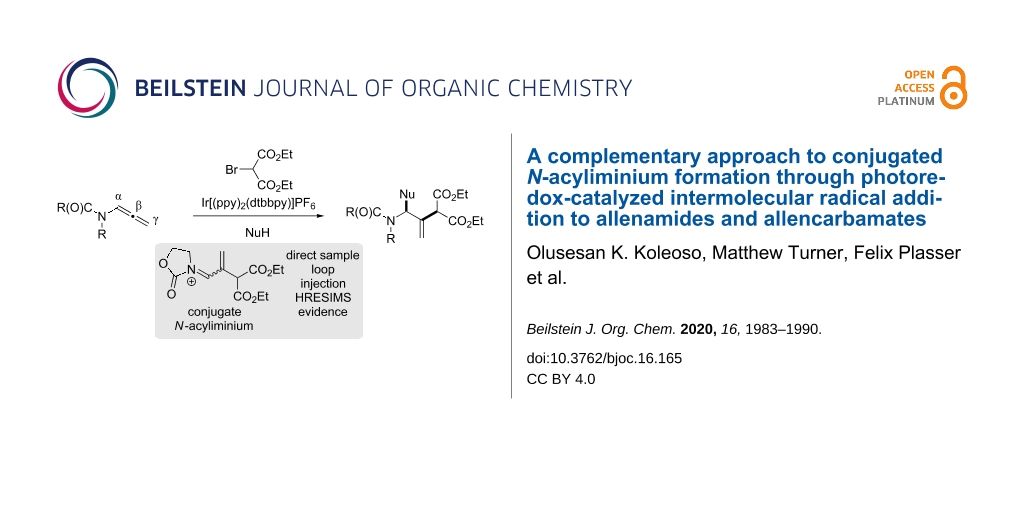
Abstract
An intermolecular radical addition, using photoredox catalysis, to allenamides and allencarbamates is reported. This transformation synthesizes N-acyl-N’-aryl-N,N’-allylaminals, and proceeds by a conjugated N-acyliminium intermediate that previously has principally been generated by electrophilic activation methods. The radical adds to the central carbon of the allene giving a conjugated N-acyliminium that undergoes nucleophilic addition by arylamines and alcohols.

Graphical Abstract
Introduction
Allenamides (Scheme 1, 1) and their congeners have attracted considerable attention over the past 15 years due to their characteristic reactivity profiles [1-4]. The reactivity that an allenamide can display is distinct from a traditional allene due to the presence of an amide unit attached at the α-carbon. This substituent can donate electron density into the allene, principally onto the central β-carbon, that can be harnessed in subsequent chemical transformations leading to regiochemical confidence in the resulting products (Scheme 1).
![[1860-5397-16-165-i1]](/bjoc/content/inline/1860-5397-16-165-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Electrophilic activation of allenamides.
Scheme 1: Electrophilic activation of allenamides.
Importantly, this unique reactivity has underpinned allenamide chemistry and led to the development of a number of innovative transformations, including cycloadditions [5-9], intramolecular cyclizations and intermolecular addition reactions [10-18], as well as the use of the allenamide building block in natural product synthesis [1].
Addition reactions of allenamides, which can also encompass intramolecular cyclizations, are typically promoted by the electrophilic activation of the β-carbon of the allenamide. This can be achieved using various electrophilic methods, including the use of Brönsted acids [15,19-22], halogenation sources [16,18,23-28], by means of oxidation [20,29-31] or through the use of a transition metal such as Au(I) [8,10-16,32-36]. The reaction of the allenamide with an electrophilic source promotes the formation of a conjugated N-acyliminium intermediate 2 [37-40] that subsequently undergoes an addition reaction with a suitable nucleophile (Scheme 1).
Importantly, the electrophilic mediated formation of this key intermediate 2 have been the cornerstone for allenamide chemistry over the past 15 years. Therefore, given the importance of the allenamide building block we sought a new synthetic methodology that could generate this conjugated N-acyliminium intermediate 2, that would not be dependent on conventional electrophilic activation modes (Scheme 2).
![[1860-5397-16-165-i2]](/bjoc/content/inline/1860-5397-16-165-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The planned intramolecular radical addition to allenamides generating the conjugated N-acyliminium intermediate 14.
Scheme 2: The planned intramolecular radical addition to allenamides generating the conjugated N-acyliminium ...
In principle, a new method for its generation could greatly enhance the value of the allenamide building block, as well as potentially unlocking new chemical reactivity yet unseen in the context of electrophilic activation. We envisaged that a route to the conjugated N-acyliminium intermediate could be achieved via an intermolecular addition of an electrophilic radical to an allenamide (1) followed by an oxidation process on the subsequently formed radical (Scheme 3) [41]. The thought process behind this approach is based on three observations; (i) Akita and co-workers disclosure on the photoredox-catalyzed oxytrifluoromethylation of allenes 6 to give 2-trifluoromethylated allyl acetates 7 [42-44]; (ii) that intramolecular radical cyclization of allenamides have been reported by Hsung and co-workers, where the radical adds principally to the central carbon of the allene (10) [45]; and (iii) the photoredox-catalyzed addition of radicals to enamides reported by Masson [46-49], and more recently by our own laboratory in the synthesis of N,N’-aminals [50]. Therefore, photoredox-catalysis would be employed to generate an electrophilic radical that would add to the central carbon of the allenamide 1 to give a transient radical 13, whose oxidation, facilitated by the photoredox catalyst [47,48], would provide the conjugated N-acyliminium 14. The iminium 14 could then undergo traditional nucleophilic addition giving the addition product. Given that the iridium complex Ir[(ppy)2(dtbbpy)]PF6 had been effective [47,48,50] in this radical/cationic pathway with enamides we would now like to report our preliminary findings on the performance of allenamides. Specifically, the electron-withdrawing group on the allenamide and the nucleophile is examined. We provide evidence for the formation of the N-acyliminium intermediate through direct sample loop and flow injection analysis by HRESIMS, and DFT analysis of the N-acyliminium intermediate is provided to explain the addition product distribution.
