Abstract
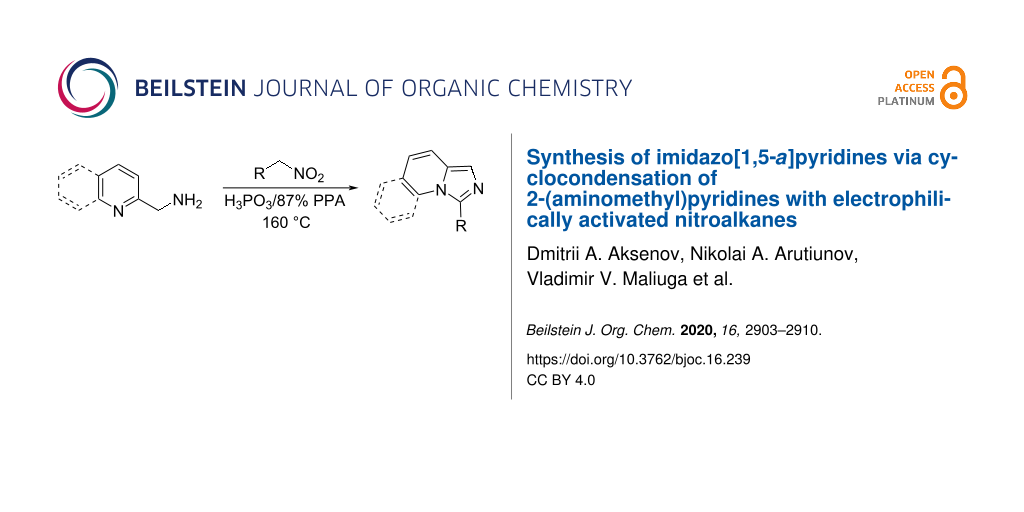
Imidazo[1,5-a]pyridines were efficiently prepared via the cyclization of 2-picolylamines with nitroalkanes electrophilically activated in the presence of phosphorous acid in polyphosphoric acid (PPA) medium.

Graphical Abstract
Introduction
It is hard to overstate the importance of imidazo[1,5-a]pyridines in modern organic and medicinal chemistry. Several natural alkaloids possessing this core were isolated from marine sponges, for example, cribrostatin 6 (Figure 1) [1-3]. The imidazo[1,5-a]pyridine core is also considered to be one of the privileged pharmacophoric scaffolds and can be found in many biologically active compounds, for example, the potent antitumor agent C 1311 inhibiting topoisomerase II [4-9] or pirmagrel, a cytotoxic immunosuppressant and DNA synthesis inhibitor (Figure 1) [10-12]. In addition, compounds with this structure were investigated as photoluminescent sensors [13] and have been employed to generate pincer and heterocyclic carbene ligands for transition metal catalysis [14,15]. A lot of efforts have been dedicated to the development of efficient synthetic methods to access imidazo[1,5-a]pyridines, with more than 120,000 individual compounds prepared to date. Most synthetic approaches rely on various cyclocondensations of nucleophilic (2-aminomethyl)pyridine precursors, introducing a new five-membered ring. Typically, carboxylic acids [16-20], acyl anhydrides [21-23], acyl chlorides [24-27], esters [28], thioamides [29-31], dithionates [32,33], or thiocarbamates [34] are employed as electrophilic components, but oxidative cyclocondensations with aldehydes have also been showcased [35-37]. However, given the importance of these targets, new preparative methods are still in high demand. Herein we demonstrate a new synthetic approach towards imidazo[1,5-a]pyridines, taking advantage of the unusual electrophilic properties of nitroalkanes activated by PPA.
![[1860-5397-16-239-1]](/bjoc/content/figures/1860-5397-16-239-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active imidazo[1,5-a]pyridines.
Figure 1: Biologically active imidazo[1,5-a]pyridines.
Results and Discussion
Within the last few years, we have developed and implemented a number of novel synthetic methodologies in our laboratories, involving acid-mediated cascade transformations of nitroalkenes and nitroalkanes, which target materials science and medicinal chemistry applications. It was demonstrated that upon heating in polyphosphoric acid, the nitroalkanes 1 convert into the phosphorylated nitronates 2, which exhibit strong electrophilic properties. This allowed for the employment of these species in reactions wherein electron-rich arenes serve as carbon-based nucleophilies [38-40]. It was also discovered that the nucleophilic amines 3 can be successfully employed in this type of transformations as well, providing the amidinium intermediates 4, which are susceptible to a variety of subsequent cyclizations. This approach opens up a novel avenue by which to access the benzoxazoles 5 [40], benzimidazoles 6 [40,41], diazines 7 [42,43], or imidazolines 8 (Scheme 1) [44]. We have also shown that a nucleophilic attack on the phosphorylated nitronate species 2 can be carried out with the participation of N-acylhydrazides or thiosemicarbazides to afford the 1,3,4-oxadiazoles 9 [45] and the 1,3,4-thiadiazoles 10 [46], respectively (Scheme 1). Finally, it was found that the reaction of 2-hydrazinylpyridine with electrophilically activated nitroalkanes provides the corresponding triazolopyridines 11 (Scheme 1) [47].
![[1860-5397-16-239-i1]](/bjoc/content/inline/1860-5397-16-239-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Activation of nitroalkanes towards nucleophilic attack by amines.
Scheme 1: Activation of nitroalkanes towards nucleophilic attack by amines.
These results prompted the desire to implement this general scheme in order to access highly important imidazo[1,5-a]pyridines. It was imagined that the initial nucleophilic attack of the 2-(aminomethyl)pyridine (12) on the nitronate 2 would provide an amidinium species 13, which is very well suited for a 5-exo-trig cyclization involving the masked imine moiety of the pyridine ring, providing a 2,3-dihydro-1H-imidazo[1,5-a]pyridin-4-ium ion 14. After deprotonation, the latter would form the 2,3-dihydroimidazo[1,5-a]pyridine 15, which after the elimination of O-phosphorylated hydroxylamine, would afford the imidazo[1,5-a]pyridines 16 (Scheme 2).
![[1860-5397-16-239-i2]](/bjoc/content/inline/1860-5397-16-239-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
To test this idea, nitroethane (1а, 5 equiv) was heated at 110 °C with 85% PPA for 30 min, and 2-(aminomethyl)pyridine (12) was added in small portions. After 3 h of stirring at 110 °C, the reaction mixture was quenched with ice-cold water, neutralized with aqueous ammonia, and extracted with ethyl acetate. The organic layer was analyzed by GC and 1H NMR to reveal that the desired product 16a was formed in a disappointingly low yield (Table 1, entry 1). Heating of the reaction mixture to 130 °C, together with the employment of even more highly concentrated 87% PPA, resulted in an improved conversion, but only insignificantly so (Table 1, entries 2 and 3). To the contrary, an attempt to carry out the reaction in 80% PPA provided a conversion of only 6% even at 140 °C (Table 1, entry 4), whereas no reaction was observed in 100% orthophosphoric acid (Table 1, entry 5). Evidently, under acidic conditions, protonation of the primary amino group in 12 attenuates the nucleophilicity of the molecule, rendering the attack on the relatively bulky phosphorylated nitronate 2 inefficient. To prove this, we decided to moderate the basicity of the amine function by protection with a sulfonyl group. Gratifyingly, the N-tosylamine 17 [48] reacted smoothly with 1-nitropropane (1b), 1-nitrobutane (1c), and 1-nitrohexane (1d) in the presence of 87% PPA, although this required heating to 160 °C to bring the reaction to completion. The corresponding imidazo[1,5-a]pyridines 16b–d were isolated in good practical yield in all three cases (Scheme 3).
