreover, these β-carboline-linked benzothiophenones displayed excellent fluorescence properties with quantum yields (ΦF) of up to 47%. Detailed studies to synthesize novel fluorophores with improved optical properties which can easily find application in materials science are underway in our laboratory
Experimental
General experimental procedure for the synthesis of β-carboline-based 2-nitrochalcone derivatives (1aA, 1bA, 1dA and 1hA) as exemplified for compound 1bA. To a stirred solution of KOH (0.033 g, 0.587 mmol) in dry MeOH (4 mL), 2-nitroacetophenone (A, 0.080 mL, 0.587 mmol) was added at room temperature, and the reaction mixture was stirred for 15 min. Thereafter, 1b (0.15 g, 0.560 mmol) was added portionwise and the reaction mixture was allowed to stir for an additional 1 h at room temperature. After completion of the reaction (as monitored by TLC), the precipitate was filtered through a sintered funnel, washed twice with anhydrous MeOH, and dried in vacuum to obtain the analytically pure product 1bA, 0.20 g (86%) as yellow solid.
General experimental procedure for the synthesis of β-carboline C-1-substituted benzothiophenone derivatives (2aA, 2bA, 2dA, and 2hA) as exemplified for compound 2bA. A 10 mL round-bottomed flask was charged with 2-nitrochalcone 1bA (0.20 g, 0.482 mmol), Et3N (0.336 mL, 2.41 mmol), sulfur powder (0.077 g, 2.41 mmol), DMSO (1 mL), and the reaction mixture was stirred at 70 °C for 20 min. After completion of the reaction, as analyzed by TLC, the crude product was directly purified by silica gel column chromatography (CHCl3/MeOH 95:5, v/v) without aqueous treatment to afford 0.15 g of 2bA (78%) as orange solid.
One-pot experimental procedure for the synthesis of β-carboline C-1(3)-substituted benzothiophenone derivatives (2aA–nA, 2bB, 2hB, 4aA–gA, and 4eB) as exemplified for compound 2bA. To a stirred solution of KOH (0.033 g, 0.587 mmol) in dry MeOH (4 mL) in a 10 mL round-bottomed flask; 2-nitroacetophenone (0.080 mL, 0.587 mmol) was added at room temperature and the reaction mixture was stirred for 15 min. Thereafter, methyl 1-formyl-9-methyl-9H-pyrido[3,4-b]indole-3-carboxylate (1b, 0.15 g, 0.560 mmol) was added portionwise and the reaction mixture was allowed to stir for an additional 1 h at room temperature. After completion of the reaction (as detected by TLC), the reaction content was allowed to settle for 5 min, MeOH was decanted, and evaporated under reduced pressure. Thereafter, DMSO (1.5 mL) was added to the crude product 1bA (nitrochalcone) followed by the sequential addition of sulfur powder (0.089 g, 2.80 mmol) and Et3N (0.390 mL, 2.80 mmol) at room temperature. The reaction mixture was stirred at 70 °C for 20 min. After completion of the reaction (as analyzed by TLC), the product 2bA was directly purified through column chromatography on silica gel (CHCl3/MeOH 95:5, v/v) to afford the analytically pure product 2bA as orange solid in 74% yield (two step yield).
One-pot experimental procedure for the synthesis of methyl 1-(benzo[b]thiophene-2-carbonyl)-9H-pyrido[3,4-b]indole-3-carboxylate (6C). To a stirred suspension of Cs2CO3 (0.182 g, 0.560 mmol) in dry THF (4 mL) in a 10 mL round-bottomed flask, methyl 1-acetyl-9-benzyl-9H-pyrido[3,4-b]indole-3-carboxylate (5, 0.10 g, 0.373 mmol) was added and the mixture was stirred for 10 min. Thereafter, 2-nitrobenzaldehyde (C, 0.062 g, 0.410 mmol) was added and the reaction mixture was stirred for additional 2 h at room temperature. After completion of the reaction (TLC), THF was evaporated under reduced pressure. Next, the crude nitrochalcone 5C was re-dissolved in DMSO (1 mL) followed by the addition of sulfur powder (0.060 g, 1.86 mmol) and Et3N (0.260 mL, 1.86 mmol) at room temperature. The reaction mixture was stirred at 70 °C for 1 h. After completion of the reaction, the product was directly purified by silica gel column chromatography (hexane/EtOAc 60:40, v/v) to afford 0.056 g (39%) of 6C as light brown solid (two step yield).
Supporting Information
| Supporting Information File 1: General information, experimental procedures, spectroscopic data, photophysical data, and copies of spectra. | ||
| Format: PDF | Size: 6.4 MB | Download |
Acknowledgements
We are grateful to Dr. Shivani Nain (BITS Pilani, India) for recording some spectral data reported in this paper.
