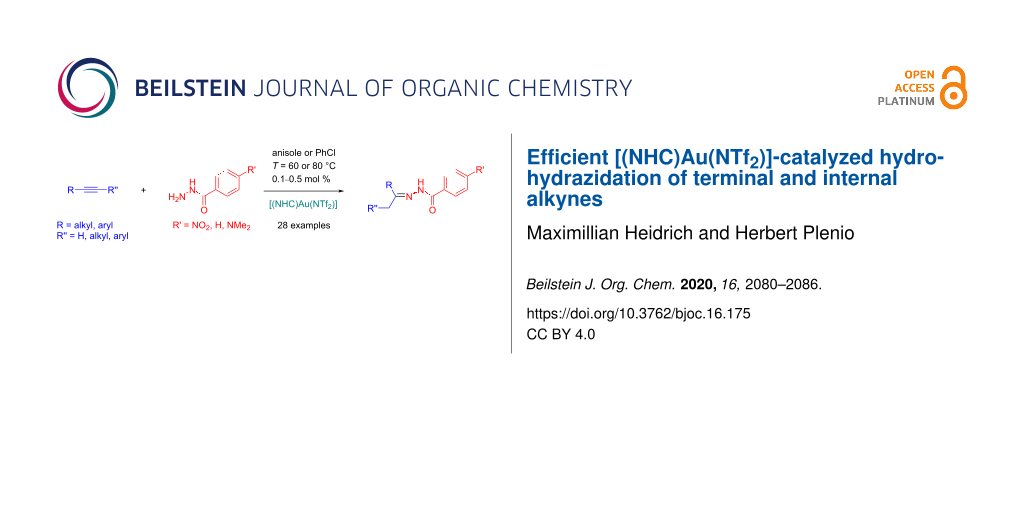
Abstract
The efficient hydrohydrazidation of terminal (6a–r, 18 examples, 0.1–0.2 mol % [(NHC)Au(NTf2)], T = 60 °C) and internal alkynes (7a–j, 10 examples, 0.2–0.5 mol % [(NHC)Au(NTf2)], T = 60–80 °C) utilizing a complex with a sterically demanding bispentiptycenyl-substituted NHC ligand and the benign reaction solvent anisole, is reported.

Graphical Abstract
Introduction
Cationic gold complexes with weakly coordinating counterions (which are often considered to be solvent-separated ions) [1-3], render powerful catalysts for the transformation of organic substrates [4-6], specifically the reactions of alkynes with a variety of heteroatom nucleophiles (Scheme 1) [7] such as water or alcohol [8-22], primary or secondary amines [23-28], or hydrazine [29-31]. These reactions are synthetically useful since gold catalysis is characterized by a good functional group tolerance for oxygen and nitrogen-containing molecules, which tend to be more difficult for catalytic transformations utilizing other transition metals [32-36]. The [LAu(NTf2)]-catalyzed reaction can be described by a general mechanism (Scheme 1), in which the coordination of LAu+ by the alkyne [37,38] leads to the activation of the triple bond for a nucleophilic attack, generating a vinylgold complex [39,40]. This is followed by the substitution of LAu+ with an electrophile E, corresponding (for E = H+) to a protodeauration and is leading to the release of the desired organic product [41]. The activity of gold complexes in such reactions tends to be higher, when using sterically demanding ligands (NHC, phosphine) [41].
![[1860-5397-16-175-i1]](/bjoc/content/inline/1860-5397-16-175-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Simplified mechanism of the hydrohydrazidation (NuH= ArCONHNH2) of alkynes.
Scheme 1: Simplified mechanism of the hydrohydrazidation (NuH= ArCONHNH2) of alkynes.
Recently, the hydrohydrazidation of substituted phenylacetylenes using [(Ph3P)Au(NTf2)] as the catalyst was reported by Rassadin, Kukushkin et al. [42]. Catalyst loadings of 6 mol % at 60 °C were required to obtain the respective addition product in yields of 66–93%. Internal alkynes were much less reactive and even at 12 mol % catalyst loading the reaction of 3-hexyne and PhCONHNH2 yielded the respective addition product in only 32% yield, with diphenylacetylene only 12% of the addition product were obtained. We recently reported the excellent activity of the very bulky bispentiptycenyl-substituted (NHC)Au complexes in the hydration of terminal and internal alkynes [43] as well as in other transition-metal-catalyzed transformations [44,45]. We were now interested, whether catalysts with such ligands also display high activities in other gold-catalyzed reactions.