![[1860-5397-16-165-i3]](/bjoc/content/inline/1860-5397-16-165-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Photoredox Ir-catalyzed intermolecular addition of bromide 18 and aniline 16 to allenamide 15.
Scheme 3: Photoredox Ir-catalyzed intermolecular addition of bromide 18 and aniline 16 to allenamide 15.
Results and Discussion
We began our study by using the iridium catalyst Ir[(ppy)2(dtbbpy)]PF6 (17) and the reaction conditions used in the addition of amine nucleophiles to enamides [48,50]. By exploiting these reaction conditions, using diethyl bromomalonate (18), 4-anisidine (16) as the amine nucleophile, and allenamide 15 we were able to isolate N-acyl-N’-aryl-N,N’-allylaminal 19 as the sole identifiable product in 54% yield.
All of the starting allenamide 15 was consumed in the reaction, but the isolation of 19 proved very challenging and we can account for this instability by the identification of 14a in the 1H NMR spectrum of the crude reaction mixture. A comparable instability profile was observed in the formation of N-acyl-N’-aryl-N,N’-aminals derived from enamides [50]; however, to our knowledge, there are no reports of the isolation of analogous N-acyl-N’-aryl-N,N’-allylaminal substrates in the literature, consequently, these specific conditions were not optimized [51]. The evidence for the formation of 19 was the appearance of the methylene protons in 1H NMR at δ 5.46 (d, J = 1.6 Hz, 1H) and δ 5.45 (d, J = 2.0 Hz, 1H) ppm, respectively. Crucially, it is apparent that using the photoredox conditions described in Scheme 3, the aniline nucleophile adds primarily at the α-position of the allenamide 15; this is in contrast to the archetypal electrophilic activation modes where comparable nucleophiles add to the γ-position [37,39,40].
An examination of the allenamide unit under these conditions is shown in Scheme 4, and the six allenylamides/sulfonamides (15, 21–25) were prepared using known conditions [52,53]. The allenamides derived from pyrrolidinone (21), piperidinone (22) and oxazolidinone (15) with 2,4-dimethylaniline all performed adequately in this reaction giving their N,N’-allylaminal products 26, 27 and 28, respectively. The isolated yields once again reflected the sensitivity of the N,N’-allylaminal functional group. The aminosulfonyl allenyl 23 failed to provide any discernable product, with only a complex mixture, as identified by 1H NMR, being isolated. Significantly, in the 1H NMR spectra of this complex mixture we observed complete fragmentation of the sulfonamide unit. Two chiral allenamides (24 and 25) were exposed to the photoredox conditions with 2,4-dimethylaniline, and it was observed that the predominant product in each case was Z-30 and Z-31, respectively, which presumably result from γ-addition of the nucleophile. This was confirmed by 1H NMR NOE analysis of Z-30, where an enhancement between the enamide proton and the methylene proton were observed, as shown in Scheme 4.
![[1860-5397-16-165-i4]](/bjoc/content/inline/1860-5397-16-165-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction scope (a) allenamide; (b) arylamine nucleophile; (c) alcohol nucleophile.
Scheme 4: Reaction scope (a) allenamide; (b) arylamine nucleophile; (c) alcohol nucleophile.
Given our interest in the N,N’-aminal functionality [50], we also explored variation of the arylamine nucleophile. 2,4-Dimethylaniline, o-anisole, 2-trifluoromethoxyaniline and 3,5-ditrifluoromethylaniline were all effective in this transformation, giving their labile N,N’-allylaminal products (32 to 35) in moderate to good isolated yields.
4-Bromo-2-fluoroaniline was also examined as a nucleophile, as we had previously shown this to be an effective aniline platform for developing linezolid analogues, and this delivered two N,N’-allylaminals 36 and 37, respectively. Masson had previously explored oxygen nucleophiles in their photoredox-catalyzed addition to enamides [48]. Consequently, the use of excess methanol provided the addition product 38, resulting from α-addition, in a modest isolated yield. The stability of product 38 was marginal, but an improved stability of the N,O’-allyl product was observed when ethanol was used as the nucleophile giving product 39 in 52% isolated yield. In contrast, isopropanol gave an inseparable mixture of the α- and γ-addition products, 40a/b in 33% isolated yield. As with the case for the formation of 19, all of the starting allenes were consumed in each reaction shown in Scheme 4.
A tentative mechanism for this transformation is described in Scheme 5a. Excitation of the Ir(III) complex 17 provides *Ir(III) that subsequently undergoes reductive quenching by Et3N, delivering Ir(II) [48]. Single electron transfer from Ir(II) to 18 then generates an electrophilic radical R• together with stoichiometric bromide ion; R• subsequently adds to the β-carbon of the allenamide 15, providing an α-aminoallyl radical 41. To support this hypothesis the calculated HOMO of allenamide 15 is shown in Scheme 5b, signifying the increased nucleophilicity at the β-carbon [54]. Mechanistically, the formation of the key conjugated N-acyliminum 14a from α-aminoallyl radical 41 is analogous to the photoredox initiated addition of radicals to enamides [46,48,49]. Consequently, the formation of 14a can results from a SET event between the oxidized Ir(III) [55] and the α-aminoallyl radical 41; a radical propagation pathway through the reduction of 18 was discounted, as the reaction required continuous irradiation.