![[1860-5397-16-239-i3]](/bjoc/content/inline/1860-5397-16-239-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of the N-tosylate 17 with electrophilic nitroalkanes.
Scheme 3: Reaction of the N-tosylate 17 with electrophilic nitroalkanes.
Table 1: Optimization of the reaction conditions for the cyclization of 2-(aminomethyl)pyridine (12) with nitroethane (1a).
| entry | mediuma | T, °C (t, h) | yield, %b |
| 1 | PPA 85% 1 g/mmol | 110 (3) | 4 |
| 2 | PPA 85% 1 g/mmol | 130 (3) | 13 |
| 3 | PPA 87% 1 g/mmol | 130 (3) | 15 |
| 4 | PPA 80% | 140 (3) | 6 |
| 5 | H3PO4 100% | 140 (5) | 0 |
| 6 | PPA 87% 0.5 g/H3PO3 0.25 g/mmol | 110 (5) | 22 |
| 7 | PPA 87% 0.5 g/H3PO3 0.25 g/mmol | 140 (2) | 43 |
| 8 | PPA 87% 0.5 g/H3PO3 0.5 g/mmol | 140 (1.5) | 62 |
| 9 | PPA 87% 0.5 g/H3PO3 0.5 g/mmol | 160 (2) | 77c |
aAmounts are provided in grams per mmol of substrate 12. bNMR yields, unless specified otherwise. cIsolated yield of purified material.
This approach, however, seems less attractive as it requires an additional step for the proper modification of the starting material. An alternative, more appealing method would capitalize on the in situ generation of a less sterically hindered, and therefore more reactive electrophilic nitronate. As we have recently demonstrated, this type of species can be efficiently generated upon the treatment of nitroalkanes with anhydrous H3PO4 or in combination with 87% PPA [44]. We were pleased to find that the performance of the direct transformation of 12 into 16a was significantly improved after doping the PPA medium with H3PO3. Indeed, in a mixture of 87% PPA and H3PO3 2:1 (mass/mass ratio), the yield reached 22% and 43% at 110 °C and 140 °C, respectively (Table 1, entries 6 and 7). A further increase of the H3PO3 concentration and temperature improved the result. In the 1:1 87% PPA/H3PO3 mixture at 140 °C, the yield of 16a improved to 62% (Table 1, entry 8), while at 160 °C, it reached 77% (Table 1, entry 9). It should be pointed out that the reported yield of 77% is the isolated yield given complete conversion, with no detected side products aside from easily removable polymeric resins.
With optimized reaction conditions in hand, we proceeded with the examination of the scope of, and tolerance towards the differrent nitroalkanes 1b–f. Overall, the reaction proceeded smoothly, providing moderate yields of the corresponding imidazo[1,5-a]pyridines 16b–f (Scheme 4). The only exception to this general trend was the reaction with α-nitrotoluene (1g), which proceeded sluggishly and provided a disappointingly low yield of 3-phenylimidazo[1,5-a]pyridine (16g, Scheme 4).
![[1860-5397-16-239-i4]](/bjoc/content/inline/1860-5397-16-239-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of 2-(aminomethyl)pyridine (12) with electrophilic nitroalkanes.
Scheme 4: Reaction of 2-(aminomethyl)pyridine (12) with electrophilic nitroalkanes.
Next, the scope of nucleophilic substrates was investigated. The readily available 2-(aminomethyl)quinolines 18 bearing various substituents at C-5, C-6, and C-1' were tested in reactions with the nitroalkanes 1a–c and 1e. All of these reactions proceeded uneventfully, affording the corresponding imidazo[1,5-a]pyridines in moderate to good yield (Scheme 5).
![[1860-5397-16-239-i5]](/bjoc/content/inline/1860-5397-16-239-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Reaction of the 2-(aminomethyl)quinolines 18 with electrophilic nitroalkanes.
Scheme 5: Reaction of the 2-(aminomethyl)quinolines 18 with electrophilic nitroalkanes.
We also attempted to address the issue of the low reactivity of α-nitrotoluene (1g). We have previously demonstrated that in the reaction with nucleophilic 1,2-diamines, α-nitroacetophenone (1h) can be employed as a much more versatile synthetic equivalent to afford the corresponding 2-phenylimidazolines [44]. Gratifyingly, this approach also worked well with the substrates 12, 18a, and 18b. The corresponding imidazo[1,5-a]pyridines 16g, 19ag, and 18bg were obtained in a highly efficient manner (Scheme 6). In addition, α-nitroacetic ester (1i) was probed as an electrophile in the reaction with substrate 12 in order to demonstrate that the phosphorylated nitronate function is a superior electrophile in comparison to the ester function. Indeed, this bielectrophilic reagent reacted only at the nitro group, providing the amide 20 as the sole product. The yield was quite marginal, primarily due to decomposition of the fragile ester functionality under the harsh reaction conditions employed. Unfortunately, cyclization did not take place (Scheme 6).
![[1860-5397-16-239-i6]](/bjoc/content/inline/1860-5397-16-239-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reactivity of α-nitroacetophenone (1h) and α-nitroacetic ester (1i).
Scheme 6: Reactivity of α-nitroacetophenone (1h) and α-nitroacetic ester (1i).
Conclusion
In conclusion, a new method to access imidazo[1,5-a]pyridines via the unusual cyclization of 2-(aminomethyl)pyridines or 2-(aminomethyl)quinolines with nitroalkanes that are electrophilically activated in the presence of phosphorous acid in polyphosphoric acid medium was described. The reaction is quite sensitive to steric factors and affords medium to good yields of the products, although relatively harsh reaction conditions are required. The introduction of a phenyl substituent proved problematic in the reactions involving α-nitrotoluene, but this could be easily circumvented by replacing it with the better-performing α-nitroacetophenone (1h).
Experimental
General information. 1H and 13C NMR spectra were recorded on a Bruker Avance-III spectrometer (400 or 100 MHz, respectively) equipped with a BBO probe in CDCl3 or DMSO-d6, using TMS as an internal standard. High-resolution mass spectra were obtained using a Bruker Maxis spectrometer (electrospray ionization, MeCN solution, using HCO2Na/HCO2H for calibration). Melting points were measured with a Stuart smp30 apparatus. All reactions were performed in oven-dried flasks equipped with reflux condensers and magnetic stir bars. All reactions were followed by thin-layer chromatography (TLC) using Silufol UV-254 plates, which were visualized under UV light (254 nm), with acetone, hexane/acetone, or hexane/ethanol/acetone mixtures as eluent. Polyphosphoric acid (87%) was obtained by dissolving a precise amount of P2O5 in 85% orthophosphoric acid according to the published protocols [49,50]. All other reagents and solvents were purchased from commercial vendors and used as received.