Funding
DS and VK acknowledge the financial assistance in the form of Senior Research fellowship (SRF) from Council of Scientific & Industrial Research (CSIR) and MHRD, New Delhi (India). VS gratefully acknowledges the financial support in the form of a research grant from Science and Engineering Research Board, New Delhi (EMR/2017/000155 and SB/FT/CS-188/2011) and Council of Scientific & Industrial Research (02(0356)/19/EMR-II) New Delhi (India). We also acknowledge the research grant (DST-FIST (CSI-228/2011) provided by Department of Science and Technology, New Delhi.
References
-
Devi, N.; Kumar, S.; Pandey, S. K.; Singh, V. Asian J. Org. Chem. 2018, 7, 6–36. doi:10.1002/ajoc.201700477
Return to citation in text: [1] [2] -
Chen, Y.-F.; Kuo, P.-C.; Chan, H.-H.; Kuo, I.-J.; Lin, F.-W.; Su, C.-R.; Yang, M.-L.; Li, D.-T.; Wu, T.-S. J. Nat. Prod. 2010, 73, 1993–1998. doi:10.1021/np1003627
Return to citation in text: [1] -
Wong, S.-L.; Chang, H.-S.; Wang, G.-J.; Chiang, M. Y.; Huang, H.-Y.; Chen, C.-H.; Tsai, S.-C.; Lin, C.-H.; Chen, I.-S. J. Nat. Prod. 2011, 74, 2489–2496. doi:10.1021/np100874f
Return to citation in text: [1] -
Tran, T. D.; Pham, N. B.; Ekins, M.; Hooper, J. N. A.; Quinn, R. J. Mar. Drugs 2015, 13, 4556–4575. doi:10.3390/md13074556
Return to citation in text: [1] -
Kumar, S.; Singh, A.; Kumar, K.; Kumar, V. Eur. J. Med. Chem. 2017, 142, 48–73. doi:10.1016/j.ejmech.2017.05.059
Return to citation in text: [1] -
Cao, R.; Peng, W.; Wang, Z.; Xu, A. Curr. Med. Chem. 2007, 14, 479–500. doi:10.2174/092986707779940998
Return to citation in text: [1] -
Chen, H.; Gao, P.; Zhang, M.; Liao, W.; Zhang, J. New J. Chem. 2014, 38, 4155–4166. doi:10.1039/c4nj00262h
Return to citation in text: [1] -
Zhang, M.; Sun, D. Anti-Cancer Agents Med. Chem. 2015, 15, 537–547. doi:10.2174/1871520614666141128121812
Return to citation in text: [1] -
He, L.; Li, Y.; Tan, C.-P.; Ye, R.-R.; Chen, M.-H.; Cao, J.-J.; Ji, L.-N.; Mao, Z.-W. Chem. Sci. 2015, 6, 5409–5418. doi:10.1039/c5sc01955a
Return to citation in text: [1] -
Ashok, P.; Ganguly, S.; Murugesan, S. Mini-Rev. Med. Chem. 2013, 13, 1778–1791. doi:10.2174/1389557511313120008
Return to citation in text: [1] -
Fourtillan, J. B.; Fourtillan, M.; Karam, O.; Zunino, F.; Jacquesy, J. C.; Tafani, J. P. Dihydroimidazo[5,1-a]b-carboline derivatives, method for preparing same and use thereof as medicine. U.S. Pat. Appl. US20060089372A1, April 27, 2006.
Return to citation in text: [1] -
Reixach, N.; Crooks, E.; Ostresh, J. M.; Houghten, R. A.; Blondelle, S. E. J. Struct. Biol. 2000, 130, 247–258. doi:10.1006/jsbi.2000.4245
Return to citation in text: [1] -
Ostresh, J. M.; Houghten, R. A. Combinatorial libraries of imidazol-pyrido-indole and imidazol-pyrido-benzothiophene derivatives, methods of making the libraries and compounds therein. U.S. Pat. Appl. US5,856,107A, Jan 5, 1999.
Return to citation in text: [1] -
Sorbera, L. A.; Martin, L.; Leeson, P. A.; Castaner, J. Drugs Future 2001, 26, 15–19. doi:10.1358/dof.2001.026.01.606182
Return to citation in text: [1] -
Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.-C.; Coste, H.; Kirilovsky, J.; Hyafil, F.; Labaudinière, R. J. Med. Chem. 2003, 46, 4525–4532. doi:10.1021/jm030056e
Return to citation in text: [1] -
He, L.; Liao, S. Y.; Tan, C. P.; Lu, Y. Y.; Xu, C. X.; Ji, L. N.; Mao, Z. W. Chem. Commun. 2014, 50, 5611–5614. doi:10.1039/c4cc01461h
Return to citation in text: [1] -
Bai, B.; Shen, L.; Ren, J.; Zhu, H. J. Adv. Synth. Catal. 2012, 354, 354–358. doi:10.1002/adsc.201100592
Return to citation in text: [1] -
Swami, S.; Behera, D.; Agarwala, A.; Verma, V. P.; Shrivastava, R. New J. Chem. 2018, 42, 10317–10326. doi:10.1039/c8nj01851k
Return to citation in text: [1] -
Das, A.; Dighe, S. U.; Das, N.; Batra, S.; Sen, P. Spectrochim. Acta, Part A 2019, 220, 117099. doi:10.1016/j.saa.2019.05.004
Return to citation in text: [1] -
Clayden, J.; MacLellan, P. Beilstein J. Org. Chem. 2011, 7, 582–595. doi:10.3762/bjoc.7.68
Return to citation in text: [1] -
Damani, L. A., Ed. Sulphur-containing drugs and related organic compounds: Chemistry, biochemistry, and toxicology; Ellis Horwood: Chichester, UK, 1989; Vol. 1, Part B.