![[1860-5397-16-165-i5]](/bjoc/content/inline/1860-5397-16-165-i5.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: (a) Tentative mechanism for the photoredox-catalyzed formation of the conjugated N-acyliminium intermediate; (b) the calculated HOMO [54] for allenamide 15; (c) the identification of intermediate 14a by direct sample loop and flow injection HRESIMS analysis.
Scheme 5: (a) Tentative mechanism for the photoredox-catalyzed formation of the conjugated N-acyliminium inte...
To further support this mechanistic hypothesis, we used ESIMS to identify the formation of the conjugated N-acyliminium 14a. The addition of 18 to allenamide 15 under the Ir-catalyzed photoredox conditions in the presence of 4-bromoaniline was monitored by direct sample loop and flow injection analysis by HRESIMS [56,57]. After 5 minutes we observed a peak at m/z 284.1129 that corresponded satisfactorily to the expected iminium complex 14a (m/z 284.1129: calcd for [C13H18NO6]+); this peak persisted at 15, 30, 60 and 120 min time intervals, respectively.
Upon the oxidation of 41 two plausible iminium stereoisomers can be formed, Z-42 and E-42, respectively, with each of these iminium stereoisomers existing in two further conformers designated E-42’ and Z-42’. DFT calculations [54] were performed on all four of these proposed structures, where it was observed that Z-42 was approx. 6 kcal/mol higher in energy, relative to the most stable isomer E-42, which was 1–2 kcal/mol lower in energy than E-42’ and Z-42’, respectively. It is feasible that both E-42 and Z-42’ undergo nucleophilic addition at the α-position giving the observed N,N’-allylaminal product 43. Conversely, addition of a nucleophile at the γ-position of E-42 gives the observed Z-enamide 44; and addition at the γ-position of Z-42’ gives the same observed Z-enamide 44 after C–N bond rotation.
Conclusion
In conclusion, in this letter we have demonstrated the first intermolecular addition of an electrophilic radical, generated under photoredox conditions, to an allenamide building block. The addition of the radical occurs at the central carbon of the allene, giving a conjugated N-acyliminium intermediate after subsequent oxidation. We have established that the conjugated N-acyliminium intermediate can be formed from a broad range of allenamide precursors; additionally, the conjugated N-acyliminium intermediate can undergo nucleophilic addition with an arylamine or alcohol nucleophiles at the α- or γ-position, with the regioselectivity of the addition being controlled by steric factors. Significantly, the formation of the key conjugated N-acyliminium intermediate using these photoredox conditions can be seen as complementary to the well-developed electrophilic activation modes of allenamides. We are currently examining the full mechanism of this transformation, expanding the scope of substrates that can be used in the radical addition step, and alternative fates for the α-N-acyl radical 13 [58-60].
Supporting Information
| Supporting Information File 1: Experimental details, analytical (1H NMR, 13C NMR) and ESIMS data. | ||
| Format: PDF | Size: 3.8 MB | Download |
References
-
Lu, T.; Lu, Z.; Ma, Z.-X.; Zhang, Y.; Hsung, R. P. Chem. Rev. 2013, 113, 4862–4904. doi:10.1021/cr400015d
Return to citation in text: [1] [2] -
Mascareñas, J. L.; Varela, I.; López, F. Acc. Chem. Res. 2019, 52, 465–479. doi:10.1021/acs.accounts.8b00567
Return to citation in text: [1] -
Praveen, C. Coord. Chem. Rev. 2019, 392, 1–34. doi:10.1016/j.ccr.2019.04.010
Return to citation in text: [1] -
Standen, P. E.; Kimber, M. C. Curr. Opin. Drug Discovery Dev. 2010, 13, 645.
Return to citation in text: [1] -
Yan, F.; Liang, H.; Ai, B.; Liang, W.; Jiao, L.; Yao, S.; Zhao, P.; Liu, Q.; Dong, Y.; Liu, H. Org. Biomol. Chem. 2019, 17, 2651–2656. doi:10.1039/c8ob03072c
Return to citation in text: [1] -