Results and Discussion
Catalyst screening: Five different [(NHC)Au(NTf2)] complexes 1, 2, 3, 4, and 5 with bulky NHC ligands were tested in the hydrohydrazidation of phenylacetylene with benzohydrazide (Scheme 2; T = 60 °C, solvent chlorobenzene, reaction time 24 h, catalyst loading 0.2 mol %). The following conversions were observed: complex 1 >99%, complex 2 16%, complex 3 38%, complex 4 31%, and complex 5 60%. Complex 1 having a bispentiptycene-NHC ligand was the clear best among the five NHC-gold complexes. In this group of gold complexes with sterically demanding NHC ligands, the NHC ligands with intermediate bulk provided the best catalytic performance. The use of the bulkiest complex 2 [46], resulted only in modest substrate conversion. Nonetheless, all of the tested complexes performed much better in hydrohydrazidation reactions than the previously reported [(Ph3P)Au(NTf2)] [42].
![[1860-5397-16-175-i2]](/bjoc/content/inline/1860-5397-16-175-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: [(NHC)Au(NTf2)] complexes tested in hydrohydrazidation reactions of phenylacetylene.
Scheme 2: [(NHC)Au(NTf2)] complexes tested in hydrohydrazidation reactions of phenylacetylene.
Solvent screening: Among the solvents tested in the previous study by Rassadin, Kukushkin et al., chlorobenzene was found to be the best solvent concerning the catalytic activity [42]. However, according to the CHEM21 consortium chlorobenzene is a problematic solvent, whose use should be avoided [47]. We therefore performed an extensive solvent screening focusing on “greener” solvents for the reactions of benzohydrazide with either phenylacetylene or 4-methoxyphenylacetylene (Table 1). It was found that the catalytic performance in anisole was comparable to that in chlorobenzene. According to both the CHEM21 consortium and the GSK solvent sustainability guide the use of anisole is highly favorable [48]. To clarify the role of chlorobenzene and anisole, most substrate screening reactions were performed in both solvents (see also Scheme 3 and Scheme 4) and based on this the use of anisole as a reaction solvent could be recommended.
Table 1: Solvent screening for two hydrohydrazidation reactions using complex 1.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-175-i5.svg?max-width=637&scale=1.0)
|
||||
| product | solvent | time (h) | conversion (%)a | |
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-175-i6.svg?max-width=637&scale=1.0)
|
PhCl | 24b, 6c | 81b | >99c |
| anisole | 24b, 6c | 80b | >99c | |
| xylene | 24b | 46b | ||
| MeNO2 | 24b, 6c | 45b | 82c | |
| dioxane | 24b, 6c | 31b | 67c | |
| MeCN | 6c | 64c | ||
| THF | 6c | 53c | ||
| glyme | 6c | 68c | ||
aGC-based conversion (internal standard mesitylene); bsubstrate a; csubstrate b.
Reaction time vs catalyst loading: In order to optimize catalyst loading and reaction times the hydrohydrazidation of phenylacetylene was carried out at successively lower catalyst loading (Table 2). The obvious consequence being a decrease in the substrate conversion.
Table 2: Catalyst loading vs. reaction time in the hydrohydrazidation.
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-175-i7.svg?max-width=637&scale=1.0)
|
|||
| entry | complex 1 (mol %) | time (h) | conversion (%)a |
| 1 | 0.2 | 24 | >99 |
| 2 | 0.1 | 24/48 | 79/93 |
| 3 | 0.05 | 24/48/96 | 51/70/>99 |
| 4 | 0.02 | 24/48 | 22/29 |
| 5 | 0.01 | 24/48 | 2/4 |
| 6 | zero | 24 | 0 |
aGC-based conversion (internal standard mesitylene).
This, however, could be compensated to some extent by longer reaction times. Based on this observation catalyst decomposition appeared to be negligible and it seemed, that the role of gold is primarily that of a Lewis acid activating the alkyne. Consequently, even at 0.05 mol % catalyst loading virtually quantitative substrate conversion was observed after extending the reaction time to 96 h (Table 2, entry 3). In the absence of the Au catalyst, no product was formed.
Hydrohydrazidation reactions of terminal alkynes: Following the initial optimization, a variety of hydrohydrazidation reactions of terminal and internal alkynes with benzohydrazides was studied (Scheme 3). Various combinations of alkylalkynes (1-octyne, cyclohexylacetylene, tert-butylacetylene), and arylalkynes (phenylacetylene, 2,4,6-trimethylphenylacetylene, 2,4,6-triisopropylphenylacetylene, and para-substituted 4-R-phenylacetylenes (R = NMe2, OMe, CF3)) with electronically variable benzohydrazides (4-R = NMe2, H, NO2) were tested, to cover a range of steric and electronic effects.