Return to citation in text: [1] -
De Oliveira Silva, A.; McQuade, J.; Szostak, M. Adv. Synth. Catal. 2019, 361, 3050–3067. doi:10.1002/adsc.201900072
Return to citation in text: [1] -
Réamonn, L. S. S.; O'Sullivan, W. I. J. Chem. Soc., Perkin Trans. 1 1977, 1009–1012. doi:10.1039/p19770001009
Return to citation in text: [1] -
Xiong, R.; Patel, H. K.; Gutgesell, L. M.; Zhao, J.; Delgado-Rivera, L.; Pham, T. N. D.; Zhao, H.; Carlson, K.; Martin, T.; Katzenellenbogen, J. A.; Moore, T. W.; Tonetti, D. A.; Thatcher, G. R. J. J. Med. Chem. 2016, 59, 219–237. doi:10.1021/acs.jmedchem.5b01276
Return to citation in text: [1] -
Malamas, M. S.; Sredy, J.; Moxham, C.; Katz, A.; Xu, W.; McDevitt, R.; Adebayo, F. O.; Sawicki, D. R.; Seestaller, L.; Sullivan, D.; Taylor, J. R. J. Med. Chem. 2000, 43, 1293–1310. doi:10.1021/jm990560c
Return to citation in text: [1] -
Qin, Z.; Kastrati, I.; Chandrasena, R. E. P.; Liu, H.; Yao, P.; Petukhov, P. A.; Bolton, J. L.; Thatcher, G. R. J. J. Med. Chem. 2007, 50, 2682–2692. doi:10.1021/jm070079j
Return to citation in text: [1] -
Fukuda, N.; Ikemoto, T. Synthesis 2015, 47, 3467–3472. doi:10.1055/s-0034-1378746
Return to citation in text: [1] -
Eggers, K.; Fyles, T. M.; Montoya-Pelaez, P. J. J. Org. Chem. 2001, 66, 2966–2977. doi:10.1021/jo0056848
Return to citation in text: [1] -
Steinle, W.; Rück-Braun, K. Org. Lett. 2003, 5, 141–144. doi:10.1021/ol027102+
Return to citation in text: [1] -
Zweig, J. E.; Newhouse, T. R. J. Am. Chem. Soc. 2017, 139, 10956–10959. doi:10.1021/jacs.7b04448
Return to citation in text: [1] -
Hoffmann, K.; Guentner, M.; Mayer, P.; Dube, H. Org. Chem. Front. 2019, 6, 1244–1252. doi:10.1039/c9qo00202b
Return to citation in text: [1] -
Jia, J.; Yu, A.; Ma, S.; Zhang, Y.; Li, K.; Meng, X. Org. Lett. 2017, 19, 6084–6087. doi:10.1021/acs.orglett.7b02916
Return to citation in text: [1] -
Zhang, Y.; Yu, A.; Jia, J.; Ma, S.; Li, K.; Wei, Y.; Meng, X. Chem. Commun. 2017, 53, 10672–10675. doi:10.1039/c7cc04466f
Return to citation in text: [1] -
Géant, P.-Y.; Urban, M.; Remeš, M.; Císařová, I.; Veselý, J. Eur. J. Org. Chem. 2013, 7979–7988. doi:10.1002/ejoc.201300931
Return to citation in text: [1] -
Rostami, A.; Rostami, A.; Ghaderi, A. J. Org. Chem. 2015, 80, 8694–8704. doi:10.1021/acs.joc.5b01248
Return to citation in text: [1] -
Zheng, X.; Ding, J.; Chen, J.; Gao, W.; Liu, M.; Wu, H. Org. Lett. 2011, 13, 1726–1729. doi:10.1021/ol200251x
Return to citation in text: [1] -
Yadav, M. R.; Nagaoka, M.; Kashihara, M.; Zhong, R.-L.; Miyazaki, T.; Sakaki, S.; Nakao, Y. J. Am. Chem. Soc. 2017, 139, 9423–9426. doi:10.1021/jacs.7b03159
Return to citation in text: [1] -
Yan, J.; Xu, J.; Zhou, Y.; Chen, J.; Song, Q. Org. Chem. Front. 2018, 5, 1483–1487. doi:10.1039/c8qo00147b
Return to citation in text: [1] -
Zheng, G.; Ma, X.; Liu, B.; Dong, Y.; Wang, M. Adv. Synth. Catal. 2014, 356, 743–748. doi:10.1002/adsc.201300815
Return to citation in text: [1] -
Wang, X.; Gensch, T.; Glorius, F. Org. Chem. Front. 2016, 3, 1619–1623. doi:10.1039/c6qo00477f
Return to citation in text: [1] -
Xia, X.-F.; He, W.; Zhang, G.-W.; Wang, D. Org. Chem. Front. 2019, 6, 342–346. doi:10.1039/c8qo01190g