Zhong, X.; Tang, Q.; Zhou, P.; Zhong, Z.; Dong, S.; Liu, X.; Feng, X. Chem. Commun. 2018, 54, 10511–10514. doi:10.1039/c8cc06416d
Return to citation in text: [1] -
Ayres, J. N.; Williams, M. T. J.; Tizzard, G. J.; Coles, S. J.; Ling, K. B.; Morrill, L. C. Org. Lett. 2018, 20, 5282–5285. doi:10.1021/acs.orglett.8b02225
Return to citation in text: [1] -
De, N.; Song, C. E.; Ryu, D. H.; Yoo, E. J. Chem. Commun. 2018, 54, 6911–6914. doi:10.1039/c8cc02570c
Return to citation in text: [1] [2] -
Zheng, W.-F.; Sun, G.-J.; Chen, L.; Kang, Q. Adv. Synth. Catal. 2018, 360, 1790–1794. doi:10.1002/adsc.201800021
Return to citation in text: [1] -
Zhang, P.-C.; Han, J.; Zhang, J. Angew. Chem., Int. Ed. 2019, 58, 11444–11448. doi:10.1002/anie.201904805
Return to citation in text: [1] [2] -
Pirovano, V.; Brambilla, E.; Rizzato, S.; Abbiati, G.; Bozzi, M.; Rossi, E. J. Org. Chem. 2019, 84, 5150–5166. doi:10.1021/acs.joc.9b00143
Return to citation in text: [1] [2] -
Cui, J.; Meng, L.; Chi, X.; Liu, Q.; Zhao, P.; Zhang, D.-p.; Chen, L.; Li, X.; Dong, Y.; Liu, H. Chem. Commun. 2019, 55, 4355–4358. doi:10.1039/c9cc00797k
Return to citation in text: [1] [2] -
Banerjee, S.; Senthilkumar, B.; Patil, N. T. Org. Lett. 2019, 21, 180–184. doi:10.1021/acs.orglett.8b03651
Return to citation in text: [1] [2] -
An, J.; Lombardi, L.; Grilli, S.; Bandini, M. Org. Lett. 2018, 20, 7380–7383. doi:10.1021/acs.orglett.8b03018
Return to citation in text: [1] [2] -
Yang, K.; Bao, X.; Liu, S.; Xu, J.; Qu, J.; Wang, B. Eur. J. Org. Chem. 2018, 6469–6473. doi:10.1002/ejoc.201801039
Return to citation in text: [1] [2] [3] -
Li, Y.; Luo, G. L.; Li, X. X.; Zhao, Z. G. New J. Chem. 2018, 42, 16940–16947. doi:10.1039/c8nj03641a
Return to citation in text: [1] [2] [3] -
Hédouin, J.; Schneider, C.; Gillaizeau, I.; Hoarau, C. Org. Lett. 2018, 20, 6027–6032. doi:10.1021/acs.orglett.8b02365
Return to citation in text: [1] -
Yuan, X.; Re He Man, X. J. A. T.; Li, X.-X.; Zhao, Z.-G. Tetrahedron 2018, 74, 5674–5682. doi:10.1016/j.tet.2018.07.045
Return to citation in text: [1] [2] -
Yang, B.; Zhai, X.; Feng, S.; Hu, D.; Deng, Y.; Shao, Z. Org. Lett. 2019, 21, 330–334. doi:10.1021/acs.orglett.8b03934
Return to citation in text: [1] -
Villar, L.; Uria, U.; Martínez, J. I.; Prieto, L.; Reyes, E.; Carrillo, L.; Vicario, J. L. Angew. Chem., Int. Ed. 2017, 56, 10535–10538. doi:10.1002/anie.201704804
Return to citation in text: [1] [2] -
Yang, X.; Toste, F. D. Chem. Sci. 2016, 7, 2653–2656. doi:10.1039/c5sc04202j
Return to citation in text: [1] -
Manoni, E.; Gualandi, A.; Mengozzi, L.; Bandini, M.; Cozzi, P. G. RSC Adv. 2015, 5, 10546–10550. doi:10.1039/c4ra14942d
Return to citation in text: [1] -
Li, H.-H.; Li, X.-X.; Zhao, Z.-G.; Lin, C.-B.; Ma, T.; Sun, C.-Y.; Yang, B.-W.; Fu, X.-L. Tetrahedron Lett. 2016, 57, 4640–4644. doi:10.1016/j.tetlet.2016.09.020
Return to citation in text: [1] -
Li, H.; Li, X.; Zhao, Z.; Ma, T.; Sun, C.; Yang, B. Chem. Commun. 2016, 52, 10167–10170. doi:10.1039/c6cc05046h
Return to citation in text: [1] -
Michihiko, N.; Hiroshi, O.; Masanori, W.; Kumi, O.; Osamu, N. Tetrahedron 1996, 52, 6581–6590. doi:10.1016/0040-4020(96)00314-6
Return to citation in text: [1] -
Zhu, Y.; Yin, G.; Hong, D.; Lu, P.; Wang, Y. Org. Lett. 2011, 13, 1024–1027. doi:10.1021/ol103074d
Return to citation in text: [1] -
Zhu, Y.; Yin, G.; Sun, L.; Lu, P.; Wang, Y. Tetrahedron 2012, 68, 10194–10200. doi:10.1016/j.tet.2012.09.092
Return to citation in text: [1] -
Hyland, C. J. T.; Hegedus, L. S. J. Org. Chem. 2006, 71, 8658–8660. doi:10.1021/jo061340r
Return to citation in text: [1] -
Rameshkumar, C.; Xiong, H.; Tracey, M. R.; Berry, C. R.; Yao, L. J.; Hsung, R. P. J. Org. Chem. 2002, 67, 1339–1345. doi:10.1021/jo011048d
Return to citation in text: [1] -
Xiong, H.; Hsung, R. P.; Berry, C. R.; Rameshkumar, C. J. Am. Chem. Soc. 2001, 123, 7174–7175. doi:10.1021/ja0108638
Return to citation in text: [1] -