1-(6-Bromoquinolin-2-yl)methanamine (18c): typical procedure 1
First, 6-bromo-2-(bromomethyl)quinoline (21c) was prepared from commercially available 6-bromo-2-methylquinoline (22c, via radical bromination in the presence of NBS and dibenzoyl peroxide [51]. To this end, the methylquinoline 22c (3.33 g, 15 mmol) was dissolved in carbon tetrachloride (30 mL), and N-bromosuccinimide (2.94 g, 16.5 mmol) was added, followed by dibenzoyl peroxide (194 mg, 0.80 mmol). The mixture was refluxed for 5–7 h while the reaction progress was monitored by TLC. Upon completion, the reaction mixture was cooled to room temperature, the formed precipitate of succinimide was filtered off, and the filtrate was concentrated in vacuum. The crude product was purified by flash column chromatography eluting with EtOAc/petroleum ether 1:6. Then, the amine was afforded via a Gabriel synthesis using a modified protocol described in the literature [52]. To a stirred solution of 6-bromo-2-(bromomethyl)quinoline (3.01 g, 10 mmol) in DMF (16 mL) was added potassium phthalimide (1.85 g, 10 mmol) in portions. The mixture was stirred at room temperature for 5 h. Then, water (30 mL) was added, and the formed precipitate was collected by filtration and washed with water. Recrystallization from ethanol afforded colorless crystals of intermediate 2-((6-bromoquinolin-2-yl)methyl)isoindoline-1,3-dione. This material (3.05 g, 8.3 mmol) was suspended in ethanol, and hydrazine hydrate (50%, 1.0 g, 10 mmol) was added. The reaction mixture was refluxed for 5 h and then cooled down to 0 °C. The precipitated phthalyl hydrazide was filtered off and rinsed several times with cold ethanol. The combined filtrates were concentrated under reduced pressure to provide the crude amine, which was purified by flash column chromatography on silica gel, eluting with a mixture of dichloromethane/ethanol/triethylamine, gradient 80:10:1–80:30:1. The titled compound was obtained as colorless powder, overall yield 80% (1.89 g, 8.00 mmol). Overall yield 1.89 g (8.00 mmol, 80%). Mp 73–74 °C; Rf 0.50 (CH2Cl2/EtOH/NEt3 80:30:1, v/v/v); 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 1H, 9-H), 7.95 (d, J = 1.0 Hz, 1H, 4-H), 7.91 (d, J = 9.0 Hz, 1H, 6-H), 7.75 (dd, J = 8.8; 1.5 Hz, 1H, 8-H), 7.41 (d, J = 8.5 Hz, 1H, 3-H), 4.17 (s, 2H, CH2), 2.49 (s, 2H, NH); 13C NMR (101 MHz, CDCl3) δ 162.2, 146.4, 135.7, 133.1, 130.8, 129.8, 128.5, 120.8, 120.0, 48.1; ATR-FTIR (ZnSe) νmax: 3293, 2920, 2597, 1908, 1735, 1594, 1489, 1314, 1186, 1061 cm−1; HRESIMS (TOF) m/z: [M + H]+ calcd for C10H10BrN2, 237.0022; found, 237.0022.
3-Methylimidazo[1,5-a]pyridine (16a) [53]: typical procedure 2
A 5 mL Erlenmeyer flask equipped with a magnetic stirring bar was charged with nitroethane (1a, 150 mg, 2.00 mmol), 2-picolylamine (12, 108 mg, 1.00 mmol), 87% polyphosphoric acid (500 mg), and phosphorous acid (500 mg). The flask was capped with a septum, placed into an oil bath preheated to 160 °C, and stirred for 2 h. Then, the mixture was poured into ice-cold water (20 mL), neutralized with aqueous ammonia, and extracted with ethyl acetate (4 × 20 mL). The combined organic extracts were concentrated, the residue dried in vacuum, and then purified by preparative column chromatography on silica gel, eluting with a mixture of petroleum ether and ethyl acetate. The titled compound was obtained as yellow solid (lit. [53] yellow crystals), yield 77% (101 mg, 0.77 mmol). Mp 50–52 °C (lit. [53] mp 49–50 °C, cyclohexane); Rf 0.32 (EtOAc); 1H NMR (400 MHz, DMSO-d6) δ 8.04 (dd, J = 7.1; 1.0 Hz, 1H, 5-H), 7.48 (dt, J = 9.1; 1.1 Hz, 1H, 8-H), 7.24 (d, J = 0.6 Hz, 1H, 1-H), 6.69 (ddd, J = 9.1; 6.3; 0.8 Hz, 1H, 7-H), 6.61 (ddd, 1H, 6-H), 2.57 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 134.8, 129.7, 121.7, 117.9, 117.72, 117.69, 111.8, 12.21. ATR-FTIR (ZnSe) νmax: 3041, 2924, 2857, 1635, 1494, 1442, 1400, 1357, 1322, 1267, 1248, 1170 cm−1; HRESIMS (TOF) m/z: [M + H]+ calcd for C8H9N2, 133.0760; found, 133.0758.
3-Ethylimidazo[1,5-a]pyridine (16b) [54]: typical procedure 3
A 5 mL Erlenmeyer flask equipped with a magnetic stirring bar was charged with 1-nitropropane (1b, 178 mg, 2.00 mmol), 4-methyl-N-(pyridin-2-ylmethyl)benzenesulfonamide (17, 262 mg, 1.00 mmol), and 87% polyphosphoric acid (2.00 g). The flask was capped with a septum, placed into an oil bath preheated to 160 °C, and stirred for 2 h. Upon reaction completion, the aqueous work-up, isolation, and purification of the product was performed in the same way as described in the typical procedure 1. The titled compound was obtained as yellow solid (lit. [54] yellow crystals), yield 66% (96 mg, 0.66 mmol). Alternatively, the same compound was obtained via the typical procedure 1 starting with 1-nitropropane (1b, 178 mg, 2.00 mmol) and 2-picolylamine (12, 108 mg, 1.00 mmol), yield 53% (77 mg, 0.53 mmol). Mp 59–62 °C (lit. [54] mp 61 °C, cyclohexane); Rf 0.27 (EtOAc/petroleum ether 1:1, v/v); 1H NMR (400 MHz, DMSO-d6) δ 8.10 (d, J = 6.6 Hz, 1H, 5-H), 7.49 (d, J = 8.9 Hz, 1H, 8-H), 7.26 (s, 1H, 1-H), 6.70 (t, 1H, 7-H), 6.61 (t, J = 6.0 Hz, 1H, 6-H), 2.96 (dd, J = 14.7; 7.3 Hz, 2H, CH2, 3-Et), 1.30 (t, J = 7.4 Hz, 3H, CH3, 3-Et); 13C NMR (101 MHz, DMSO-d6) δ 139.5, 129.8, 121.6, 118.1, 117.9, 117.6, 111.8, 19.1, 11.2; ATR-FTIR (ZnSe) νmax: 2975, 1638, 1504, 1491, 1454, 1367, 1326, 1241 cm−1; HRESIMS (TOF) m/z: [M + H]+ calcd for C9H11N2, 147.0917; found, 147.0913.
