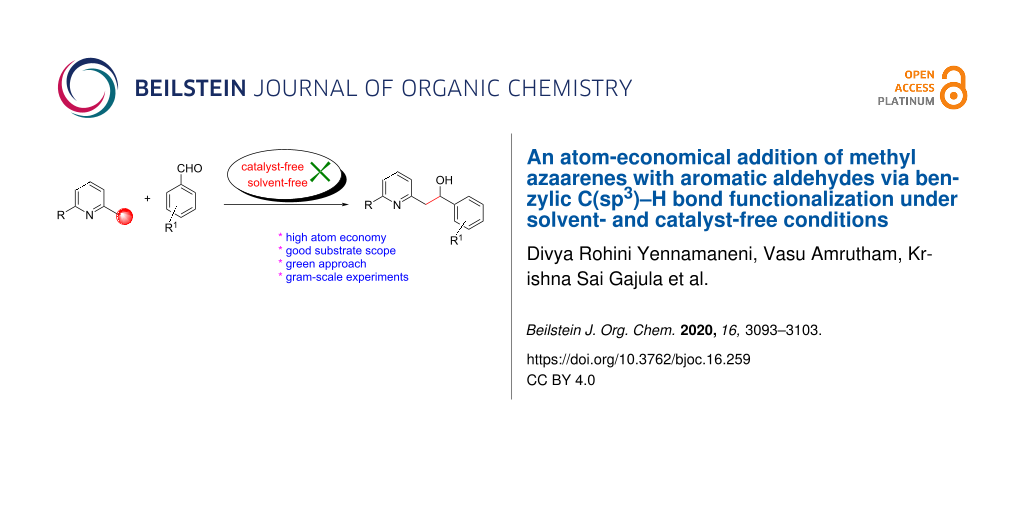
Abstract
A convenient practical approach for the synthesis of 2-(pyridin-2-yl)ethanols by direct benzylic addition of azaarenes and aldehydes under catalyst- and solvent-free conditions is reported. This reaction is metal-free, green, and was carried out in a facile operative environment without using any hazardous transition metal catalysts or any other coupling reagents. Different aromatic aldehydes and azaarenes were monitored, and the yields of the resulting products were moderate to excellent. We accomplished several azaarene derivatives under neat conditions through a highly atom-economical pathway. To evaluate the preparative potential of this process, gram-scale reactions were performed up to a 10 g scale.

Graphical Abstract
Introduction
Azaarenes are a distinct class of heterocyclic compounds possessing wide compatibility in the field of synthetic organic chemistry. The recent advancements in nitrogen-containing carbon compounds have marked them as an unusual moiety due to their attractive applications in biology and as materials [1-4]. Among various nitrogen-containing heterocyclic compounds, pyridine and quinolines are readily found in bioactive compounds [5]. The functionalization of alkylpyridines and quinolines is significant and plays a remarkable role in the efficient drug design [6-8]. Due to their conformational diversity, these compounds constitute a motif in various natural alkaloid products, such as chimanine and those derived from lobelia, sedum, etc. These compounds act as anti-HIV and anti-asthma drugs [9-11]. Mainly, 2-substituted quinolines and their analogues exhibits magnificent bioactivity [12,13]. The synthesis of higher cores of nitrogen-containing heterocyclic compounds through C(sp3)–H functionalization of simple compounds like methyl azaarenes allows direct transformation without any critical reaction conditions. Thus, in this regard the further development of this approach is still in need to be developed [14].