Krenske, E. H.; Houk, K. N.; Lohse, A. G.; Antoline, J. E.; Hsung, R. P. Chem. Sci. 2010, 1, 387–392. doi:10.1039/c0sc00280a
Return to citation in text: [1] -
Wang, Y.; Zhang, P.; Di, X.; Dai, Q.; Zhang, Z.-M.; Zhang, J. Angew. Chem., Int. Ed. 2017, 56, 15905–15909. doi:10.1002/anie.201709595
Return to citation in text: [1] -
Fernández-Casado, J.; Nelson, R.; Mascareñas, J. L.; López, F. Chem. Commun. 2016, 52, 2909–2912. doi:10.1039/c5cc09533f
Return to citation in text: [1] -
Wang, Y.; Zhang, P.; Liu, Y.; Xia, F.; Zhang, J. Chem. Sci. 2015, 6, 5564–5570. doi:10.1039/c5sc01827g
Return to citation in text: [1] -
Rocchigiani, L.; Jia, M.; Bandini, M.; Macchioni, A. ACS Catal. 2015, 5, 3911–3915. doi:10.1021/acscatal.5b00502
Return to citation in text: [1] -
Marcote, D. C.; Varela, I.; Fernández-Casado, J.; Mascareñas, J. L.; López, F. J. Am. Chem. Soc. 2018, 140, 16821–16833. doi:10.1021/jacs.8b10388
Return to citation in text: [1] -
Slater, N. H.; Brown, N. J.; Elsegood, M. R. J.; Kimber, M. C. Org. Lett. 2014, 16, 4606–4609. doi:10.1021/ol502178v
Return to citation in text: [1] [2] -
Singh, S.; Elsegood, M. R. J.; Kimber, M. C. Synlett 2012, 23, 565–568. doi:10.1055/s-0031-1290335
Return to citation in text: [1] -
Hill, A. W.; Elsegood, M. R. J.; Kimber, M. C. J. Org. Chem. 2010, 75, 5406–5409. doi:10.1021/jo101035n
Return to citation in text: [1] [2] -
Kimber, M. C. Org. Lett. 2010, 12, 1128–1131. doi:10.1021/ol1001494
Return to citation in text: [1] [2] -
Liu, L.; Ward, R. M.; Schomaker, J. M. Chem. Rev. 2019, 119, 12422–12490. doi:10.1021/acs.chemrev.9b00312
Return to citation in text: [1] -
Tomita, R.; Koike, T.; Akita, M. Chem. Commun. 2017, 53, 4681–4684. doi:10.1039/c7cc01759f
Return to citation in text: [1] -
Lu, N.; Zhang, Z.; Ma, N.; Wu, C.; Zhang, G.; Liu, Q.; Liu, T. Org. Lett. 2018, 20, 4318–4322. doi:10.1021/acs.orglett.8b01765
Return to citation in text: [1] -
Dai, X.; Mao, R.; Guan, B.; Xu, X.; Li, X. RSC Adv. 2015, 5, 55290–55294. doi:10.1039/c5ra10491b
Return to citation in text: [1] -
Shen, L.; Hsung, R. P. Org. Lett. 2005, 7, 775–778. doi:10.1021/ol047572z
Return to citation in text: [1] -
Carboni, A.; Dagousset, G.; Magnier, E.; Masson, G. Org. Lett. 2014, 16, 1240–1243. doi:10.1021/ol500374e
Return to citation in text: [1] [2] -
Courant, T.; Masson, G. J. Org. Chem. 2016, 81, 6945–6952. doi:10.1021/acs.joc.6b01058
Return to citation in text: [1] [2] [3] -
Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Dagousset, G.; Carboni, A.; Magnier, E.; Masson, G. Org. Lett. 2014, 16, 4340–4343. doi:10.1021/ol5021477
Return to citation in text: [1] [2] -
Koleoso, O. K.; Elsegood, M. R. J.; Teat, S. J.; Kimber, M. C. Org. Lett. 2018, 20, 1003–1006. doi:10.1021/acs.orglett.7b03946
Return to citation in text: [1] [2] [3] [4] [5] -
Given the instability of these reaction products, this and all subsequent reactions were performed on a 1.00 mmol scale.
Return to citation in text: [1] -
Bousfield, T. W.; Kimber, M. C. Tetrahedron Lett. 2015, 56, 350–352. doi:10.1016/j.tetlet.2014.11.093
Return to citation in text: [1] -
Wei, L.-L.; Xiong, H.; Douglas, C. J.; Hsung, R. P. Tetrahedron Lett. 1999, 40, 6903–6907. doi:10.1016/s0040-4039(99)01354-4
Return to citation in text: [1] -
DFT geometry optimisations calculations were performed at the PBEh-3c level of theory; please see Supporting Information File 1.
Return to citation in text: [1] [2] [3] -
Slinker, J. D.; Gorodetsky, A. A.; Lowry, M. S.; Wang, J.; Parker, S.; Rohl, R.; Bernhard, S.; Malliaras, G. G. J. Am. Chem. Soc. 2004, 126, 2763–2767. doi:10.1021/ja0345221
Return to citation in text: [1] -
Al-Jawaheri, Y.; Turner, M.; Kimber, M. C. Synthesis 2018, 50, 2329–2336. doi:10.1055/s-0036-1591580
Return to citation in text: [1] -
Qian, R.; Guo, H.; Liao, Y.; Guo, Y.; Ma, S. Angew. Chem., Int. Ed. 2005, 44, 4771–4774. doi:10.1002/anie.200463101
Return to citation in text: [1] -