![[1860-5397-16-175-i3]](/bjoc/content/inline/1860-5397-16-175-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Hydrohydrazidation of terminal alkynes in chlorobenzene and anisole using complex 1 (first line solvent chlorobenzene, second line solvent anisole: catalyst loading, reaction time, conversion % (isolated yield %), reaction temperature 60 °C). aPartial decomposition during column chromatography observed.
Scheme 3: Hydrohydrazidation of terminal alkynes in chlorobenzene and anisole using complex 1 (first line sol...
Most hydrohydrazidation reactions showed nearly quantitative product conversion to 6 and provided isolated yields in excess of 90% using as little as 0.1–0.2 mol % of complex 1. Just like in related reactions, the nucleophile exclusively added to the β-carbon of the triple bond. A single substituent in the ortho-position of the alkyne did not exert a strong steric effect. Even the hydrohydrazidation of the sterically highly demanding 2,4,6-triisopropylphenylacetylene provided 82% isolated yield based on 90% substrate conversion (Scheme 3, compound 6i). There was a pronounced electronic effect and electron-rich arylalkynes reacted faster than those with electron-withdrawing groups. The products derived from benzohydrazides with electron-rich aromatic groups tended to be somewhat unstable during chromatographic purification (6j, 6q). In such cases it is advisable to facilitate product purification by choosing modified reaction conditions (e.g., higher catalyst loading or longer reaction times), to ensure virtually quantitative product formation.
Hydrohydrazidation of internal alkynes: Previously, internal alkynes could not be converted efficiently into the respective hydrohydrazidation products via gold catalysis [42], while product formation was claimed in a thermal reaction [49]. However, in the presence of complex 1 internal alkynes provided the respective addition products 7 in excellent yields using catalyst loadings in the 0.2–0.5 mol % range at slightly elevated temperature (T = 60–80 °C, Scheme 4). A range of dialkyl-, alkyl-, aryl-, and diarylacetylenes was reacted with electronically variable benzohydrazides (4-R = NMe2, H, NO2), to cover a range of steric and electronic substituents. In case shorter reaction times were desired, this could be compensated by increasing the amount of catalyst or the reaction temperature. The order of the alkyne reactivity in the present study was as follows: RCCH > alkyl-CC-alkyl > aryl-CC-alkyl ≥ aryl-CC-aryl.
![[1860-5397-16-175-i4]](/bjoc/content/inline/1860-5397-16-175-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Hydrohydrazidation of internal alkynes in chlorobenzene and anisole using complex 1. Reaction temperature was 60 °C unless otherwise noted (first line solvent chlorobenzene, second line solvent anisole: catalyst loading, reaction time, conversion % (isolated yield %). a80 °C reaction temperature; bisolated yield after 1 w; cisomeric ratio determined via NMR spectroscopy.
Scheme 4: Hydrohydrazidation of internal alkynes in chlorobenzene and anisole using complex 1. Reaction tempe...
Disubstituted acetylenes with different substituents tended to produce isomers in the hydrohydrazidation reaction (Scheme 4, products 7b, c, d, i, and j). No preference for either isomer was observed – not even when using aryl-, alkylacetylenes, or electronically different aryl groups in tolanes. However, the steric bulk of a single methyl group in 2-methyltolane led to the selective formation of the single addition product 7h, but even such sterically hindered tolanes (synthesized via Sonogashira coupling) [50] show reasonable substrate conversion. To assess the electronic effect of substituents on the hydrazide-substituted RC6H4-CONHNH2 relatives (R= NMe2, NO2) were tested leading to 7f and 7g. With a view to the larger distance of the functional group from the reactive center, the electronic effect was weaker, but nonetheless the conversion of the electron-deficient hydrazides (R = NO2) was slightly more efficient than with the electron-rich hydrazides (R = NMe2).
Conclusion
In conclusion, we have demonstrated the efficient hydrohydrazidation of various terminal and internal alkynes utilizing the bispentiptycenyl-substituted [(NHC)Au(NTf2)] complex 1 using catalyst loadings between 0.1–0.5 mol % at temperatures between 60–80 °C in chlorobenzene, or anisole as a green reaction solvent.
Experimental
All reagents were obtained from commercial providers (Sigma-Aldrich, Alfa Aesar, TCI Europe) and were used without further purification. All solvents were dried over CaH2 and distilled. All screening reactions were performed in Schlenk tubes under normal atmosphere unless otherwise noted. 1H and 13C NMR spectra were recorded at the NMR department of the Techincal University Darmstadt at 300 MHz (1H) and 75 MHz (13C) or at 500 MHz (1H) and 126 MHz (13C), respectively. Chemical shifts are given in parts per million (ppm) on the delta scale (δ) and are referred either to tetramethylsilane (1H; 13C NMR = 0 ppm) or the residual solvent peak. Thin-layer chromatography (TLC) was performed using silica gel 60 F254 (0.2 mm) on alumina plates. For preparative chromatography silica gel 60 (70–200 µm) was used. Measurements of the high resolution mass spectra (HRMS) were performed on a Bruker Daltonics Autoflex Speed TOF. Gas chromatographic data was obtained on a Clarus 500 gas chromatography system.