3-Phenylimidazo[1,5-a]pyridine (16g) [36]: typical procedure 4
A 5 mL Erlenmeyer flask equipped with a magnetic stirring bar was charged with 2-nitro-1-phenylethan-1-one (1h, 330 mg, 2.00 mmol), 2-picolylamine (12, 108 mg, 1.00 mmol), 87% polyphosphoric acid (500 mg), and phosphorous acid (500 mg). The flask was capped with a septum, placed into an oil bath preheated to 160 °C, and stirred for 2 h. Upon reaction completion, the aqueous work-up, isolation, and purification of the product were performed in the same way as described in the typical procedure 1. The titled compound was obtained as yellow solid (lit. [36] yellow solid), yield 76% (147 mg, 0.76 mmol). Alternatively, the same compound was obtained via the typical procedure 2 starting with (nitromethyl)benzene (274 mg, 2.00 mmol) and 2-picolylamine (12, 108 mg, 1.00 mmol), yield 12% (24 mg, 0.12 mmol). Mp 108–110 °C (lit. [36] mp 105–110 °C); Rf 0.51 (EtOAc/petroleum ether 1:1, v/v); 1H NMR (400 MHz, DMSO-d6) δ 8.44 (d, J = 7.2 Hz, 1H, 5-H), 7.83 (d, J = 7.3 Hz, 2H, 2,6-H 3-Ph), 7.64 (d, J = 9.1 Hz, 1H, 8-H), 7.55 (dd, J = 9.1; 6.1 Hz, 3H, 3,4,5-H 3-Ph), 7.46 (t, J = 7.4 Hz, 1H, 1-H), 6.83 (dd, J = 9.0; 6.4 Hz, 1H, 7-H), 6.71 (t, J = 6.8 Hz, 1H, 6-H); 13C NMR (101 MHz, DMSO-d6) δ 137.3, 131.4, 130.2, 129.0 (2C), 128.4, 127.6 (2C), 121.8, 120.4, 119.3, 118.5, 113.5; ATR-FTIR (ZnSe) νmax: 3066, 1749, 1676, 1603, 1473, 1458, 1433, 1358, 1305, 1251, 1267, 1120 cm−1; HRESIMS (TOF) m/z: [M + H]+ calcd for C13H11N2, 195.0917; found, 195.0918.
6-Bromo-7-methoxyimidazo[1,5-a]quinoline (19de)
The title compound was obtained via the typical procedure 2, starting from nitromethane (1e, 122 mg, 2.00 mmol) and (5-bromo-6-methoxyquinolin-2-yl)methanamine (18d, 267 mg, 1.00 mmol). Pale yellow solid, yield 72% (198 mg, 0.72 mmol). Mp 177–179 °C; Rf 0.43 (acetone/petroleum ether 1:1, v/v); 1H NMR (400 MHz, DMSO-d6) δ 9.11 (d, J = 0.8 Hz, 1H, 1-H), 8.42 (dd, J = 9.2; 0.7 Hz, 1H, 9-H), 7.48 (s, 1H, 3-H), 7.45 (d, J = 9.2 Hz, 1H, 4-H), 7.44 (s, 1H, 5-H), 7.37 (d, J = 9.8 Hz, 1H, 8-H), 3.96 (s, 3H, CH3); 13C NMR (101 MHz, DMSO-d6) δ 153.4, 129.7, 127.4, 125.7, 124.0, 123.0, 119.3, 118.8, 115.9, 113.4, 109.9, 56.9; ATR-FTIR (ZnSe) νmax: 3106, 2938, 2846, 1603, 1466, 1436, 1423, 1273, 1208, 1129, 1069 cm−1; HRESIMS (TOF) m/z: [M + H]+ calcd for C12H10BrN2O, 276.9971; found, 276.9974.
Supporting Information
| Supporting Information File 1: Synthetic procedures and characterization data for compounds 16c–f, 18b and 18d, 19aa, 19ab, 19ac, 19ag, 19bb, 19bg, 19ce, and 20 as well as 1H NMR, 13C NMR, and HRMS spectral charts for all new compounds. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Calcul, L.; Longeon, A.; Al Mourabit, A.; Guyot, M.; Bourguet-Kondracki, M.-L. Tetrahedron 2003, 59, 6539–6544. doi:10.1016/s0040-4020(03)01069-x
Return to citation in text: [1] -
Pettit, G. R.; Collins, J. C.; Knight, J. C.; Herald, D. L.; Nieman, R. A.; Williams, M. D.; Pettit, R. K. J. Nat. Prod. 2003, 66, 544–547. doi:10.1021/np020012t
Return to citation in text: [1] -
Yu, H.-B.; Yang, F.; Sun, F.; Ma, G.-Y.; Gan, J.-H.; Hu, W.-Z.; Han, B.-N.; Jiao, W.-H.; Lin, H.-W. J. Nat. Prod. 2014, 77, 2124–2129. doi:10.1021/np500583z
Return to citation in text: [1] -
Augustin, E.; Pawłowska, M.; Polewska, J.; Potega, A.; Mazerska, Z. Cell Biol. Int. 2013, 37, 109–120. doi:10.1002/cbin.10018
Return to citation in text: [1] -
Isambert, N.; Campone, M.; Bourbouloux, E.; Drouin, M.; Major, A.; Yin, W.; Loadman, P.; Capizzi, R.; Grieshaber, C.; Fumoleau, P. Eur. J. Cancer 2010, 46, 729–734. doi:10.1016/j.ejca.2009.12.005
Return to citation in text: [1] -
Paradziej-Łukowicz, J.; Skwarska, A.; Peszyńska-Sularz, G.; Brillowska-Dąbrowska, A.; Konopa, J. Cancer Biol. Ther. 2011, 12, 586–597. doi:10.4161/cbt.12.7.15980
Return to citation in text: [1] -
Polewska, J.; Skwarska, A.; Augustin, E.; Konopa, J. J. Pharmacol. Exp. Ther. 2013, 346, 393–405. doi:10.1124/jpet.113.203851
Return to citation in text: [1] -
Skwarska, A.; Augustin, E.; Konopa, J. Apoptosis 2007, 12, 2245–2257. doi:10.1007/s10495-007-0144-y
Return to citation in text: [1] -
Zaffaroni, N.; De Marco, C.; Villa, R.; Riboldi, S.; Daidone, M. G.; Double, J. A. Eur. J. Cancer 2001, 37, 1953–1962. doi:10.1016/s0959-8049(01)00227-1
Return to citation in text: [1] -
Gresele, P.; Blockmans, D.; Deckmyn, H.; Vermylen, J. J. Pharmacol. Exp. Ther. 1988, 246, 301–307.