Return to citation in text: [1] -
Nguyen, T. B. Adv. Synth. Catal. 2017, 359, 1066–1130. doi:10.1002/adsc.201601329
Return to citation in text: [1] -
Huang, Y.; Zhou, P.; Wu, W.; Jiang, H. J. Org. Chem. 2018, 83, 2460–2466. doi:10.1021/acs.joc.7b03118
Return to citation in text: [1] -
Jiang, X., Ed. Sulfur Chemistry; Topics in Current Chemistry Collections; Springer: Berlin, Germany, 2019. doi:10.1007/978-3-030-25598-5
Return to citation in text: [1] -
Wang, M.; Dai, Z.; Jiang, X. Nat. Commun. 2019, 10, 2661. doi:10.1038/s41467-019-10651-w
Return to citation in text: [1] -
Wang, M.; Fan, Q.; Jiang, X. Org. Lett. 2016, 18, 5756–5759. doi:10.1021/acs.orglett.6b03078
Return to citation in text: [1] -
Ohmoto, T.; Koike, K. Chem. Pharm. Bull. 1984, 32, 3579–3583. doi:10.1248/cpb.32.3579
Return to citation in text: [1] -
Singh, D.; Tiwari, S. K.; Singh, V. New J. Chem. 2019, 43, 93–102. doi:10.1039/c8nj04294b
Return to citation in text: [1] -
Singh, D.; Devi, N.; Kumar, V.; Malakar, C. C.; Mehra, S.; Rattan, S.; Rawal, R. K.; Singh, V. Org. Biomol. Chem. 2016, 14, 8154–8166. doi:10.1039/c6ob01216g
Return to citation in text: [1] -
Devi, N.; Singh, D.; Honey, H.; Mor, S.; Chaudhary, S.; Rawal, R. K.; Kumar, V.; Chowdhury, A. K.; Singh, V. RSC Adv. 2016, 6, 43881–43891. doi:10.1039/c6ra04841b
Return to citation in text: [1] -
Devi, N.; Singh, D.; Kaur, G.; Mor, S.; Putta, V. P. R. K.; Polina, S.; Malakar, C. C.; Singh, V. New J. Chem. 2017, 41, 1082–1093. doi:10.1039/c6nj03210a
Return to citation in text: [1] [2] -
Singh, D.; Devi, N.; Kumar, V.; Malakar, C. C.; Mehra, S.; Rawal, R. K.; Kaith, B. S.; Singh, V. RSC Adv. 2016, 6, 88066–88076. doi:10.1039/c6ra15875g
Return to citation in text: [1] -
Singh, D.; Hazra, C. K.; Malakar, C. C.; Pandey, S. K.; Kaith, B. S.; Singh, V. ChemistrySelect 2018, 3, 4859–4864. doi:10.1002/slct.201800006
Return to citation in text: [1] -
Singh, D.; Sharma, P.; Kumar, R.; Pandey, S. K.; Malakar, C. C.; Singh, V. Asian J. Org. Chem. 2018, 7, 383–394. doi:10.1002/ajoc.201700545
Return to citation in text: [1] -
Singh, D.; Kumar, V.; Devi, N.; Malakar, C. C.; Shankar, R.; Singh, V. Adv. Synth. Catal. 2017, 359, 1213–1226. doi:10.1002/adsc.201600970
Return to citation in text: [1] -
Singh, D.; Sharma, S.; Kumar, M.; Kaur, I.; Shankar, R.; Pandey, S. K.; Singh, V. Org. Biomol. Chem. 2019, 17, 835–844. doi:10.1039/c8ob02705f
Return to citation in text: [1] -
Meunier, B. Acc. Chem. Res. 2008, 41, 69–77. doi:10.1021/ar7000843
Return to citation in text: [1] -
Abdolmaleki, A.; Ghasemi, J. B. Curr. Top. Med. Chem. 2017, 17, 1096–1114. doi:10.2174/1568026616666160927151144
Return to citation in text: [1] -
Nguyen, T. B.; Retailleau, P. Org. Lett. 2018, 20, 186–189. doi:10.1021/acs.orglett.7b03547
Return to citation in text: [1] [2] -
Nguyen, T. B.; Retailleau, P. Org. Lett. 2017, 19, 4858–4860. doi:10.1021/acs.orglett.7b02321
Return to citation in text: [1] -
Nguyen, T. B.; Retailleau, P. Org. Lett. 2017, 19, 3879–3882. doi:10.1021/acs.orglett.7b01766
Return to citation in text: [1] -
Singh, M.; Awasthi, P.; Singh, V. Eur. J. Org. Chem. 2020, 1023–1041. doi:10.1002/ejoc.201901908