Synthesis of gold complex 1. To a solution of the (NHC)AuCl complex 1a (60 mg, 0.043 mmol, 1 equiv) in DCM (5 mL) silver bis(trifluoromethanesulfonyl)amide (18 mg, 0.043 mmol, 1 equiv) was added and the mixture was stirred at rt for 30 min. The formed suspension was filtered through a plug of celite and the celite washed with a small amount of DCM. The volatiles of the filtrate were evaporated under reduced pressure and the residue dried in vacuo, to yield complex 1 (62 mg, 0.039 mmol, 92% yield) as white solid. 1H NMR (500 MHz, CD2Cl2) δ 7.54–7.48 (m, 8H), 7.46–7.39 (m, 8H), 7.08–6.93 (m, 16H), 5.90 (s, 4H), 5.19 (s, 4H), 4.12 (t, J = 6.8 Hz, 4H), 2.11–2.05 (m, 4H), 2.03 (s, 6H), 1.83–1.74 (m, 4H), 1.19 (td, J = 7.4, 1.7 Hz, 6H); 13C NMR (126 MHz, CD2Cl2) δ 166.41, 151.25, 146.21, 145.15, 144.71, 144.08, 142.03, 138.12, 128.93, 126.38, 125.98, 125.48, 124.67, 124.04, 123.98, 123.94, 76.83, 51.06, 48.99, 48.92, 33.13, 20.23, 14.44, 10.38; HRESIMS (m/z): [NHC(Au) + CH3CN]+ calcd for C83H67AuN3O2, 1334.48935; found, 1334.48923.
General procedure for the hydrohydrazidation of alkynes. In a small Schlenk flask equipped with a small stirring bar the corresponding alkyne (0.5 mmol) and the corresponding benzhydrazide (0.5 mmol) were mixed with anisole or chlorobenzene (2 mL). For the internal standard mesitylene (69 µL, 0.5 mmol) was added. A stock solution of the corresponding gold-triflimide catalyst (0.005 M in anisole or chlorobenzene, 200 µL, 0.001 mmol, 0.2 mol %) was introduced to the mixture. The reaction mixture was stirred at 60 °C or 80 °C for the respective time and the progress of the reaction was monitored by GC. After the reaction was completed, the solvent was removed in vacuo and the residue triturated in pentane. Column chromatography (DCM/ethyl acetate 5:1) afforded the respective benzohydrazone in good to excellent yields.
Hydrohydrazidation of phenylacetylene with benzhydrazide on a larger scale. In a 50 mL Schlenk flask equipped with a stirring bar phenylacetylene (5.00 mmol, 511 mg, 1 equiv) and benzhydrazide (5.00 mmol, 681 mg, 1 equiv) were mixed with anisole (20 mL). Gold-triflimide catalyst 1 (15.5 mg, 0.01 mmol, 0.2 mol %) was added and the reaction mixture was stirred at 60 °C for 24 h. The mixture was cooled to 0 °C and the formed precipitate was collected and washed with a small amount of pentane to afford N'-(1-phenylethylidene)benzohydrazide (6d, 1.15 g, 4.83 mmol, 97% yield) as colorless needles.