Return to citation in text: [1] -
Huang, A.; Sun, D.; Koller, A. Hypertension 1993, 22, 913–921. doi:10.1161/01.hyp.22.6.913
Return to citation in text: [1] -
Reilly, I. A.; Doran, J. B.; Smith, B.; FitzGerald, G. A. Circulation 1986, 73, 1300–1309. doi:10.1161/01.cir.73.6.1300
Return to citation in text: [1] -
Volpi, G.; Garino, C.; Priola, E.; Magistris, C.; Chierotti, M. R.; Barolo, C. Dyes Pigm. 2019, 171, 107713. doi:10.1016/j.dyepig.2019.107713
Return to citation in text: [1] -
Cui, Y.; Ge, Y.; Li, Y.; Tao, J.; Yao, J.; Dong, Y. Struct. Chem. 2020, 31, 547–555. doi:10.1007/s11224-019-01429-3
Return to citation in text: [1] -
Park, D.-A.; Byun, S.; Ryu, J. Y.; Lee, J.; Lee, J.; Hong, S. ACS Catal. 2020, 10, 5443–5453. doi:10.1021/acscatal.0c00802
Return to citation in text: [1] -
Volpi, G.; Lace, B.; Garino, C.; Priola, E.; Artuso, E.; Cerreia Vioglio, P.; Barolo, C.; Fin, A.; Genre, A.; Prandi, C. Dyes Pigm. 2018, 157, 298–304. doi:10.1016/j.dyepig.2018.04.037
Return to citation in text: [1] -
Volpi, G.; Garino, C.; Priola, E.; Diana, E.; Gobetto, R.; Buscaino, R.; Viscardi, G.; Barolo, C. Dyes Pigm. 2017, 143, 284–290. doi:10.1016/j.dyepig.2017.04.034
Return to citation in text: [1] -
Singh, D.; Kumar, V.; Devi, N.; Malakar, C. C.; Shankar, R.; Singh, V. Adv. Synth. Catal. 2017, 359, 1213–1226. doi:10.1002/adsc.201600970
Return to citation in text: [1] -
Buil, M. A.; Calbet, M.; Castillo, M.; Castro, J.; Esteve, C.; Ferrer, M.; Forns, P.; González, J.; López, S.; Roberts, R. S.; Sevilla, S.; Vidal, B.; Vidal, L.; Vilaseca, P. Eur. J. Med. Chem. 2016, 113, 102–133. doi:10.1016/j.ejmech.2016.02.023
Return to citation in text: [1] -
Chino, A.; Masuda, N.; Amano, Y.; Honbou, K.; Mihara, T.; Yamazaki, M.; Tomishima, M. Bioorg. Med. Chem. 2014, 22, 3515–3526. doi:10.1016/j.bmc.2014.04.023
Return to citation in text: [1] -
Amini-Rentsch, L.; Vanoli, E.; Richard-Bildstein, S.; Marti, R.; Vilé, G. Ind. Eng. Chem. Res. 2019, 58, 10164–10171. doi:10.1021/acs.iecr.9b01906
Return to citation in text: [1] -
Bosch, P.; García, V.; Bilen, B. S.; Sucunza, D.; Domingo, A.; Mendicuti, F.; Vaquero, J. J. Dyes Pigm. 2017, 138, 135–146. doi:10.1016/j.dyepig.2016.11.041
Return to citation in text: [1] -
Tverdiy, D. O.; Chekanov, M. O.; Savitskiy, P. V.; Syniugin, A. R.; Yarmoliuk, S. M.; Fokin, A. A. Synthesis 2016, 48, 4269–4277. doi:10.1055/s-0035-1561489
Return to citation in text: [1] -
Kurhade, S.; Konstantinidou, M.; Sutanto, F.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Dömling, A. Eur. J. Org. Chem. 2019, 2029–2034. doi:10.1002/ejoc.201801880
Return to citation in text: [1] -
Nirogi, R.; Mohammed, A. R.; Shinde, A. K.; Bogaraju, N.; Gagginapalli, S. R.; Ravella, S. R.; Kota, L.; Bhyrapuneni, G.; Muddana, N. R.; Benade, V.; Palacharla, R. C.; Jayarajan, P.; Subramanian, R.; Goyal, V. K. Eur. J. Med. Chem. 2015, 103, 289–301. doi:10.1016/j.ejmech.2015.08.051
Return to citation in text: [1] -
Roberts, L. R.; Bradley, P. A.; Bunnage, M. E.; England, K. S.; Fairman, D.; Fobian, Y. M.; Fox, D. N. A.; Gymer, G. E.; Heasley, S. E.; Molette, J.; Smith, G. L.; Schmidt, M. A.; Tones, M. A.; Dack, K. N. Bioorg. Med. Chem. Lett. 2011, 21, 6515–6518. doi:10.1016/j.bmcl.2011.08.071
Return to citation in text: [1] -
Trotter, B. W.; Nanda, K. K.; Burgey, C. S.; Potteiger, C. M.; Deng, J. Z.; Green, A. I.; Hartnett, J. C.; Kett, N. R.; Wu, Z.; Henze, D. A.; Della Penna, K.; Desai, R.; Leitl, M. D.; Lemaire, W.; White, R. B.; Yeh, S.; Urban, M. O.; Kane, S. A.; Hartman, G. D.; Bilodeau, M. T. Bioorg. Med. Chem. Lett. 2011, 21, 2354–2358. doi:10.1016/j.bmcl.2011.02.082
Return to citation in text: [1] -
Kurhade, S.; Diekstra, E.; Sutanto, F.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Dömling, A. Org. Lett. 2018, 20, 3871–3874. doi:10.1021/acs.orglett.8b01452
Return to citation in text: [1] -
Fuse, S.; Ohuchi, T.; Asawa, Y.; Sato, S.; Nakamura, H. Bioorg. Med. Chem. Lett. 2016, 26, 5887–5890. doi:10.1016/j.bmcl.2016.11.009
Return to citation in text: [1] -
Murai, T.; Nagaya, E.; Shibahara, F.; Maruyama, T. Org. Biomol. Chem. 2012, 10, 4943–4953. doi:10.1039/c2ob25438g
Return to citation in text: [1] -
Tahara, S.; Shibahara, F.; Maruyama, T.; Murai, T. Chem. Commun. 2009, 7009–7011. doi:10.1039/b910172a
Return to citation in text: [1] -
Ramesha, A. B.; Pavan Kumar, C. S.; Sandhya, N. C.; Kumara, M. N.; Mantelingu, K.; Rangappa, K. S. RSC Adv. 2016, 6, 48375–48378. doi:10.1039/c6ra03771b
Return to citation in text: [1] -
Ramesha, A. B.; Sandhya, N. C.; Pavan Kumar, C. S.; Hiremath, M.; Mantelingu, K.; Rangappa, K. S. New J. Chem. 2016, 40, 7637–7642. doi:10.1039/c6nj01038e