Return to citation in text: [1] [2] -
Kumar, V.; Singh, D.; Paul, A. K.; Shrivastava, R.; Singh, V. New J. Chem. 2019, 43, 18304–18315. doi:10.1039/c9nj04256c
Return to citation in text: [1] -
Sangeetha, S.; Sekar, G. Org. Lett. 2017, 19, 1670–1673. doi:10.1021/acs.orglett.7b00462
Return to citation in text: [1] -
Sangeetha, S.; Muthupandi, P.; Sekar, G. Org. Lett. 2015, 17, 6006–6009. doi:10.1021/acs.orglett.5b02977
Return to citation in text: [1] -
Hayashi, Y. Chem. Sci. 2016, 7, 866–880. doi:10.1039/c5sc02913a
Return to citation in text: [1] -
Battini, N.; Padala, A. K.; Mupparapu, N.; Vishwakarma, R. A.; Ahmed, Q. N. RSC Adv. 2014, 4, 26258–26263. doi:10.1039/c4ra01387e
Return to citation in text: [1] -
Mori, T.; Hoshino, S.; Sahashi, S.; Wakimoto, T.; Matsui, T.; Morita, H.; Abe, I. Chem. Biol. 2015, 22, 898–906. doi:10.1016/j.chembiol.2015.06.006
Return to citation in text: [1] -
Zhang, G.; Yi, H.; Chen, H.; Bian, C.; Liu, C.; Lei, A. Org. Lett. 2014, 16, 6156–6159. doi:10.1021/ol503015b
Return to citation in text: [1] [2] -
Liu, J.; Liu, C.; He, W. Curr. Org. Chem. 2013, 17, 564–579. doi:10.2174/1385272811317060003
Return to citation in text: [1] -
Demas, J. N.; Crosby, G. A. J. Phys. Chem. 1971, 75, 991–1024. doi:10.1021/j100678a001
Return to citation in text: [1] -
Pardo, A.; Reyman, D.; Poyato, J. M. L.; Medina, F. J. Lumin. 1992, 51, 269–274. doi:10.1016/0022-2313(92)90077-m
Return to citation in text: [1] -
Rasmussen, S. C.; Sattler, D. J.; Mitchell, K. A.; Maxwell, J. J. Lumin. 2004, 109, 111–119. doi:10.1016/j.jlumin.2004.01.088
Return to citation in text: [1] [2] -
Fujitsuka, M.; Sato, T.; Sezaki, F.; Tanaka, K.; Watanabe, A.; Ito, O. J. Chem. Soc., Faraday Trans. 1998, 94, 3331–3337. doi:10.1039/a806072j
Return to citation in text: [1] [2]
| 67. | Battini, N.; Padala, A. K.; Mupparapu, N.; Vishwakarma, R. A.; Ahmed, Q. N. RSC Adv. 2014, 4, 26258–26263. doi:10.1039/c4ra01387e |
| 68. | Mori, T.; Hoshino, S.; Sahashi, S.; Wakimoto, T.; Matsui, T.; Morita, H.; Abe, I. Chem. Biol. 2015, 22, 898–906. doi:10.1016/j.chembiol.2015.06.006 |
| 69. | Zhang, G.; Yi, H.; Chen, H.; Bian, C.; Liu, C.; Lei, A. Org. Lett. 2014, 16, 6156–6159. doi:10.1021/ol503015b |
| 69. | Zhang, G.; Yi, H.; Chen, H.; Bian, C.; Liu, C.; Lei, A. Org. Lett. 2014, 16, 6156–6159. doi:10.1021/ol503015b |
| 1. | Devi, N.; Kumar, S.; Pandey, S. K.; Singh, V. Asian J. Org. Chem. 2018, 7, 6–36. doi:10.1002/ajoc.201700477 |
| 2. | Chen, Y.-F.; Kuo, P.-C.; Chan, H.-H.; Kuo, I.-J.; Lin, F.-W.; Su, C.-R.; Yang, M.-L.; Li, D.-T.; Wu, T.-S. J. Nat. Prod. 2010, 73, 1993–1998. doi:10.1021/np1003627 |
| 3. | Wong, S.-L.; Chang, H.-S.; Wang, G.-J.; Chiang, M. Y.; Huang, H.-Y.; Chen, C.-H.; Tsai, S.-C.; Lin, C.-H.; Chen, I.-S. J. Nat. Prod. 2011, 74, 2489–2496. doi:10.1021/np100874f |
| 4. | Tran, T. D.; Pham, N. B.; Ekins, M.; Hooper, J. N. A.; Quinn, R. J. Mar. Drugs 2015, 13, 4556–4575. doi:10.3390/md13074556 |
| 16. | He, L.; Liao, S. Y.; Tan, C. P.; Lu, Y. Y.; Xu, C. X.; Ji, L. N.; Mao, Z. W. Chem. Commun. 2014, 50, 5611–5614. doi:10.1039/c4cc01461h |