The formation of new C–C bonds through direct C–H bond functionalization in organic chemistry is attractive [15]. Such methodologies are omnipresent and facilitate sustainable organic transformations for the synthesis of complex natural products and pharmaceuticals. In the past years, C−H bond functionalization catalyzed by transition metals received a strong emphasis, and other different catalytic systems have also been encouraged [16-20]. Huang and co-workers first realized the addition of alkylazaarenes directly to unsaturated bonds via C(sp3)–H functionalization [21,22]. The synthesis of pyridine and related azaarene derivatives involve the C(sp3)–H activation of 2-methylpyridines using different transition-metal compounds, Lewis acids, and Brønsted acids [23-28]. Recently, C(sp3)–H functionalizations of methylazaarenes with isatins and malononitrile under catalyst-free conditions have been reported [29]. Wang et al. reported the functionalization of benzylic C–H bonds of 2-methylazaarenes by nucleophilic addition to aromatic aldehydes catalyzed by acetic acid using harmful chlorinated solvent, and this reaction suffers from longer reaction times [30]. Rao et al. performed similar reactions without catalyst under microwave irradiation in the presence of water as a solvent [31], but when considering an industrial scale, there are numerous factors that serves as obstacles for the usage of microwave reactors, such as escalated heat loss, variations in the absorption, an inadequate penetrating ability of the radiation into the reaction medium, and further reflection of the microwaves. Nevertheless, the solvent was water, which required long extraction processes compared to solvent-free conditions. Castro and co-workers have also reported the synthesis of 1-phenyl-2-(2-pyridyl)ethanol and 1-phenyl-2-(2-pyridyl)ethene under catalyst- and solvent-free conditions. Despite the low yield of the product of around 4%, this work described crystallographic data and the influence of reaction conditions such as the temperature and the reaction time on the stability of 1-phenyl-2-(2-pyridyl)ethanol [32]. Different catalysts such as ionic liquids [33], CuFe2O4 [34], and Ca(OTf)2 [35] were also utilized. Due to the abundant importance of these products, several different approaches were reported (Scheme 1 and Scheme 2). These higher azaarene products were also achieved from other starting materials, such as 2-methyl-6-(phenylethynyl)pyridine [36], 2,6-dimethylpyridine (1a) and (E)-N-benzylidene-4-methylbenzenesulfonamide [22], 2-bromo-6-methylpyridine and (E)-styrylboronic acid [37], as well as 2-methylpyridine and phenylmethanamine [38]. Most recently, Yaragorla et al. published a review on C(sp3)–H bond functionalization of 2-methylazaarenes [39]. These strategies are proficient, but due to the involvement of drastic reaction conditions, the use of expensive reagents, toxic metals, harmful solvents, and tedious workup procedures, they need to be reevaluated. Therefore, alternatives with environmentally benign reagents are much in focus.
![[1860-5397-16-259-i1]](/bjoc/content/inline/1860-5397-16-259-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Benzylic addition of aldehydes to azaarenes using different catalysts.
Scheme 1: Benzylic addition of aldehydes to azaarenes using different catalysts.
![[1860-5397-16-259-i2]](/bjoc/content/inline/1860-5397-16-259-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of azaarene derivatives from different precursors.
Scheme 2: Synthesis of azaarene derivatives from different precursors.
Correspondingly, considering the exemplar shift from conventional synthetic methodologies towards green chemistry, there have been some alternatives or replacements of toxic catalysts and hazardous solvents in chemical reactions [40]. These conversions play a vital role for the syntheses of active pharmaceutical ingredients as these approaches reduce the contamination in an industrial setting. In addition, the replacement of toxic reagents by environmentally benign reagents can decrease pollution to some extent [41,42].
However, to develop advantageous eco-friendly, atom-economical, simple, and efficient synthetic-organic processes under solvent- and catalyst-free conditions in order to synthesize highly demanding pharmaceuticals or natural products can be quite backbreaking [43-45]. From this perspective, we herein disclose environmentally benign, atom-economical, catalyst-free nucleophilic additions of benzylic-like azaarene C–H groups to various benzaldehydes under neat conditions (Scheme 3).
![[1860-5397-16-259-i3]](/bjoc/content/inline/1860-5397-16-259-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Our work: catalyst- and solvent-free benzylic addition of aldehydes to azaarenes.