Return to citation in text: [1] -
Späth, A.; Gonschor, J.; König, B. Monatsh. Chem. 2011, 142, 1289–1308. doi:10.1007/s00706-011-0660-x
Return to citation in text: [1] -
Bori, J.; Behera, N.; Mahata, S.; Manivannan, V. ChemistrySelect 2017, 2, 11727–11731. doi:10.1002/slct.201702420
Return to citation in text: [1] -
Li, M.; Xie, Y.; Ye, Y.; Zou, Y.; Jiang, H.; Zeng, W. Org. Lett. 2014, 16, 6232–6235. doi:10.1021/ol503165b
Return to citation in text: [1] [2] [3] [4] -
Shibahara, F.; Sugiura, R.; Yamaguchi, E.; Kitagawa, A.; Murai, T. J. Org. Chem. 2009, 74, 3566–3568. doi:10.1021/jo900415y
Return to citation in text: [1] -
Aksenov, A. V.; Aksenov, N. A.; Nadein, O. N.; Aksenova, I. V. Synlett 2010, 2628–2630. doi:10.1055/s-0030-1258767
Return to citation in text: [1] -
Aksenov, A. V.; Aksenov, N. A.; Orazova, N. A.; Aksenov, D. A.; Dmitriev, M. V.; Rubin, M. RSC Adv. 2015, 5, 84849–84855. doi:10.1039/c5ra17668a
Return to citation in text: [1] -
Aksenov, N. A.; Aksenov, A. V.; Nadein, O. N.; Aksenov, D. A.; Smirnov, A. N.; Rubin, M. RSC Adv. 2015, 5, 71620–71626. doi:10.1039/c5ra15128g
Return to citation in text: [1] [2] [3] -
Aksenov, A. V.; Smirnov, A. N.; Aksenov, N. A.; Bijieva, A. S.; Aksenova, I. V.; Rubin, M. Org. Biomol. Chem. 2015, 13, 4289–4295. doi:10.1039/c5ob00131e
Return to citation in text: [1] -
Aksenov, A. V.; Aksenov, N. A.; Ovcharov, D. S.; Aksenov, D. A.; Griaznov, G.; Voskressensky, L. G.; Rubin, M. RSC Adv. 2016, 6, 82425–82431. doi:10.1039/c6ra17269e
Return to citation in text: [1] -
Aksenov, A. V.; Ovcharov, D. S.; Aksenov, N. A.; Aksenov, D. A.; Nadein, O. N.; Rubin, M. RSC Adv. 2017, 7, 29927–29932. doi:10.1039/c7ra04751g
Return to citation in text: [1] -
Aksenov, A. V.; Aksenov, N. A.; Arutiunov, N. A.; Malyuga, V. V.; Ovcharov, S. N.; Rubin, M. RSC Adv. 2019, 9, 39458–39465. doi:10.1039/c9ra08630g
Return to citation in text: [1] [2] [3] -
Aksenov, A. V.; Khamraev, V.; Aksenov, N. A.; Kirilov, N. K.; Domenyuk, D. A.; Zelensky, V. A.; Rubin, M. RSC Adv. 2019, 9, 6636–6642. doi:10.1039/c9ra00976k
Return to citation in text: [1] -
Aksenov, N. A.; Arutiunov, N. A.; Kirillov, N. K.; Aksenov, D. A.; Aksenov, A. V.; Rubin, M. Chem. Heterocycl. Compd. 2020, 56, 1067–1072. doi:10.1007/s10593-020-02775-5
Return to citation in text: [1] -
Aksenov, N. A.; Aksenov, A. V.; Kirilov, N. K.; Arutiunov, N. A.; Aksenov, D. A.; Maslivetc, V.; Zhao, Z.; Du, L.; Rubin, M.; Kornienko, A. Org. Biomol. Chem. 2020, 18, 6651–6664. doi:10.1039/d0ob01007c
Return to citation in text: [1] -
Klingele, M. H.; Moubaraki, B.; Murray, K. S.; Brooker, S. Chem. – Eur. J. 2005, 11, 6962–6973. doi:10.1002/chem.200500387
Return to citation in text: [1] -
Huhti, A.-L.; Gartaganis, P. A. Can. J. Chem. 1956, 34, 785–797. doi:10.1139/v56-102
Return to citation in text: [1] -
Uhlig, F. Angew. Chem. 1954, 66, 435–436. doi:10.1002/ange.19540661503
Return to citation in text: [1] -
Lyapchev, R.; Petrov, P.; Dangalov, M.; Vassilev, N. G. J. Organomet. Chem. 2017, 851, 194–209. doi:10.1016/j.jorganchem.2017.09.036
Return to citation in text: [1] -
Zhao, Q.; Liu, S.; Li, Y.; Wang, Q. J. Agric. Food Chem. 2009, 57, 2849–2855. doi:10.1021/jf803632t
Return to citation in text: [1] -
Vu, T. Y.; Chrostowska, A.; Huynh, T. K. X.; Khayar, S.; Dargelos, A.; Justyna, K.; Pasternak, B.; Leśniak, S.; Wentrup, C. Chem. – Eur. J. 2013, 19, 14983–14988. doi:10.1002/chem.201301663
Return to citation in text: [1] [2] [3] -
Winterfeld, K.; Franzke, H. Angew. Chem. 1963, 75, 1101–1102. doi:10.1002/ange.19630752209
Return to citation in text: [1] [2] [3]
| 53. | Vu, T. Y.; Chrostowska, A.; Huynh, T. K. X.; Khayar, S.; Dargelos, A.; Justyna, K.; Pasternak, B.; Leśniak, S.; Wentrup, C. Chem. – Eur. J. 2013, 19, 14983–14988. doi:10.1002/chem.201301663 |
| 53. | Vu, T. Y.; Chrostowska, A.; Huynh, T. K. X.; Khayar, S.; Dargelos, A.; Justyna, K.; Pasternak, B.; Leśniak, S.; Wentrup, C. Chem. – Eur. J. 2013, 19, 14983–14988. doi:10.1002/chem.201301663 |
| 53. | Vu, T. Y.; Chrostowska, A.; Huynh, T. K. X.; Khayar, S.; Dargelos, A.; Justyna, K.; Pasternak, B.; Leśniak, S.; Wentrup, C. Chem. – Eur. J. 2013, 19, 14983–14988. doi:10.1002/chem.201301663 |
| 1. | Calcul, L.; Longeon, A.; Al Mourabit, A.; Guyot, M.; Bourguet-Kondracki, M.-L. Tetrahedron 2003, 59, 6539–6544. doi:10.1016/s0040-4020(03)01069-x |
| 2. | Pettit, G. R.; Collins, J. C.; Knight, J. C.; Herald, D. L.; Nieman, R. A.; Williams, M. D.; Pettit, R. K. J. Nat. Prod. 2003, 66, 544–547. doi:10.1021/np020012t |
| 3. | Yu, H.-B.; Yang, F.; Sun, F.; Ma, G.-Y.; Gan, J.-H.; Hu, W.-Z.; Han, B.-N.; Jiao, W.-H.; Lin, H.-W. J. Nat. Prod. 2014, 77, 2124–2129. doi:10.1021/np500583z |