Trowbridge, A.; Reich, D.; Gaunt, M. J. Nature 2018, 561, 522–527. doi:10.1038/s41586-018-0537-9
Return to citation in text: [1] -
Ryder, A. S. H.; Cunningham, W. B.; Ballantyne, G.; Mules, T.; Kinsella, A. G.; Turner-Dore, J.; Alder, C. M.; Edwards, L. J.; McKay, B. S. J.; Grayson, M. N.; Cresswell, A. J. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202005294
Return to citation in text: [1] -
Trowbridge, A.; Walton, S. M.; Gaunt, M. J. Chem. Rev. 2020, 120, 2613–2692. doi:10.1021/acs.chemrev.9b00462
Return to citation in text: [1]
| 54. | DFT geometry optimisations calculations were performed at the PBEh-3c level of theory; please see Supporting Information File 1. |
| 56. | Al-Jawaheri, Y.; Turner, M.; Kimber, M. C. Synthesis 2018, 50, 2329–2336. doi:10.1055/s-0036-1591580 |
| 57. | Qian, R.; Guo, H.; Liao, Y.; Guo, Y.; Ma, S. Angew. Chem., Int. Ed. 2005, 44, 4771–4774. doi:10.1002/anie.200463101 |
| 54. | DFT geometry optimisations calculations were performed at the PBEh-3c level of theory; please see Supporting Information File 1. |
| 1. | Lu, T.; Lu, Z.; Ma, Z.-X.; Zhang, Y.; Hsung, R. P. Chem. Rev. 2013, 113, 4862–4904. doi:10.1021/cr400015d |
| 2. | Mascareñas, J. L.; Varela, I.; López, F. Acc. Chem. Res. 2019, 52, 465–479. doi:10.1021/acs.accounts.8b00567 |
| 3. | Praveen, C. Coord. Chem. Rev. 2019, 392, 1–34. doi:10.1016/j.ccr.2019.04.010 |
| 4. | Standen, P. E.; Kimber, M. C. Curr. Opin. Drug Discovery Dev. 2010, 13, 645. |
| 15. | Yang, K.; Bao, X.; Liu, S.; Xu, J.; Qu, J.; Wang, B. Eur. J. Org. Chem. 2018, 6469–6473. doi:10.1002/ejoc.201801039 |
| 19. | Yang, B.; Zhai, X.; Feng, S.; Hu, D.; Deng, Y.; Shao, Z. Org. Lett. 2019, 21, 330–334. doi:10.1021/acs.orglett.8b03934 |
| 20. | Villar, L.; Uria, U.; Martínez, J. I.; Prieto, L.; Reyes, E.; Carrillo, L.; Vicario, J. L. Angew. Chem., Int. Ed. 2017, 56, 10535–10538. doi:10.1002/anie.201704804 |
| 21. | Yang, X.; Toste, F. D. Chem. Sci. 2016, 7, 2653–2656. doi:10.1039/c5sc04202j |
| 22. | Manoni, E.; Gualandi, A.; Mengozzi, L.; Bandini, M.; Cozzi, P. G. RSC Adv. 2015, 5, 10546–10550. doi:10.1039/c4ra14942d |
| 47. | Courant, T.; Masson, G. J. Org. Chem. 2016, 81, 6945–6952. doi:10.1021/acs.joc.6b01058 |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 1. | Lu, T.; Lu, Z.; Ma, Z.-X.; Zhang, Y.; Hsung, R. P. Chem. Rev. 2013, 113, 4862–4904. doi:10.1021/cr400015d |
| 47. | Courant, T.; Masson, G. J. Org. Chem. 2016, 81, 6945–6952. doi:10.1021/acs.joc.6b01058 |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 50. | Koleoso, O. K.; Elsegood, M. R. J.; Teat, S. J.; Kimber, M. C. Org. Lett. 2018, 20, 1003–1006. doi:10.1021/acs.orglett.7b03946 |
| 10. | Zhang, P.-C.; Han, J.; Zhang, J. Angew. Chem., Int. Ed. 2019, 58, 11444–11448. doi:10.1002/anie.201904805 |
| 11. | Pirovano, V.; Brambilla, E.; Rizzato, S.; Abbiati, G.; Bozzi, M.; Rossi, E. J. Org. Chem. 2019, 84, 5150–5166. doi:10.1021/acs.joc.9b00143 |
| 12. | Cui, J.; Meng, L.; Chi, X.; Liu, Q.; Zhao, P.; Zhang, D.-p.; Chen, L.; Li, X.; Dong, Y.; Liu, H. Chem. Commun. 2019, 55, 4355–4358. doi:10.1039/c9cc00797k |
| 13. | Banerjee, S.; Senthilkumar, B.; Patil, N. T. Org. Lett. 2019, 21, 180–184. doi:10.1021/acs.orglett.8b03651 |
| 14. | An, J.; Lombardi, L.; Grilli, S.; Bandini, M. Org. Lett. 2018, 20, 7380–7383. doi:10.1021/acs.orglett.8b03018 |
| 15. | Yang, K.; Bao, X.; Liu, S.; Xu, J.; Qu, J.; Wang, B. Eur. J. Org. Chem. 2018, 6469–6473. doi:10.1002/ejoc.201801039 |
| 16. | Li, Y.; Luo, G. L.; Li, X. X.; Zhao, Z. G. New J. Chem. 2018, 42, 16940–16947. doi:10.1039/c8nj03641a |
| 17. | Hédouin, J.; Schneider, C.; Gillaizeau, I.; Hoarau, C. Org. Lett. 2018, 20, 6027–6032. doi:10.1021/acs.orglett.8b02365 |
| 18. | Yuan, X.; Re He Man, X. J. A. T.; Li, X.-X.; Zhao, Z.-G. Tetrahedron 2018, 74, 5674–5682. doi:10.1016/j.tet.2018.07.045 |