Supporting Information
| Supporting Information File 1: Characterization data and copies of NMR spectra and mass spectrometric data. | ||
| Format: PDF | Size: 7.1 MB | Download |
References
-
Jia, M.; Bandini, M. ACS Catal. 2015, 5, 1638–1652. doi:10.1021/cs501902v
Return to citation in text: [1] -
Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A. S. K. Adv. Synth. Catal. 2018, 360, 2493–2502. doi:10.1002/adsc.201800233
Return to citation in text: [1] -
Zuccaccia, D.; Belpassi, L.; Macchioni, A.; Tarantelli, F. Eur. J. Inorg. Chem. 2013, 4121–4135. doi:10.1002/ejic.201300285
Return to citation in text: [1] -
Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k
Return to citation in text: [1] -
Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. doi:10.1039/c5cs00721f
Return to citation in text: [1] -
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u
Return to citation in text: [1] -
Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e
Return to citation in text: [1] -
Weerasiri, K. C.; Chen, D.; Wozniak, D. I.; Dobereiner, G. E. Adv. Synth. Catal. 2016, 358, 4106–4113. doi:10.1002/adsc.201601013
Return to citation in text: [1] -
Xu, Y.; Hu, X.; Shao, J.; Yang, G.; Wu, Y.; Zhang, Z. Green Chem. 2015, 17, 532–537. doi:10.1039/c4gc01322k
Return to citation in text: [1] -
Biasiolo, L.; Del Zotto, A.; Zuccaccia, D. Organometallics 2015, 34, 1759–1765. doi:10.1021/acs.organomet.5b00308
Return to citation in text: [1] -
Xu, X.; Kim, S. H.; Zhang, X.; Das, A. K.; Hirao, H.; Hong, S. H. Organometallics 2013, 32, 164–171. doi:10.1021/om3009603
Return to citation in text: [1] -
Goodwin, J. A.; Aponick, A. Chem. Commun. 2015, 51, 8730–8741. doi:10.1039/c5cc00120j
Return to citation in text: [1] -
Carriedo, G. A.; López, S.; Suárez-Suárez, S.; Presa-Soto, D.; Presa-Soto, A. Eur. J. Inorg. Chem. 2011, 1442–1447. doi:10.1002/ejic.201001317
Return to citation in text: [1] -
Wang, W.; Zheng, A.; Zhao, P.; Xia, C.; Li, F. ACS Catal. 2014, 4, 321–327. doi:10.1021/cs400983y
Return to citation in text: [1] -
Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067–2074. doi:10.1021/jo802558e
Return to citation in text: [1] -
Ibrahim, H.; de Frémont, P.; Braunstein, P.; Théry, V.; Nauton, L.; Cisnetti, F.; Gautier, A. Adv. Synth. Catal. 2015, 357, 3893–3900. doi:10.1002/adsc.201500729
Return to citation in text: [1] -
Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. ACS Catal. 2016, 6, 7363–7376. doi:10.1021/acscatal.6b01626
Return to citation in text: [1] -
Xu, Y.; Hu, X.; Zhang, S.; Xi, X.; Wu, Y. ChemCatChem 2016, 8, 262–267. doi:10.1002/cctc.201501065
Return to citation in text: [1] -
Ibrahim, N.; Vilhelmsen, M. H.; Pernpointner, M.; Rominger, F.; Hashmi, A. S. K. Organometallics 2013, 32, 2576–2583. doi:10.1021/om400060s
Return to citation in text: [1] -
Rühling, A.; Galla, H.-J.; Glorius, F. Chem. – Eur. J. 2015, 21, 12291–12294. doi:10.1002/chem.201502542
Return to citation in text: [1] -
Ebule, R. E.; Malhotra, D.; Hammond, G. B.; Xu, B. Adv. Synth. Catal. 2016, 358, 1478–1481. doi:10.1002/adsc.201501079
Return to citation in text: [1] -
Yang, H.; Gabbaı̈, F. P. J. Am. Chem. Soc. 2015, 137, 13425–13432. doi:10.1021/jacs.5b07998
Return to citation in text: [1] -
Malhotra, D.; Mashuta, M. S.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2014, 53, 4456–4459. doi:10.1002/anie.201310239
Return to citation in text: [1] -
Tang, Y.; Benaissa, I.; Huynh, M.; Vendier, L.; Lugan, N.; Bastin, S.; Belmont, P.; César, V.; Michelet, V. Angew. Chem., Int. Ed. 2019, 58, 7977–7981. doi:10.1002/anie.201901090