| 14. | Cui, Y.; Ge, Y.; Li, Y.; Tao, J.; Yao, J.; Dong, Y. Struct. Chem. 2020, 31, 547–555. doi:10.1007/s11224-019-01429-3 |
| 15. | Park, D.-A.; Byun, S.; Ryu, J. Y.; Lee, J.; Lee, J.; Hong, S. ACS Catal. 2020, 10, 5443–5453. doi:10.1021/acscatal.0c00802 |
| 40. | Aksenov, N. A.; Aksenov, A. V.; Nadein, O. N.; Aksenov, D. A.; Smirnov, A. N.; Rubin, M. RSC Adv. 2015, 5, 71620–71626. doi:10.1039/c5ra15128g |
| 13. | Volpi, G.; Garino, C.; Priola, E.; Magistris, C.; Chierotti, M. R.; Barolo, C. Dyes Pigm. 2019, 171, 107713. doi:10.1016/j.dyepig.2019.107713 |
| 40. | Aksenov, N. A.; Aksenov, A. V.; Nadein, O. N.; Aksenov, D. A.; Smirnov, A. N.; Rubin, M. RSC Adv. 2015, 5, 71620–71626. doi:10.1039/c5ra15128g |
| 41. | Aksenov, A. V.; Smirnov, A. N.; Aksenov, N. A.; Bijieva, A. S.; Aksenova, I. V.; Rubin, M. Org. Biomol. Chem. 2015, 13, 4289–4295. doi:10.1039/c5ob00131e |
| 10. | Gresele, P.; Blockmans, D.; Deckmyn, H.; Vermylen, J. J. Pharmacol. Exp. Ther. 1988, 246, 301–307. |
| 11. | Huang, A.; Sun, D.; Koller, A. Hypertension 1993, 22, 913–921. doi:10.1161/01.hyp.22.6.913 |
| 12. | Reilly, I. A.; Doran, J. B.; Smith, B.; FitzGerald, G. A. Circulation 1986, 73, 1300–1309. doi:10.1161/01.cir.73.6.1300 |
| 35. | Bori, J.; Behera, N.; Mahata, S.; Manivannan, V. ChemistrySelect 2017, 2, 11727–11731. doi:10.1002/slct.201702420 |
| 36. | Li, M.; Xie, Y.; Ye, Y.; Zou, Y.; Jiang, H.; Zeng, W. Org. Lett. 2014, 16, 6232–6235. doi:10.1021/ol503165b |
| 37. | Shibahara, F.; Sugiura, R.; Yamaguchi, E.; Kitagawa, A.; Murai, T. J. Org. Chem. 2009, 74, 3566–3568. doi:10.1021/jo900415y |
| 36. | Li, M.; Xie, Y.; Ye, Y.; Zou, Y.; Jiang, H.; Zeng, W. Org. Lett. 2014, 16, 6232–6235. doi:10.1021/ol503165b |
| 4. | Augustin, E.; Pawłowska, M.; Polewska, J.; Potega, A.; Mazerska, Z. Cell Biol. Int. 2013, 37, 109–120. doi:10.1002/cbin.10018 |
| 5. | Isambert, N.; Campone, M.; Bourbouloux, E.; Drouin, M.; Major, A.; Yin, W.; Loadman, P.; Capizzi, R.; Grieshaber, C.; Fumoleau, P. Eur. J. Cancer 2010, 46, 729–734. doi:10.1016/j.ejca.2009.12.005 |
| 6. | Paradziej-Łukowicz, J.; Skwarska, A.; Peszyńska-Sularz, G.; Brillowska-Dąbrowska, A.; Konopa, J. Cancer Biol. Ther. 2011, 12, 586–597. doi:10.4161/cbt.12.7.15980 |
| 7. | Polewska, J.; Skwarska, A.; Augustin, E.; Konopa, J. J. Pharmacol. Exp. Ther. 2013, 346, 393–405. doi:10.1124/jpet.113.203851 |
| 8. | Skwarska, A.; Augustin, E.; Konopa, J. Apoptosis 2007, 12, 2245–2257. doi:10.1007/s10495-007-0144-y |
| 9. | Zaffaroni, N.; De Marco, C.; Villa, R.; Riboldi, S.; Daidone, M. G.; Double, J. A. Eur. J. Cancer 2001, 37, 1953–1962. doi:10.1016/s0959-8049(01)00227-1 |
| 38. | Aksenov, A. V.; Aksenov, N. A.; Nadein, O. N.; Aksenova, I. V. Synlett 2010, 2628–2630. doi:10.1055/s-0030-1258767 |
| 39. | Aksenov, A. V.; Aksenov, N. A.; Orazova, N. A.; Aksenov, D. A.; Dmitriev, M. V.; Rubin, M. RSC Adv. 2015, 5, 84849–84855. doi:10.1039/c5ra17668a |
| 40. | Aksenov, N. A.; Aksenov, A. V.; Nadein, O. N.; Aksenov, D. A.; Smirnov, A. N.; Rubin, M. RSC Adv. 2015, 5, 71620–71626. doi:10.1039/c5ra15128g |
| 36. | Li, M.; Xie, Y.; Ye, Y.; Zou, Y.; Jiang, H.; Zeng, W. Org. Lett. 2014, 16, 6232–6235. doi:10.1021/ol503165b |
| 28. | Kurhade, S.; Diekstra, E.; Sutanto, F.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Dömling, A. Org. Lett. 2018, 20, 3871–3874. doi:10.1021/acs.orglett.8b01452 |
| 32. | Ramesha, A. B.; Pavan Kumar, C. S.; Sandhya, N. C.; Kumara, M. N.; Mantelingu, K.; Rangappa, K. S. RSC Adv. 2016, 6, 48375–48378. doi:10.1039/c6ra03771b |
| 33. | Ramesha, A. B.; Sandhya, N. C.; Pavan Kumar, C. S.; Hiremath, M.; Mantelingu, K.; Rangappa, K. S. New J. Chem. 2016, 40, 7637–7642. doi:10.1039/c6nj01038e |
| 54. | Winterfeld, K.; Franzke, H. Angew. Chem. 1963, 75, 1101–1102. doi:10.1002/ange.19630752209 |
| 24. | Kurhade, S.; Konstantinidou, M.; Sutanto, F.; Kurpiewska, K.; Kalinowska-Tłuścik, J.; Dömling, A. Eur. J. Org. Chem. 2019, 2029–2034. doi:10.1002/ejoc.201801880 |
| 25. | Nirogi, R.; Mohammed, A. R.; Shinde, A. K.; Bogaraju, N.; Gagginapalli, S. R.; Ravella, S. R.; Kota, L.; Bhyrapuneni, G.; Muddana, N. R.; Benade, V.; Palacharla, R. C.; Jayarajan, P.; Subramanian, R.; Goyal, V. K. Eur. J. Med. Chem. 2015, 103, 289–301. doi:10.1016/j.ejmech.2015.08.051 |
| 26. | Roberts, L. R.; Bradley, P. A.; Bunnage, M. E.; England, K. S.; Fairman, D.; Fobian, Y. M.; Fox, D. N. A.; Gymer, G. E.; Heasley, S. E.; Molette, J.; Smith, G. L.; Schmidt, M. A.; Tones, M. A.; Dack, K. N. Bioorg. Med. Chem. Lett. 2011, 21, 6515–6518. doi:10.1016/j.bmcl.2011.08.071 |