| 46. | Carboni, A.; Dagousset, G.; Magnier, E.; Masson, G. Org. Lett. 2014, 16, 1240–1243. doi:10.1021/ol500374e |
| 47. | Courant, T.; Masson, G. J. Org. Chem. 2016, 81, 6945–6952. doi:10.1021/acs.joc.6b01058 |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 49. | Dagousset, G.; Carboni, A.; Magnier, E.; Masson, G. Org. Lett. 2014, 16, 4340–4343. doi:10.1021/ol5021477 |
| 5. | Yan, F.; Liang, H.; Ai, B.; Liang, W.; Jiao, L.; Yao, S.; Zhao, P.; Liu, Q.; Dong, Y.; Liu, H. Org. Biomol. Chem. 2019, 17, 2651–2656. doi:10.1039/c8ob03072c |
| 6. | Zhong, X.; Tang, Q.; Zhou, P.; Zhong, Z.; Dong, S.; Liu, X.; Feng, X. Chem. Commun. 2018, 54, 10511–10514. doi:10.1039/c8cc06416d |
| 7. | Ayres, J. N.; Williams, M. T. J.; Tizzard, G. J.; Coles, S. J.; Ling, K. B.; Morrill, L. C. Org. Lett. 2018, 20, 5282–5285. doi:10.1021/acs.orglett.8b02225 |
| 8. | De, N.; Song, C. E.; Ryu, D. H.; Yoo, E. J. Chem. Commun. 2018, 54, 6911–6914. doi:10.1039/c8cc02570c |
| 9. | Zheng, W.-F.; Sun, G.-J.; Chen, L.; Kang, Q. Adv. Synth. Catal. 2018, 360, 1790–1794. doi:10.1002/adsc.201800021 |
| 50. | Koleoso, O. K.; Elsegood, M. R. J.; Teat, S. J.; Kimber, M. C. Org. Lett. 2018, 20, 1003–1006. doi:10.1021/acs.orglett.7b03946 |
| 37. | Slater, N. H.; Brown, N. J.; Elsegood, M. R. J.; Kimber, M. C. Org. Lett. 2014, 16, 4606–4609. doi:10.1021/ol502178v |
| 38. | Singh, S.; Elsegood, M. R. J.; Kimber, M. C. Synlett 2012, 23, 565–568. doi:10.1055/s-0031-1290335 |
| 39. | Hill, A. W.; Elsegood, M. R. J.; Kimber, M. C. J. Org. Chem. 2010, 75, 5406–5409. doi:10.1021/jo101035n |
| 40. | Kimber, M. C. Org. Lett. 2010, 12, 1128–1131. doi:10.1021/ol1001494 |
| 42. | Tomita, R.; Koike, T.; Akita, M. Chem. Commun. 2017, 53, 4681–4684. doi:10.1039/c7cc01759f |
| 43. | Lu, N.; Zhang, Z.; Ma, N.; Wu, C.; Zhang, G.; Liu, Q.; Liu, T. Org. Lett. 2018, 20, 4318–4322. doi:10.1021/acs.orglett.8b01765 |
| 44. | Dai, X.; Mao, R.; Guan, B.; Xu, X.; Li, X. RSC Adv. 2015, 5, 55290–55294. doi:10.1039/c5ra10491b |
| 8. | De, N.; Song, C. E.; Ryu, D. H.; Yoo, E. J. Chem. Commun. 2018, 54, 6911–6914. doi:10.1039/c8cc02570c |
| 10. | Zhang, P.-C.; Han, J.; Zhang, J. Angew. Chem., Int. Ed. 2019, 58, 11444–11448. doi:10.1002/anie.201904805 |
| 11. | Pirovano, V.; Brambilla, E.; Rizzato, S.; Abbiati, G.; Bozzi, M.; Rossi, E. J. Org. Chem. 2019, 84, 5150–5166. doi:10.1021/acs.joc.9b00143 |
| 12. | Cui, J.; Meng, L.; Chi, X.; Liu, Q.; Zhao, P.; Zhang, D.-p.; Chen, L.; Li, X.; Dong, Y.; Liu, H. Chem. Commun. 2019, 55, 4355–4358. doi:10.1039/c9cc00797k |
| 13. | Banerjee, S.; Senthilkumar, B.; Patil, N. T. Org. Lett. 2019, 21, 180–184. doi:10.1021/acs.orglett.8b03651 |
| 14. | An, J.; Lombardi, L.; Grilli, S.; Bandini, M. Org. Lett. 2018, 20, 7380–7383. doi:10.1021/acs.orglett.8b03018 |
| 15. | Yang, K.; Bao, X.; Liu, S.; Xu, J.; Qu, J.; Wang, B. Eur. J. Org. Chem. 2018, 6469–6473. doi:10.1002/ejoc.201801039 |
| 16. | Li, Y.; Luo, G. L.; Li, X. X.; Zhao, Z. G. New J. Chem. 2018, 42, 16940–16947. doi:10.1039/c8nj03641a |
| 32. | Wang, Y.; Zhang, P.; Di, X.; Dai, Q.; Zhang, Z.-M.; Zhang, J. Angew. Chem., Int. Ed. 2017, 56, 15905–15909. doi:10.1002/anie.201709595 |
| 33. | Fernández-Casado, J.; Nelson, R.; Mascareñas, J. L.; López, F. Chem. Commun. 2016, 52, 2909–2912. doi:10.1039/c5cc09533f |
| 34. | Wang, Y.; Zhang, P.; Liu, Y.; Xia, F.; Zhang, J. Chem. Sci. 2015, 6, 5564–5570. doi:10.1039/c5sc01827g |
| 35. | Rocchigiani, L.; Jia, M.; Bandini, M.; Macchioni, A. ACS Catal. 2015, 5, 3911–3915. doi:10.1021/acscatal.5b00502 |
| 36. | Marcote, D. C.; Varela, I.; Fernández-Casado, J.; Mascareñas, J. L.; López, F. J. Am. Chem. Soc. 2018, 140, 16821–16833. doi:10.1021/jacs.8b10388 |
| 20. | Villar, L.; Uria, U.; Martínez, J. I.; Prieto, L.; Reyes, E.; Carrillo, L.; Vicario, J. L. Angew. Chem., Int. Ed. 2017, 56, 10535–10538. doi:10.1002/anie.201704804 |
| 29. | Rameshkumar, C.; Xiong, H.; Tracey, M. R.; Berry, C. R.; Yao, L. J.; Hsung, R. P. J. Org. Chem. 2002, 67, 1339–1345. doi:10.1021/jo011048d |