Scheme 3: Our work: catalyst- and solvent-free benzylic addition of aldehydes to azaarenes.
Results and Discussion
With the purpose to develop an environmentally benign process, we first examined p-nitrobenzaldehyde (2a) and 2,6-dimethylpyridine (1a) as model substrates. Our investigation started with the reaction of p-nitrobenzaldehyde (2a) and 2,6-dimethylpyridine (1a) in the presence of Hβ zeolite as a catalyst at 120 °C in toluene for 24 h in a sealed vial (Table 1, entry 1). However, the desired compound 3a was observed in traces. We thought that the catalyst pore size may be an obstruction to a higher yield and used a mesoporous material, MCM-41, as a catalyst, which slightly improved the yield (Table 1, entry 2). At the same time, increasing the temperature to 130 °C enhanced the yield of the product 3a (Table 1, entry 3). In order to address the economic cost and ecological impact, we tried this reaction without using a solvent (Table 1, entry 4). Further, this reaction was screened using various zeolites, such as Hβ, H-mordenite, H-ZSM, HY, NaY, and MCM-41 under solvent-free conditions (Table 1, entries 5–9). In addition, to justify the role of the catalyst, this reaction was carried out in the absence of a catalyst. Thankfully, the yield of the product enhanced to 79% (Table 1, entry 10). We managed to increase the yield of the desired product 3a by further screening of the reaction conditions under catalyst- as well as solvent-free conditions. As we increased the temperature to 135 °C, the yield of the product increased to 84% (Table 1, entry 11). An additional increase of the temperature to 140 °C did not exhibit any promising change, but decreasing the temperature to 120 °C reduced the yield of the product 3a (Table 1, entries 12 and 13). Hence, we carried out further reactions at 135 °C. Generally, the temperature of the reaction plays a crucial role, in formation of a uniform reaction mixture of the reactants under solvent-free conditions. Increasing the reaction time did not produce a remarkable change, but when decreasing the reaction time, a drastic effect on the yield of the product 3a was observed (Table 1, entries 14 and 15). Surprisingly, when the reaction was carried out in an open atmosphere, the yield of the required product 3a was drastically reduced (Table 1, entry 16). Different reactant stoichiometries were analyzed to maximize the yield of the desired product 3a (Table 1, entries 17–20). After these tests, we concluded the best yield of 3a to be 92% (Table 1, entry 20), which was achieved from a reaction mixture of 2 mmol 2,6-dimethylpyridine (1a) and 1 mmol p-nitrobenzaldehyde (2a) at 135 °C for 24 h in a sealed vial without any catalysts or additive under solvent-free conditions.
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-259-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | catalyst | solvent | temperature (°C) | yield (%)b |
| 1 | Hβ | toluene | 120 | 4 |
| 2 | MCM-41 | toluene | 120 | 10 |
| 3 | MCM-41 | toluene | 130 | 23 |
| 4 | MCM-41 | — | 130 | 62 |
| 5 | Hβ | — | 130 | 76 |
| 6 | H-mordenite | — | 130 | 38 |
| 7 | H-ZSM | — | 130 | 72 |
| 8 | HY | — | 130 | 30 |
| 9 | NaY | — | 130 | 6 |
| 10 | — | — | 130 | 79 |
| 11 | — | — | 135 | 84 |
| 12 | — | — | 140 | 82 |
| 13 | — | — | 120 | 36 |
| 14 | — | — | 135 | 36c |
| 15 | — | — | 135 | 80d |
| 16 | — | — | 135 | 58e |
| 17 | — | — | 135 | 63f |
| 18 | — | — | 135 | 85g |
| 19 | — | — | 135 | 85h |
| 20 | — | — | 135 | 92i |
a1 mmol 1a, 1 mmol 2a, 24 h. bBased on 2a. c18 h. d30 h. eOpen atmosphere. f0.8 mmol 1a. g1.5 mmol 1a. h1.75 mmol 1a. i2 mmol 1a.