Return to citation in text: [1] -
Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L. J. Chem. Rev. 2015, 115, 2596–2697. doi:10.1021/cr300389u
Return to citation in text: [1] -
Veguillas, M.; Rosair, G. M.; Bebbington, M. W. P.; Lee, A.-L. ACS Catal. 2019, 9, 2552–2557. doi:10.1021/acscatal.9b00249
Return to citation in text: [1] -
Couce-Rios, A.; Lledós, A.; Fernández, I.; Ujaque, G. ACS Catal. 2019, 9, 848–858. doi:10.1021/acscatal.8b03843
Return to citation in text: [1] -
Rotta-Loria, N. L.; Chisholm, A. J.; MacQueen, P. M.; McDonald, R.; Ferguson, M. J.; Stradiotto, M. Organometallics 2017, 36, 2470–2475. doi:10.1021/acs.organomet.7b00373
Return to citation in text: [1] -
Couce-Rios, A.; Kovács, G.; Ujaque, G.; Lledós, A. ACS Catal. 2015, 5, 815–829. doi:10.1021/cs501705b
Return to citation in text: [1] -
Manzano, R.; Wurm, T.; Rominger, F.; Hashmi, A. S. K. Chem. – Eur. J. 2014, 20, 6844–6848. doi:10.1002/chem.201402560
Return to citation in text: [1] -
Bailey, G. A.; Lummiss, J. A. M.; Foscato, M.; Occhipinti, G.; McDonald, R.; Jensen, V. R.; Fogg, D. E. J. Am. Chem. Soc. 2017, 139, 16446–16449. doi:10.1021/jacs.7b08578
Return to citation in text: [1] -
Ireland, B. J.; Dobigny, B. T.; Fogg, D. E. ACS Catal. 2015, 5, 4690–4698. doi:10.1021/acscatal.5b00813
Return to citation in text: [1] -
Fleckenstein, C. A.; Plenio, H. J. Org. Chem. 2008, 73, 3236–3244. doi:10.1021/jo8001886
Return to citation in text: [1] -
Schareina, T.; Zapf, A.; Mägerlein, W.; Müller, N.; Beller, M. Chem. – Eur. J. 2007, 13, 6249–6254. doi:10.1002/chem.200700079
Return to citation in text: [1] -
Farmer, J. L.; Pompeo, M.; Lough, A. J.; Organ, M. G. Chem. – Eur. J. 2014, 20, 15790–15798. doi:10.1002/chem.201404705
Return to citation in text: [1] -
Brooner, R. E. M.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2013, 52, 11714–11724. doi:10.1002/anie.201303468
Return to citation in text: [1] -
Vasiuta, R.; Plenio, H. Chem. – Eur. J. 2016, 22, 6353–6360. doi:10.1002/chem.201600264
Return to citation in text: [1] -
Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/c3cc45518a
Return to citation in text: [1] -
Halliday, C. J. V.; Lynam, J. M. Dalton Trans. 2016, 45, 12611–12626. doi:10.1039/c6dt01641c
Return to citation in text: [1] -
Lu, Z.; Hammond, G. B.; Xu, B. Acc. Chem. Res. 2019, 52, 1275–1288. doi:10.1021/acs.accounts.8b00544
Return to citation in text: [1] [2] -
Zimin, D. P.; Dar’in, D. V.; Rassadin, V. A.; Kukushkin, V. Y. Org. Lett. 2018, 20, 4880–4884. doi:10.1021/acs.orglett.8b02019
Return to citation in text: [1] [2] [3] [4] -
Heidrich, M.; Bergmann, M.; Müller-Borges, D.; Plenio, H. Adv. Synth. Catal. 2018, 360, 3572–3578. doi:10.1002/adsc.201800605
Return to citation in text: [1] -
Vasiuta, R.; Stockert, A.; Plenio, H. Chem. Commun. 2018, 54, 1706–1709. doi:10.1039/c7cc08476e
Return to citation in text: [1] -
Kanai, Y.; Foro, S.; Plenio, H. Organometallics 2019, 38, 544–551. doi:10.1021/acs.organomet.8b00836
Return to citation in text: [1] -
Weber, S. G.; Loos, C.; Rominger, F.; Straub, B. F. ARKIVOC 2012, No. iii, 226–242. doi:10.3998/ark.5550190.0013.317
Return to citation in text: [1] -
Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C. R.; Abou-Shehada, S.; Dunn, P. J. Green Chem. 2016, 18, 288–296. doi:10.1039/c5gc01008j
Return to citation in text: [1] -
Alder, C. M.; Hayler, J. D.; Henderson, R. K.; Redman, A. M.; Shukla, L.; Shuster, L. E.; Sneddon, H. F. Green Chem. 2016, 18, 3879–3890. doi:10.1039/c6gc00611f