| 30. | Xiong, H.; Hsung, R. P.; Berry, C. R.; Rameshkumar, C. J. Am. Chem. Soc. 2001, 123, 7174–7175. doi:10.1021/ja0108638 |
| 31. | Krenske, E. H.; Houk, K. N.; Lohse, A. G.; Antoline, J. E.; Hsung, R. P. Chem. Sci. 2010, 1, 387–392. doi:10.1039/c0sc00280a |
| 58. | Trowbridge, A.; Reich, D.; Gaunt, M. J. Nature 2018, 561, 522–527. doi:10.1038/s41586-018-0537-9 |
| 59. | Ryder, A. S. H.; Cunningham, W. B.; Ballantyne, G.; Mules, T.; Kinsella, A. G.; Turner-Dore, J.; Alder, C. M.; Edwards, L. J.; McKay, B. S. J.; Grayson, M. N.; Cresswell, A. J. Angew. Chem., Int. Ed. 2020, in press. doi:10.1002/anie.202005294 |
| 60. | Trowbridge, A.; Walton, S. M.; Gaunt, M. J. Chem. Rev. 2020, 120, 2613–2692. doi:10.1021/acs.chemrev.9b00462 |
| 16. | Li, Y.; Luo, G. L.; Li, X. X.; Zhao, Z. G. New J. Chem. 2018, 42, 16940–16947. doi:10.1039/c8nj03641a |
| 18. | Yuan, X.; Re He Man, X. J. A. T.; Li, X.-X.; Zhao, Z.-G. Tetrahedron 2018, 74, 5674–5682. doi:10.1016/j.tet.2018.07.045 |
| 23. | Li, H.-H.; Li, X.-X.; Zhao, Z.-G.; Lin, C.-B.; Ma, T.; Sun, C.-Y.; Yang, B.-W.; Fu, X.-L. Tetrahedron Lett. 2016, 57, 4640–4644. doi:10.1016/j.tetlet.2016.09.020 |
| 24. | Li, H.; Li, X.; Zhao, Z.; Ma, T.; Sun, C.; Yang, B. Chem. Commun. 2016, 52, 10167–10170. doi:10.1039/c6cc05046h |
| 25. | Michihiko, N.; Hiroshi, O.; Masanori, W.; Kumi, O.; Osamu, N. Tetrahedron 1996, 52, 6581–6590. doi:10.1016/0040-4020(96)00314-6 |
| 26. | Zhu, Y.; Yin, G.; Hong, D.; Lu, P.; Wang, Y. Org. Lett. 2011, 13, 1024–1027. doi:10.1021/ol103074d |
| 27. | Zhu, Y.; Yin, G.; Sun, L.; Lu, P.; Wang, Y. Tetrahedron 2012, 68, 10194–10200. doi:10.1016/j.tet.2012.09.092 |
| 28. | Hyland, C. J. T.; Hegedus, L. S. J. Org. Chem. 2006, 71, 8658–8660. doi:10.1021/jo061340r |
| 41. | Liu, L.; Ward, R. M.; Schomaker, J. M. Chem. Rev. 2019, 119, 12422–12490. doi:10.1021/acs.chemrev.9b00312 |
| 51. | Given the instability of these reaction products, this and all subsequent reactions were performed on a 1.00 mmol scale. |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 50. | Koleoso, O. K.; Elsegood, M. R. J.; Teat, S. J.; Kimber, M. C. Org. Lett. 2018, 20, 1003–1006. doi:10.1021/acs.orglett.7b03946 |
| 50. | Koleoso, O. K.; Elsegood, M. R. J.; Teat, S. J.; Kimber, M. C. Org. Lett. 2018, 20, 1003–1006. doi:10.1021/acs.orglett.7b03946 |
| 46. | Carboni, A.; Dagousset, G.; Magnier, E.; Masson, G. Org. Lett. 2014, 16, 1240–1243. doi:10.1021/ol500374e |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 49. | Dagousset, G.; Carboni, A.; Magnier, E.; Masson, G. Org. Lett. 2014, 16, 4340–4343. doi:10.1021/ol5021477 |
| 55. | Slinker, J. D.; Gorodetsky, A. A.; Lowry, M. S.; Wang, J.; Parker, S.; Rohl, R.; Bernhard, S.; Malliaras, G. G. J. Am. Chem. Soc. 2004, 126, 2763–2767. doi:10.1021/ja0345221 |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 54. | DFT geometry optimisations calculations were performed at the PBEh-3c level of theory; please see Supporting Information File 1. |
| 50. | Koleoso, O. K.; Elsegood, M. R. J.; Teat, S. J.; Kimber, M. C. Org. Lett. 2018, 20, 1003–1006. doi:10.1021/acs.orglett.7b03946 |
| 48. | Courant, T.; Masson, G. Chem. – Eur. J. 2012, 18, 423–427. doi:10.1002/chem.201103062 |
| 37. | Slater, N. H.; Brown, N. J.; Elsegood, M. R. J.; Kimber, M. C. Org. Lett. 2014, 16, 4606–4609. doi:10.1021/ol502178v |
| 39. | Hill, A. W.; Elsegood, M. R. J.; Kimber, M. C. J. Org. Chem. 2010, 75, 5406–5409. doi:10.1021/jo101035n |
| 40. | Kimber, M. C. Org. Lett. 2010, 12, 1128–1131. doi:10.1021/ol1001494 |
| 52. | Bousfield, T. W.; Kimber, M. C. Tetrahedron Lett. 2015, 56, 350–352. doi:10.1016/j.tetlet.2014.11.093 |
| 53. | Wei, L.-L.; Xiong, H.; Douglas, C. J.; Hsung, R. P. Tetrahedron Lett. 1999, 40, 6903–6907. doi:10.1016/s0040-4039(99)01354-4 |
© 2020 Koleoso et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)