Thereupon, these optimized conditions were utilized to validate the substrate scope of this direct C–C coupling reaction. A variety of aldehydes, 2a–r, was reacted with 2,6-dimethylpyridine (1a) to obtain the corresponding products 3a–r with a moderate to excellent yield (Table 2). The substrates 2a–d with nitro substituents were well reacted to achieve the desired products 3a–d in 68–95% yield (Table 2, entries 1–4). Distinctly, 2-nitrobenzaldehyde (2c) gave the expected product 3c along with the dehydrated product 4c (Table 2, entry 3). Substrates containing a halogen, such as fluorine, chlorine, and bromine, were efficiently reacted to afford the products 3e–h in successively moderate yields (Table 2, entries 5–8). Notably, 3-fluorobenzaldehyde (2e) also reacted well and provided the alcohol 3e in 55% yield along with the alkene product 4e in 19% yield (Table 2, entry 5). Exceptionally, substrates with groups such as cyano, trifluoromethyl, and formyl were reacted and provided the respective dehydrated alkenylpyridine compounds 4i–k in 77%, 71%, and 92% yield, respectively (Table 2, entries 9–11). 4-Acetamidobenzaldehyde (2l) also reacted but provided a low product yield of 22% 3l (Table 2, entry 12). Starting materials with a para- and ortho-hydroxy group provided the corresponding alkenylpyridine products 4m and 4n in 58% and 34% yield, respectively (Table 2, entries 13 and 14). 4-Methylbenzaldehyde (2o) gave the respective dehydrated product 4o in 46% yield upon reaction for 48 h (Table 2, entry 15). Simple benzaldehyde (2p) was also tolerated well and furnished the corresponding product 3p in 45% yield (Table 2, entry 16). The heteroaromatic aldehyde 2-pyridinecarbaldehyde (2q) gave the respective dehydrated product 4q in 96% yield, whereas 2-thiophene (2r) resulted in the desired product 3r but in a low yield (Table 2, entries 17 and 18).
Table 2: Variation of the aldehyde component 2.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-259-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | substrate 2 | product 3 | product 4 | yield (%)b | |
| 3 | 4 | ||||
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-259-i8.svg?max-width=637&scale=1.0)
2a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-259-i9.svg?max-width=637&scale=1.0)
3a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-259-i10.svg?max-width=637&scale=1.0)
4a |
92 | 0 |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-259-i11.svg?max-width=637&scale=1.0)
2b |
![[Graphic 7]](/bjoc/content/inline/1860-5397-16-259-i12.svg?max-width=637&scale=1.0)
3b |
![[Graphic 8]](/bjoc/content/inline/1860-5397-16-259-i13.svg?max-width=637&scale=1.0)
4b |
68 | 0 |
| 3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-16-259-i14.svg?max-width=637&scale=1.0)
2c |
![[Graphic 10]](/bjoc/content/inline/1860-5397-16-259-i15.svg?max-width=637&scale=1.0)
3c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-16-259-i16.svg?max-width=637&scale=1.0)
4c |