| 17. | Bai, B.; Shen, L.; Ren, J.; Zhu, H. J. Adv. Synth. Catal. 2012, 354, 354–358. doi:10.1002/adsc.201100592 |
| 18. | Swami, S.; Behera, D.; Agarwala, A.; Verma, V. P.; Shrivastava, R. New J. Chem. 2018, 42, 10317–10326. doi:10.1039/c8nj01851k |
| 42. | Nguyen, T. B. Adv. Synth. Catal. 2017, 359, 1066–1130. doi:10.1002/adsc.201601329 |
| 43. | Huang, Y.; Zhou, P.; Wu, W.; Jiang, H. J. Org. Chem. 2018, 83, 2460–2466. doi:10.1021/acs.joc.7b03118 |
| 44. | Jiang, X., Ed. Sulfur Chemistry; Topics in Current Chemistry Collections; Springer: Berlin, Germany, 2019. doi:10.1007/978-3-030-25598-5 |
| 45. | Wang, M.; Dai, Z.; Jiang, X. Nat. Commun. 2019, 10, 2661. doi:10.1038/s41467-019-10651-w |
| 46. | Wang, M.; Fan, Q.; Jiang, X. Org. Lett. 2016, 18, 5756–5759. doi:10.1021/acs.orglett.6b03078 |
| 14. | Sorbera, L. A.; Martin, L.; Leeson, P. A.; Castaner, J. Drugs Future 2001, 26, 15–19. doi:10.1358/dof.2001.026.01.606182 |
| 15. | Daugan, A.; Grondin, P.; Ruault, C.; Le Monnier de Gouville, A.-C.; Coste, H.; Kirilovsky, J.; Hyafil, F.; Labaudinière, R. J. Med. Chem. 2003, 46, 4525–4532. doi:10.1021/jm030056e |
| 47. | Ohmoto, T.; Koike, K. Chem. Pharm. Bull. 1984, 32, 3579–3583. doi:10.1248/cpb.32.3579 |
| 10. | Ashok, P.; Ganguly, S.; Murugesan, S. Mini-Rev. Med. Chem. 2013, 13, 1778–1791. doi:10.2174/1389557511313120008 |
| 11. | Fourtillan, J. B.; Fourtillan, M.; Karam, O.; Zunino, F.; Jacquesy, J. C.; Tafani, J. P. Dihydroimidazo[5,1-a]b-carboline derivatives, method for preparing same and use thereof as medicine. U.S. Pat. Appl. US20060089372A1, April 27, 2006. |
| 12. | Reixach, N.; Crooks, E.; Ostresh, J. M.; Houghten, R. A.; Blondelle, S. E. J. Struct. Biol. 2000, 130, 247–258. doi:10.1006/jsbi.2000.4245 |
| 13. | Ostresh, J. M.; Houghten, R. A. Combinatorial libraries of imidazol-pyrido-indole and imidazol-pyrido-benzothiophene derivatives, methods of making the libraries and compounds therein. U.S. Pat. Appl. US5,856,107A, Jan 5, 1999. |
| 35. | Rostami, A.; Rostami, A.; Ghaderi, A. J. Org. Chem. 2015, 80, 8694–8704. doi:10.1021/acs.joc.5b01248 |
| 36. | Zheng, X.; Ding, J.; Chen, J.; Gao, W.; Liu, M.; Wu, H. Org. Lett. 2011, 13, 1726–1729. doi:10.1021/ol200251x |
| 37. | Yadav, M. R.; Nagaoka, M.; Kashihara, M.; Zhong, R.-L.; Miyazaki, T.; Sakaki, S.; Nakao, Y. J. Am. Chem. Soc. 2017, 139, 9423–9426. doi:10.1021/jacs.7b03159 |
| 5. | Kumar, S.; Singh, A.; Kumar, K.; Kumar, V. Eur. J. Med. Chem. 2017, 142, 48–73. doi:10.1016/j.ejmech.2017.05.059 |
| 6. | Cao, R.; Peng, W.; Wang, Z.; Xu, A. Curr. Med. Chem. 2007, 14, 479–500. doi:10.2174/092986707779940998 |
| 7. | Chen, H.; Gao, P.; Zhang, M.; Liao, W.; Zhang, J. New J. Chem. 2014, 38, 4155–4166. doi:10.1039/c4nj00262h |
| 8. | Zhang, M.; Sun, D. Anti-Cancer Agents Med. Chem. 2015, 15, 537–547. doi:10.2174/1871520614666141128121812 |
| 9. | He, L.; Li, Y.; Tan, C.-P.; Ye, R.-R.; Chen, M.-H.; Cao, J.-J.; Ji, L.-N.; Mao, Z.-W. Chem. Sci. 2015, 6, 5409–5418. doi:10.1039/c5sc01955a |
| 38. | Yan, J.; Xu, J.; Zhou, Y.; Chen, J.; Song, Q. Org. Chem. Front. 2018, 5, 1483–1487. doi:10.1039/c8qo00147b |
| 39. | Zheng, G.; Ma, X.; Liu, B.; Dong, Y.; Wang, M. Adv. Synth. Catal. 2014, 356, 743–748. doi:10.1002/adsc.201300815 |