Return to citation in text: [1] -
Al-Farkh, Y. A.; Al-Hajjar, F. H.; Hamoud, H. S. J. Chem. Eng. Data 1978, 23, 345–349. doi:10.1021/je60079a004
Return to citation in text: [1] -
Schilz, M.; Plenio, H. J. Org. Chem. 2012, 77, 2798–2807. doi:10.1021/jo202644g
Return to citation in text: [1]
| 48. | Alder, C. M.; Hayler, J. D.; Henderson, R. K.; Redman, A. M.; Shukla, L.; Shuster, L. E.; Sneddon, H. F. Green Chem. 2016, 18, 3879–3890. doi:10.1039/c6gc00611f |
| 42. | Zimin, D. P.; Dar’in, D. V.; Rassadin, V. A.; Kukushkin, V. Y. Org. Lett. 2018, 20, 4880–4884. doi:10.1021/acs.orglett.8b02019 |
| 47. | Prat, D.; Wells, A.; Hayler, J.; Sneddon, H.; McElroy, C. R.; Abou-Shehada, S.; Dunn, P. J. Green Chem. 2016, 18, 288–296. doi:10.1039/c5gc01008j |
| 1. | Jia, M.; Bandini, M. ACS Catal. 2015, 5, 1638–1652. doi:10.1021/cs501902v |
| 2. | Schießl, J.; Schulmeister, J.; Doppiu, A.; Wörner, E.; Rudolph, M.; Karch, R.; Hashmi, A. S. K. Adv. Synth. Catal. 2018, 360, 2493–2502. doi:10.1002/adsc.201800233 |
| 3. | Zuccaccia, D.; Belpassi, L.; Macchioni, A.; Tarantelli, F. Eur. J. Inorg. Chem. 2013, 4121–4135. doi:10.1002/ejic.201300285 |
| 23. | Yang, H.; Gabbaı̈, F. P. J. Am. Chem. Soc. 2015, 137, 13425–13432. doi:10.1021/jacs.5b07998 |
| 24. | Malhotra, D.; Mashuta, M. S.; Hammond, G. B.; Xu, B. Angew. Chem., Int. Ed. 2014, 53, 4456–4459. doi:10.1002/anie.201310239 |
| 25. | Tang, Y.; Benaissa, I.; Huynh, M.; Vendier, L.; Lugan, N.; Bastin, S.; Belmont, P.; César, V.; Michelet, V. Angew. Chem., Int. Ed. 2019, 58, 7977–7981. doi:10.1002/anie.201901090 |
| 26. | Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L. J. Chem. Rev. 2015, 115, 2596–2697. doi:10.1021/cr300389u |
| 27. | Veguillas, M.; Rosair, G. M.; Bebbington, M. W. P.; Lee, A.-L. ACS Catal. 2019, 9, 2552–2557. doi:10.1021/acscatal.9b00249 |
| 28. | Couce-Rios, A.; Lledós, A.; Fernández, I.; Ujaque, G. ACS Catal. 2019, 9, 848–858. doi:10.1021/acscatal.8b03843 |
| 46. | Weber, S. G.; Loos, C.; Rominger, F.; Straub, B. F. ARKIVOC 2012, No. iii, 226–242. doi:10.3998/ark.5550190.0013.317 |
| 8. | Marion, N.; Ramón, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448–449. doi:10.1021/ja809403e |
| 9. | Weerasiri, K. C.; Chen, D.; Wozniak, D. I.; Dobereiner, G. E. Adv. Synth. Catal. 2016, 358, 4106–4113. doi:10.1002/adsc.201601013 |
| 10. | Xu, Y.; Hu, X.; Shao, J.; Yang, G.; Wu, Y.; Zhang, Z. Green Chem. 2015, 17, 532–537. doi:10.1039/c4gc01322k |
| 11. | Biasiolo, L.; Del Zotto, A.; Zuccaccia, D. Organometallics 2015, 34, 1759–1765. doi:10.1021/acs.organomet.5b00308 |
| 12. | Xu, X.; Kim, S. H.; Zhang, X.; Das, A. K.; Hirao, H.; Hong, S. H. Organometallics 2013, 32, 164–171. doi:10.1021/om3009603 |
| 13. | Goodwin, J. A.; Aponick, A. Chem. Commun. 2015, 51, 8730–8741. doi:10.1039/c5cc00120j |
| 14. | Carriedo, G. A.; López, S.; Suárez-Suárez, S.; Presa-Soto, D.; Presa-Soto, A. Eur. J. Inorg. Chem. 2011, 1442–1447. doi:10.1002/ejic.201001317 |
| 15. | Wang, W.; Zheng, A.; Zhao, P.; Xia, C.; Li, F. ACS Catal. 2014, 4, 321–327. doi:10.1021/cs400983y |
| 16. | Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067–2074. doi:10.1021/jo802558e |
| 17. | Ibrahim, H.; de Frémont, P.; Braunstein, P.; Théry, V.; Nauton, L.; Cisnetti, F.; Gautier, A. Adv. Synth. Catal. 2015, 357, 3893–3900. doi:10.1002/adsc.201500729 |
| 18. | Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. ACS Catal. 2016, 6, 7363–7376. doi:10.1021/acscatal.6b01626 |