74 | 17 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-16-259-i17.svg?max-width=637&scale=1.0)
2d |
![[Graphic 13]](/bjoc/content/inline/1860-5397-16-259-i18.svg?max-width=637&scale=1.0)
3d |
![[Graphic 14]](/bjoc/content/inline/1860-5397-16-259-i19.svg?max-width=637&scale=1.0)
4d |
95 | 0 |
| 5 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-16-259-i20.svg?max-width=637&scale=1.0)
2e |
![[Graphic 16]](/bjoc/content/inline/1860-5397-16-259-i21.svg?max-width=637&scale=1.0)
3e |
![[Graphic 17]](/bjoc/content/inline/1860-5397-16-259-i22.svg?max-width=637&scale=1.0)
4e |
55 | 19 |
| 6 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-16-259-i23.svg?max-width=637&scale=1.0)
2f |
![[Graphic 19]](/bjoc/content/inline/1860-5397-16-259-i24.svg?max-width=637&scale=1.0)
3f |
![[Graphic 20]](/bjoc/content/inline/1860-5397-16-259-i25.svg?max-width=637&scale=1.0)
4f |
52 | 0 |
| 7 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-16-259-i26.svg?max-width=637&scale=1.0)
2g |
![[Graphic 22]](/bjoc/content/inline/1860-5397-16-259-i27.svg?max-width=637&scale=1.0)
3g |
![[Graphic 23]](/bjoc/content/inline/1860-5397-16-259-i28.svg?max-width=637&scale=1.0)
4g |
48 | 0 |
| 8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-16-259-i29.svg?max-width=637&scale=1.0)
2h |
![[Graphic 25]](/bjoc/content/inline/1860-5397-16-259-i30.svg?max-width=637&scale=1.0)
3h |
![[Graphic 26]](/bjoc/content/inline/1860-5397-16-259-i31.svg?max-width=637&scale=1.0)
4h |
42 | 0 |
| 9 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-16-259-i32.svg?max-width=637&scale=1.0)
2i |
![[Graphic 28]](/bjoc/content/inline/1860-5397-16-259-i33.svg?max-width=637&scale=1.0)
3i |
![[Graphic 29]](/bjoc/content/inline/1860-5397-16-259-i34.svg?max-width=637&scale=1.0)
4i |
0 | 77 |
| 10 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-16-259-i35.svg?max-width=637&scale=1.0)
2j |
![[Graphic 31]](/bjoc/content/inline/1860-5397-16-259-i36.svg?max-width=637&scale=1.0)
3j |
![[Graphic 32]](/bjoc/content/inline/1860-5397-16-259-i37.svg?max-width=637&scale=1.0)
4j |
0 | 71 |
| 11 |
![[Graphic 33]](/bjoc/content/inline/1860-5397-16-259-i38.svg?max-width=637&scale=1.0)
2k |
![[Graphic 34]](/bjoc/content/inline/1860-5397-16-259-i39.svg?max-width=637&scale=1.0)
3k |
![[Graphic 35]](/bjoc/content/inline/1860-5397-16-259-i40.svg?max-width=637&scale=1.0)
4k |
0 | 92 |
| 12 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-16-259-i41.svg?max-width=637&scale=1.0)
2l |
![[Graphic 37]](/bjoc/content/inline/1860-5397-16-259-i42.svg?max-width=637&scale=1.0)
3l |
![[Graphic 38]](/bjoc/content/inline/1860-5397-16-259-i43.svg?max-width=637&scale=1.0)
4l |
22 | 0 |
| 13 |
![[Graphic 39]](/bjoc/content/inline/1860-5397-16-259-i44.svg?max-width=637&scale=1.0)
2m |
![[Graphic 40]](/bjoc/content/inline/1860-5397-16-259-i45.svg?max-width=637&scale=1.0)
3m |
![[Graphic 41]](/bjoc/content/inline/1860-5397-16-259-i46.svg?max-width=637&scale=1.0)
4m |
0 | 58 |
| 14 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-16-259-i47.svg?max-width=637&scale=1.0)
2n |