| 40. | Wang, X.; Gensch, T.; Glorius, F. Org. Chem. Front. 2016, 3, 1619–1623. doi:10.1039/c6qo00477f |
| 41. | Xia, X.-F.; He, W.; Zhang, G.-W.; Wang, D. Org. Chem. Front. 2019, 6, 342–346. doi:10.1039/c8qo01190g |
| 24. | Xiong, R.; Patel, H. K.; Gutgesell, L. M.; Zhao, J.; Delgado-Rivera, L.; Pham, T. N. D.; Zhao, H.; Carlson, K.; Martin, T.; Katzenellenbogen, J. A.; Moore, T. W.; Tonetti, D. A.; Thatcher, G. R. J. J. Med. Chem. 2016, 59, 219–237. doi:10.1021/acs.jmedchem.5b01276 |
| 28. | Eggers, K.; Fyles, T. M.; Montoya-Pelaez, P. J. J. Org. Chem. 2001, 66, 2966–2977. doi:10.1021/jo0056848 |
| 29. | Steinle, W.; Rück-Braun, K. Org. Lett. 2003, 5, 141–144. doi:10.1021/ol027102+ |
| 30. | Zweig, J. E.; Newhouse, T. R. J. Am. Chem. Soc. 2017, 139, 10956–10959. doi:10.1021/jacs.7b04448 |
| 31. | Hoffmann, K.; Guentner, M.; Mayer, P.; Dube, H. Org. Chem. Front. 2019, 6, 1244–1252. doi:10.1039/c9qo00202b |
| 73. | Rasmussen, S. C.; Sattler, D. J.; Mitchell, K. A.; Maxwell, J. J. Lumin. 2004, 109, 111–119. doi:10.1016/j.jlumin.2004.01.088 |
| 74. | Fujitsuka, M.; Sato, T.; Sezaki, F.; Tanaka, K.; Watanabe, A.; Ito, O. J. Chem. Soc., Faraday Trans. 1998, 94, 3331–3337. doi:10.1039/a806072j |
| 23. | Réamonn, L. S. S.; O'Sullivan, W. I. J. Chem. Soc., Perkin Trans. 1 1977, 1009–1012. doi:10.1039/p19770001009 |
| 32. | Jia, J.; Yu, A.; Ma, S.; Zhang, Y.; Li, K.; Meng, X. Org. Lett. 2017, 19, 6084–6087. doi:10.1021/acs.orglett.7b02916 |
| 33. | Zhang, Y.; Yu, A.; Jia, J.; Ma, S.; Li, K.; Wei, Y.; Meng, X. Chem. Commun. 2017, 53, 10672–10675. doi:10.1039/c7cc04466f |
| 34. | Géant, P.-Y.; Urban, M.; Remeš, M.; Císařová, I.; Veselý, J. Eur. J. Org. Chem. 2013, 7979–7988. doi:10.1002/ejoc.201300931 |
| 20. | Clayden, J.; MacLellan, P. Beilstein J. Org. Chem. 2011, 7, 582–595. doi:10.3762/bjoc.7.68 |
| 21. | Damani, L. A., Ed. Sulphur-containing drugs and related organic compounds: Chemistry, biochemistry, and toxicology; Ellis Horwood: Chichester, UK, 1989; Vol. 1, Part B. |
| 22. | De Oliveira Silva, A.; McQuade, J.; Szostak, M. Adv. Synth. Catal. 2019, 361, 3050–3067. doi:10.1002/adsc.201900072 |
| 70. | Liu, J.; Liu, C.; He, W. Curr. Org. Chem. 2013, 17, 564–579. doi:10.2174/1385272811317060003 |
| 71. | Demas, J. N.; Crosby, G. A. J. Phys. Chem. 1971, 75, 991–1024. doi:10.1021/j100678a001 |
| 72. | Pardo, A.; Reyman, D.; Poyato, J. M. L.; Medina, F. J. Lumin. 1992, 51, 269–274. doi:10.1016/0022-2313(92)90077-m |
| 19. | Das, A.; Dighe, S. U.; Das, N.; Batra, S.; Sen, P. Spectrochim. Acta, Part A 2019, 220, 117099. doi:10.1016/j.saa.2019.05.004 |
| 25. | Malamas, M. S.; Sredy, J.; Moxham, C.; Katz, A.; Xu, W.; McDevitt, R.; Adebayo, F. O.; Sawicki, D. R.; Seestaller, L.; Sullivan, D.; Taylor, J. R. J. Med. Chem. 2000, 43, 1293–1310. doi:10.1021/jm990560c |
| 26. | Qin, Z.; Kastrati, I.; Chandrasena, R. E. P.; Liu, H.; Yao, P.; Petukhov, P. A.; Bolton, J. L.; Thatcher, G. R. J. J. Med. Chem. 2007, 50, 2682–2692. doi:10.1021/jm070079j |
| 27. | Fukuda, N.; Ikemoto, T. Synthesis 2015, 47, 3467–3472. doi:10.1055/s-0034-1378746 |