| 19. | Xu, Y.; Hu, X.; Zhang, S.; Xi, X.; Wu, Y. ChemCatChem 2016, 8, 262–267. doi:10.1002/cctc.201501065 |
| 20. | Ibrahim, N.; Vilhelmsen, M. H.; Pernpointner, M.; Rominger, F.; Hashmi, A. S. K. Organometallics 2013, 32, 2576–2583. doi:10.1021/om400060s |
| 21. | Rühling, A.; Galla, H.-J.; Glorius, F. Chem. – Eur. J. 2015, 21, 12291–12294. doi:10.1002/chem.201502542 |
| 22. | Ebule, R. E.; Malhotra, D.; Hammond, G. B.; Xu, B. Adv. Synth. Catal. 2016, 358, 1478–1481. doi:10.1002/adsc.201501079 |
| 42. | Zimin, D. P.; Dar’in, D. V.; Rassadin, V. A.; Kukushkin, V. Y. Org. Lett. 2018, 20, 4880–4884. doi:10.1021/acs.orglett.8b02019 |
| 7. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u |
| 43. | Heidrich, M.; Bergmann, M.; Müller-Borges, D.; Plenio, H. Adv. Synth. Catal. 2018, 360, 3572–3578. doi:10.1002/adsc.201800605 |
| 4. | Hashmi, A. S. K. Chem. Rev. 2007, 107, 3180–3211. doi:10.1021/cr000436x |
| 5. | Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k |
| 6. | Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331–1367. doi:10.1039/c5cs00721f |
| 44. | Vasiuta, R.; Stockert, A.; Plenio, H. Chem. Commun. 2018, 54, 1706–1709. doi:10.1039/c7cc08476e |
| 45. | Kanai, Y.; Foro, S.; Plenio, H. Organometallics 2019, 38, 544–551. doi:10.1021/acs.organomet.8b00836 |
| 39. | Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/c3cc45518a |
| 40. | Halliday, C. J. V.; Lynam, J. M. Dalton Trans. 2016, 45, 12611–12626. doi:10.1039/c6dt01641c |
| 41. | Lu, Z.; Hammond, G. B.; Xu, B. Acc. Chem. Res. 2019, 52, 1275–1288. doi:10.1021/acs.accounts.8b00544 |
| 50. | Schilz, M.; Plenio, H. J. Org. Chem. 2012, 77, 2798–2807. doi:10.1021/jo202644g |
| 37. | Brooner, R. E. M.; Widenhoefer, R. A. Angew. Chem., Int. Ed. 2013, 52, 11714–11724. doi:10.1002/anie.201303468 |
| 38. | Vasiuta, R.; Plenio, H. Chem. – Eur. J. 2016, 22, 6353–6360. doi:10.1002/chem.201600264 |
| 42. | Zimin, D. P.; Dar’in, D. V.; Rassadin, V. A.; Kukushkin, V. Y. Org. Lett. 2018, 20, 4880–4884. doi:10.1021/acs.orglett.8b02019 |
| 32. | Bailey, G. A.; Lummiss, J. A. M.; Foscato, M.; Occhipinti, G.; McDonald, R.; Jensen, V. R.; Fogg, D. E. J. Am. Chem. Soc. 2017, 139, 16446–16449. doi:10.1021/jacs.7b08578 |
| 33. | Ireland, B. J.; Dobigny, B. T.; Fogg, D. E. ACS Catal. 2015, 5, 4690–4698. doi:10.1021/acscatal.5b00813 |
| 34. | Fleckenstein, C. A.; Plenio, H. J. Org. Chem. 2008, 73, 3236–3244. doi:10.1021/jo8001886 |
| 35. | Schareina, T.; Zapf, A.; Mägerlein, W.; Müller, N.; Beller, M. Chem. – Eur. J. 2007, 13, 6249–6254. doi:10.1002/chem.200700079 |
| 36. | Farmer, J. L.; Pompeo, M.; Lough, A. J.; Organ, M. G. Chem. – Eur. J. 2014, 20, 15790–15798. doi:10.1002/chem.201404705 |
| 42. | Zimin, D. P.; Dar’in, D. V.; Rassadin, V. A.; Kukushkin, V. Y. Org. Lett. 2018, 20, 4880–4884. doi:10.1021/acs.orglett.8b02019 |
| 29. | Rotta-Loria, N. L.; Chisholm, A. J.; MacQueen, P. M.; McDonald, R.; Ferguson, M. J.; Stradiotto, M. Organometallics 2017, 36, 2470–2475. doi:10.1021/acs.organomet.7b00373 |
| 30. | Couce-Rios, A.; Kovács, G.; Ujaque, G.; Lledós, A. ACS Catal. 2015, 5, 815–829. doi:10.1021/cs501705b |
| 31. | Manzano, R.; Wurm, T.; Rominger, F.; Hashmi, A. S. K. Chem. – Eur. J. 2014, 20, 6844–6848. doi:10.1002/chem.201402560 |
| 41. | Lu, Z.; Hammond, G. B.; Xu, B. Acc. Chem. Res. 2019, 52, 1275–1288. doi:10.1021/acs.accounts.8b00544 |
| 49. | Al-Farkh, Y. A.; Al-Hajjar, F. H.; Hamoud, H. S. J. Chem. Eng. Data 1978, 23, 345–349. doi:10.1021/je60079a004 |
© 2020 Heidrich and Plenio; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)