![[Graphic 43]](/bjoc/content/inline/1860-5397-16-259-i48.svg?max-width=637&scale=1.0)
3n |
![[Graphic 44]](/bjoc/content/inline/1860-5397-16-259-i49.svg?max-width=637&scale=1.0)
4n |
0 | 34 |
| 15 |
![[Graphic 45]](/bjoc/content/inline/1860-5397-16-259-i50.svg?max-width=637&scale=1.0)
2o |
![[Graphic 46]](/bjoc/content/inline/1860-5397-16-259-i51.svg?max-width=637&scale=1.0)
3o |
![[Graphic 47]](/bjoc/content/inline/1860-5397-16-259-i52.svg?max-width=637&scale=1.0)
4o |
0 | 46c |
| 16 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-16-259-i53.svg?max-width=637&scale=1.0)
2p |
![[Graphic 49]](/bjoc/content/inline/1860-5397-16-259-i54.svg?max-width=637&scale=1.0)
3p |
![[Graphic 50]](/bjoc/content/inline/1860-5397-16-259-i55.svg?max-width=637&scale=1.0)
4p |
45 | 0 |
| 17 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-16-259-i56.svg?max-width=637&scale=1.0)
2q |
![[Graphic 52]](/bjoc/content/inline/1860-5397-16-259-i57.svg?max-width=637&scale=1.0)
3q |
![[Graphic 53]](/bjoc/content/inline/1860-5397-16-259-i58.svg?max-width=637&scale=1.0)
4q |
0 | 96 |
| 18 |
![[Graphic 54]](/bjoc/content/inline/1860-5397-16-259-i59.svg?max-width=637&scale=1.0)
2r |
![[Graphic 55]](/bjoc/content/inline/1860-5397-16-259-i60.svg?max-width=637&scale=1.0)
3r |
![[Graphic 56]](/bjoc/content/inline/1860-5397-16-259-i61.svg?max-width=637&scale=1.0)
4r |
29c | 0 |
a2 mmol 1a, 1 mmol 2, 24 h. bBased on 2. c48 h.
Subsequently, several azaarenes in combination with 4-nitrobenzaldehyde (2a) were also evaluated under optimized conditions to extend the substrate scope. Firstly 2,4,6-collidine (1b) was successfully reacted for 48 h to obtain the successive product 5b in 87% yield (Table 3, entry 1). Next, 2-ethylpyridine (1c) was made to react under standard reaction conditions. To our surprise, 1-(4-nitrophenyl)-2-(pyridin-2-yl)propan-1-ol (5c) was formed in 53% yield, and the formation of this product displays the reactivity of the benzylic C–H group attached in the ortho position of the pyridine ring (Table 3, entry 2). The reaction of 2-methylpyridine (1d) with 2a for 48 h gave the desired alcohol 5d in 42% yield along with the corresponding dehydrated alkenylpyridine compound 6d in 16% yield (Table 3, entry 3). Different from the 2,6-substitution pattern, 3,5-dimethylpyridine (1e) was also tested under standard reaction conditions upon increasing the reaction time to 48 h, which did not furnish the corresponding products (Table 3, entry 4). Later, to screen the compatibility of the azaarenes, various quinolines were screened (Table 3, entries 5–8). Among these, only 2-methylquinoline (1f) and 6-fluoro-2-methylquinoline (1g) gave the dehydrated alkenylazaarenes 6f and 6g as the products in 48% and 45% yield, respectively (Table 3, entries 5 and 6). We also tried this reaction with 6-chloro-2-methylquinoline (1h) and 6-bromo-2-methylquinoline (1g), but unfortunately the reactions did not proceed well, and the desired products 6h and 6i were not formed (Table 3, entries 7 and 8).