| 73. | Rasmussen, S. C.; Sattler, D. J.; Mitchell, K. A.; Maxwell, J. J. Lumin. 2004, 109, 111–119. doi:10.1016/j.jlumin.2004.01.088 |
| 74. | Fujitsuka, M.; Sato, T.; Sezaki, F.; Tanaka, K.; Watanabe, A.; Ito, O. J. Chem. Soc., Faraday Trans. 1998, 94, 3331–3337. doi:10.1039/a806072j |
| 1. | Devi, N.; Kumar, S.; Pandey, S. K.; Singh, V. Asian J. Org. Chem. 2018, 7, 6–36. doi:10.1002/ajoc.201700477 |
| 48. | Singh, D.; Tiwari, S. K.; Singh, V. New J. Chem. 2019, 43, 93–102. doi:10.1039/c8nj04294b |
| 49. | Singh, D.; Devi, N.; Kumar, V.; Malakar, C. C.; Mehra, S.; Rattan, S.; Rawal, R. K.; Singh, V. Org. Biomol. Chem. 2016, 14, 8154–8166. doi:10.1039/c6ob01216g |
| 50. | Devi, N.; Singh, D.; Honey, H.; Mor, S.; Chaudhary, S.; Rawal, R. K.; Kumar, V.; Chowdhury, A. K.; Singh, V. RSC Adv. 2016, 6, 43881–43891. doi:10.1039/c6ra04841b |
| 51. | Devi, N.; Singh, D.; Kaur, G.; Mor, S.; Putta, V. P. R. K.; Polina, S.; Malakar, C. C.; Singh, V. New J. Chem. 2017, 41, 1082–1093. doi:10.1039/c6nj03210a |
| 52. | Singh, D.; Devi, N.; Kumar, V.; Malakar, C. C.; Mehra, S.; Rawal, R. K.; Kaith, B. S.; Singh, V. RSC Adv. 2016, 6, 88066–88076. doi:10.1039/c6ra15875g |
| 53. | Singh, D.; Hazra, C. K.; Malakar, C. C.; Pandey, S. K.; Kaith, B. S.; Singh, V. ChemistrySelect 2018, 3, 4859–4864. doi:10.1002/slct.201800006 |
| 54. | Singh, D.; Sharma, P.; Kumar, R.; Pandey, S. K.; Malakar, C. C.; Singh, V. Asian J. Org. Chem. 2018, 7, 383–394. doi:10.1002/ajoc.201700545 |
| 55. | Singh, D.; Kumar, V.; Devi, N.; Malakar, C. C.; Shankar, R.; Singh, V. Adv. Synth. Catal. 2017, 359, 1213–1226. doi:10.1002/adsc.201600970 |
| 56. | Singh, D.; Sharma, S.; Kumar, M.; Kaur, I.; Shankar, R.; Pandey, S. K.; Singh, V. Org. Biomol. Chem. 2019, 17, 835–844. doi:10.1039/c8ob02705f |
| 51. | Devi, N.; Singh, D.; Kaur, G.; Mor, S.; Putta, V. P. R. K.; Polina, S.; Malakar, C. C.; Singh, V. New J. Chem. 2017, 41, 1082–1093. doi:10.1039/c6nj03210a |
| 64. | Sangeetha, S.; Sekar, G. Org. Lett. 2017, 19, 1670–1673. doi:10.1021/acs.orglett.7b00462 |
| 65. | Sangeetha, S.; Muthupandi, P.; Sekar, G. Org. Lett. 2015, 17, 6006–6009. doi:10.1021/acs.orglett.5b02977 |
| 62. | Singh, M.; Awasthi, P.; Singh, V. Eur. J. Org. Chem. 2020, 1023–1041. doi:10.1002/ejoc.201901908 |
| 62. | Singh, M.; Awasthi, P.; Singh, V. Eur. J. Org. Chem. 2020, 1023–1041. doi:10.1002/ejoc.201901908 |
| 63. | Kumar, V.; Singh, D.; Paul, A. K.; Shrivastava, R.; Singh, V. New J. Chem. 2019, 43, 18304–18315. doi:10.1039/c9nj04256c |
| 59. | Nguyen, T. B.; Retailleau, P. Org. Lett. 2018, 20, 186–189. doi:10.1021/acs.orglett.7b03547 |
| 57. | Meunier, B. Acc. Chem. Res. 2008, 41, 69–77. doi:10.1021/ar7000843 |
| 58. | Abdolmaleki, A.; Ghasemi, J. B. Curr. Top. Med. Chem. 2017, 17, 1096–1114. doi:10.2174/1568026616666160927151144 |
| 59. | Nguyen, T. B.; Retailleau, P. Org. Lett. 2018, 20, 186–189. doi:10.1021/acs.orglett.7b03547 |
| 60. | Nguyen, T. B.; Retailleau, P. Org. Lett. 2017, 19, 4858–4860. doi:10.1021/acs.orglett.7b02321 |
| 61. | Nguyen, T. B.; Retailleau, P. Org. Lett. 2017, 19, 3879–3882. doi:10.1021/acs.orglett.7b01766 |
© 2020 Singh et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)