| 27. | Trotter, B. W.; Nanda, K. K.; Burgey, C. S.; Potteiger, C. M.; Deng, J. Z.; Green, A. I.; Hartnett, J. C.; Kett, N. R.; Wu, Z.; Henze, D. A.; Della Penna, K.; Desai, R.; Leitl, M. D.; Lemaire, W.; White, R. B.; Yeh, S.; Urban, M. O.; Kane, S. A.; Hartman, G. D.; Bilodeau, M. T. Bioorg. Med. Chem. Lett. 2011, 21, 2354–2358. doi:10.1016/j.bmcl.2011.02.082 |
| 34. | Späth, A.; Gonschor, J.; König, B. Monatsh. Chem. 2011, 142, 1289–1308. doi:10.1007/s00706-011-0660-x |
| 36. | Li, M.; Xie, Y.; Ye, Y.; Zou, Y.; Jiang, H.; Zeng, W. Org. Lett. 2014, 16, 6232–6235. doi:10.1021/ol503165b |
| 21. | Amini-Rentsch, L.; Vanoli, E.; Richard-Bildstein, S.; Marti, R.; Vilé, G. Ind. Eng. Chem. Res. 2019, 58, 10164–10171. doi:10.1021/acs.iecr.9b01906 |
| 22. | Bosch, P.; García, V.; Bilen, B. S.; Sucunza, D.; Domingo, A.; Mendicuti, F.; Vaquero, J. J. Dyes Pigm. 2017, 138, 135–146. doi:10.1016/j.dyepig.2016.11.041 |
| 23. | Tverdiy, D. O.; Chekanov, M. O.; Savitskiy, P. V.; Syniugin, A. R.; Yarmoliuk, S. M.; Fokin, A. A. Synthesis 2016, 48, 4269–4277. doi:10.1055/s-0035-1561489 |
| 54. | Winterfeld, K.; Franzke, H. Angew. Chem. 1963, 75, 1101–1102. doi:10.1002/ange.19630752209 |
| 16. | Volpi, G.; Lace, B.; Garino, C.; Priola, E.; Artuso, E.; Cerreia Vioglio, P.; Barolo, C.; Fin, A.; Genre, A.; Prandi, C. Dyes Pigm. 2018, 157, 298–304. doi:10.1016/j.dyepig.2018.04.037 |
| 17. | Volpi, G.; Garino, C.; Priola, E.; Diana, E.; Gobetto, R.; Buscaino, R.; Viscardi, G.; Barolo, C. Dyes Pigm. 2017, 143, 284–290. doi:10.1016/j.dyepig.2017.04.034 |
| 18. | Singh, D.; Kumar, V.; Devi, N.; Malakar, C. C.; Shankar, R.; Singh, V. Adv. Synth. Catal. 2017, 359, 1213–1226. doi:10.1002/adsc.201600970 |
| 19. | Buil, M. A.; Calbet, M.; Castillo, M.; Castro, J.; Esteve, C.; Ferrer, M.; Forns, P.; González, J.; López, S.; Roberts, R. S.; Sevilla, S.; Vidal, B.; Vidal, L.; Vilaseca, P. Eur. J. Med. Chem. 2016, 113, 102–133. doi:10.1016/j.ejmech.2016.02.023 |
| 20. | Chino, A.; Masuda, N.; Amano, Y.; Honbou, K.; Mihara, T.; Yamazaki, M.; Tomishima, M. Bioorg. Med. Chem. 2014, 22, 3515–3526. doi:10.1016/j.bmc.2014.04.023 |
| 29. | Fuse, S.; Ohuchi, T.; Asawa, Y.; Sato, S.; Nakamura, H. Bioorg. Med. Chem. Lett. 2016, 26, 5887–5890. doi:10.1016/j.bmcl.2016.11.009 |
| 30. | Murai, T.; Nagaya, E.; Shibahara, F.; Maruyama, T. Org. Biomol. Chem. 2012, 10, 4943–4953. doi:10.1039/c2ob25438g |
| 31. | Tahara, S.; Shibahara, F.; Maruyama, T.; Murai, T. Chem. Commun. 2009, 7009–7011. doi:10.1039/b910172a |
| 54. | Winterfeld, K.; Franzke, H. Angew. Chem. 1963, 75, 1101–1102. doi:10.1002/ange.19630752209 |
| 45. | Aksenov, A. V.; Khamraev, V.; Aksenov, N. A.; Kirilov, N. K.; Domenyuk, D. A.; Zelensky, V. A.; Rubin, M. RSC Adv. 2019, 9, 6636–6642. doi:10.1039/c9ra00976k |
| 42. | Aksenov, A. V.; Aksenov, N. A.; Ovcharov, D. S.; Aksenov, D. A.; Griaznov, G.; Voskressensky, L. G.; Rubin, M. RSC Adv. 2016, 6, 82425–82431. doi:10.1039/c6ra17269e |
| 43. | Aksenov, A. V.; Ovcharov, D. S.; Aksenov, N. A.; Aksenov, D. A.; Nadein, O. N.; Rubin, M. RSC Adv. 2017, 7, 29927–29932. doi:10.1039/c7ra04751g |
| 44. | Aksenov, A. V.; Aksenov, N. A.; Arutiunov, N. A.; Malyuga, V. V.; Ovcharov, S. N.; Rubin, M. RSC Adv. 2019, 9, 39458–39465. doi:10.1039/c9ra08630g |
| 51. | Lyapchev, R.; Petrov, P.; Dangalov, M.; Vassilev, N. G. J. Organomet. Chem. 2017, 851, 194–209. doi:10.1016/j.jorganchem.2017.09.036 |
| 52. | Zhao, Q.; Liu, S.; Li, Y.; Wang, Q. J. Agric. Food Chem. 2009, 57, 2849–2855. doi:10.1021/jf803632t |
| 44. | Aksenov, A. V.; Aksenov, N. A.; Arutiunov, N. A.; Malyuga, V. V.; Ovcharov, S. N.; Rubin, M. RSC Adv. 2019, 9, 39458–39465. doi:10.1039/c9ra08630g |
| 49. | Huhti, A.-L.; Gartaganis, P. A. Can. J. Chem. 1956, 34, 785–797. doi:10.1139/v56-102 |
| 50. | Uhlig, F. Angew. Chem. 1954, 66, 435–436. doi:10.1002/ange.19540661503 |
| 48. | Klingele, M. H.; Moubaraki, B.; Murray, K. S.; Brooker, S. Chem. – Eur. J. 2005, 11, 6962–6973. doi:10.1002/chem.200500387 |
| 44. | Aksenov, A. V.; Aksenov, N. A.; Arutiunov, N. A.; Malyuga, V. V.; Ovcharov, S. N.; Rubin, M. RSC Adv. 2019, 9, 39458–39465. doi:10.1039/c9ra08630g |
| 46. | Aksenov, N. A.; Arutiunov, N. A.; Kirillov, N. K.; Aksenov, D. A.; Aksenov, A. V.; Rubin, M. Chem. Heterocycl. Compd. 2020, 56, 1067–1072. doi:10.1007/s10593-020-02775-5 |
| 47. | Aksenov, N. A.; Aksenov, A. V.; Kirilov, N. K.; Arutiunov, N. A.; Aksenov, D. A.; Maslivetc, V.; Zhao, Z.; Du, L.; Rubin, M.; Kornienko, A. Org. Biomol. Chem. 2020, 18, 6651–6664. doi:10.1039/d0ob01007c |
© 2020 Aksenov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)