Table 3: Variation of the azaarene component 1.a
![[Graphic 57]](/bjoc/content/inline/1860-5397-16-259-i62.svg?max-width=637&scale=1.0)
|
|||||
| entry | substrate | product 5 | product 6 | yield (%)b | |
| 5 | 6 | ||||
| 1 |
![[Graphic 58]](/bjoc/content/inline/1860-5397-16-259-i63.svg?max-width=637&scale=1.0)
1b |
![[Graphic 59]](/bjoc/content/inline/1860-5397-16-259-i64.svg?max-width=637&scale=1.0)
5b |
![[Graphic 60]](/bjoc/content/inline/1860-5397-16-259-i65.svg?max-width=637&scale=1.0)
6b |
87c | 0 |
| 2 |
![[Graphic 61]](/bjoc/content/inline/1860-5397-16-259-i66.svg?max-width=637&scale=1.0)
1c |
![[Graphic 62]](/bjoc/content/inline/1860-5397-16-259-i67.svg?max-width=637&scale=1.0)
5c |
![[Graphic 63]](/bjoc/content/inline/1860-5397-16-259-i68.svg?max-width=637&scale=1.0)
6c |
53c | 0 |
| 3 |
![[Graphic 64]](/bjoc/content/inline/1860-5397-16-259-i69.svg?max-width=637&scale=1.0)
1d |
![[Graphic 65]](/bjoc/content/inline/1860-5397-16-259-i70.svg?max-width=637&scale=1.0)
5d |
![[Graphic 66]](/bjoc/content/inline/1860-5397-16-259-i71.svg?max-width=637&scale=1.0)
6d |
42c | 16c |
| 4 |
![[Graphic 67]](/bjoc/content/inline/1860-5397-16-259-i72.svg?max-width=637&scale=1.0)
1e |
![[Graphic 68]](/bjoc/content/inline/1860-5397-16-259-i73.svg?max-width=637&scale=1.0)
5e |
![[Graphic 69]](/bjoc/content/inline/1860-5397-16-259-i74.svg?max-width=637&scale=1.0)
6e |
0 | 0 |
| 5 |
![[Graphic 70]](/bjoc/content/inline/1860-5397-16-259-i75.svg?max-width=637&scale=1.0)
1f |
![[Graphic 71]](/bjoc/content/inline/1860-5397-16-259-i76.svg?max-width=637&scale=1.0)
5f |
![[Graphic 72]](/bjoc/content/inline/1860-5397-16-259-i77.svg?max-width=637&scale=1.0)
6f |
0 | 48 |
| 6 |
![[Graphic 73]](/bjoc/content/inline/1860-5397-16-259-i78.svg?max-width=637&scale=1.0)
1g |
![[Graphic 74]](/bjoc/content/inline/1860-5397-16-259-i79.svg?max-width=637&scale=1.0)
5g |
![[Graphic 75]](/bjoc/content/inline/1860-5397-16-259-i80.svg?max-width=637&scale=1.0)
6g |
0 | 45 |
| 7 |
![[Graphic 76]](/bjoc/content/inline/1860-5397-16-259-i81.svg?max-width=637&scale=1.0)
1h |
![[Graphic 77]](/bjoc/content/inline/1860-5397-16-259-i82.svg?max-width=637&scale=1.0)
5h |
![[Graphic 78]](/bjoc/content/inline/1860-5397-16-259-i83.svg?max-width=637&scale=1.0)
6h |
0 | 0 |
| 8 |
![[Graphic 79]](/bjoc/content/inline/1860-5397-16-259-i84.svg?max-width=637&scale=1.0)
1i |
![[Graphic 80]](/bjoc/content/inline/1860-5397-16-259-i85.svg?max-width=637&scale=1.0)
5i |
![[Graphic 81]](/bjoc/content/inline/1860-5397-16-259-i86.svg?max-width=637&scale=1.0)
6i |
0 | 0 |
a2 mmol 1, 1 mmol 2a, 24 h. bBased on 2a. c48 h.
However, some dehydrated alkenyl products were obtained, which may possess wide biological activities of great demand. This reaction was also carried out on a gram scale, and the results display the potential for large-scale applications as the yields were not drastically changed when shifting from a mmol scale to a gram scale (1 g, 5 g, and 10 g). These results are shown in Scheme 4.
![[1860-5397-16-259-i4]](/bjoc/content/inline/1860-5397-16-259-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Large-scale experiments for the synthesis of 2-(6-methylpyridin-2-yl)-1-(4-nitrophenyl)ethan-1-ol (3a) from 2,6-dimethylpyridine (1a) and 4-nitrobenzaldehyde (2a).
Scheme 4: Large-scale experiments for the synthesis of 2-(6-methylpyridin-2-yl)-1-(4-nitrophenyl)ethan-1-ol (...
Based on the experimental investigations and literature reports [46], conclusions were drawn, and a plausible mechanism was suggested. Under standard reaction conditions, 2 mmol of 2,6-dimethylpyridine participates in inter reaction (1 mmol of 1a will inter react to another 1 mmol of 1a) and this inter reaction influences the nitrogen atom present in the compound to act as a Brønsted base. This resulting Brønsted base accepts benzylic C–H protons to furnish the respective enamine A, which facilitates the further nucleophilic addition to 4-nitrobenzaldehyde (2a). The product of this is the intermediate B, which favors the formation of the corresponding desired product 3a, as shown in Scheme 5.
![[1860-5397-16-259-i5]](/bjoc/content/inline/1860-5397-16-259-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism for the formation of 2-(6-methylpyridin-2-yl)-1-(4-nitrophenyl)ethan-1-ol (3a) under standard reaction conditions from 2,6-dimethylpyridine (1a) and 4-nitrobenzaldehyde (2a).
Scheme 5: Plausible mechanism for the formation of 2-(6-methylpyridin-2-yl)-1-(4-nitrophenyl)ethan-1-ol (3a) ...
Conclusion
In summary, this work provides a green and simple synthetic methodology for the synthesis of higher azaarenes from pyridines and quinolines with aromatic aldehydes, avoiding the use of solvents and a catalyst. Despite of previous reports under catalyst- and solvent-free conditions, this approach features specific traits, such as good yields and simple reaction conditions. This method requires no reagent purification and is a new and clean process. A variety of aromatic aldehydes, including electron-donating, electron-withdrawing, as well as halogen groups was screened, affording the products in moderate to excellent yields. Azaarenes such as pyridines with various substitution patterns and a few quinolines were also reacted with moderate to excellent yields. This draws the conclusion that the benzylic C–H unit of 2-alkylpyridine/quinolines actively participates in this addition reaction. However, in some cases, 2-alkenyl compounds were formed. Nevertheless, these are of equivalent significance. Moreover, this method has a high atom economy and reduces large amounts of waste generated by the unnecessary utilization of catalysts and solvents. Notably, this reaction is compatible with a gram scale, and further research is yet to be developed towards more sustainability.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, and NMR spectra. | ||
| Format: PDF | Size: 4.2 MB | Download |
References
-
Chelucci, G.; Orrù, G.; Pinna, G. A. Tetrahedron 2003, 59, 9471–9515. doi:10.1016/j.tet.2003.09.066
Return to citation in text: [1] -
Prehn, K.; Warburg, A.; Schilling, T.; Bron, M.; Schulte, K. Compos. Sci. Technol. 2009, 69, 1570–1579. doi:10.1016/j.compscitech.2008.09.006
Return to citation in text: [1] -
Kem, W. R.; Soti, F.; Rittschof, D. Biomol. Eng. 2003, 20, 355–361. doi:10.1016/s1389-0344(03)00049-2
Return to citation in text: [1] -
Friesen, R. W.; Brideau, C.; Chan, C. C.; Charleson, S.; Deschênes, D.; Dubé, D.; Ethier, D.; Fortin, R.; Gauthier, J. Y.; Girard, Y.; Gordon, R.; Greig, G. M.; Riendeau, D.; Savoie, C.; Wang, Z.; Wong, E.; Visco, D.; Xu, L. J.; Young, R. N. Bioorg. Med. Chem. Lett. 1998, 8, 2777–2782. doi:10.1016/s0960-894x(98)00499-5
Return to citation in text: [1] -
Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Nat. Rev. Drug Discovery 2002, 1, 493–502. doi:10.1038/nrd839
Return to citation in text: [1] -
Solomon, V. R.; Lee, H. Curr. Med. Chem. 2011, 18, 1488–1508. doi:10.2174/092986711795328382
Return to citation in text: [1] -
Wilson, K. J.; van Niel, M. B.; Cooper, L.; Bloomfield, D.; O’Connor, D.; Fish, L. R.; MacLeod, A. M. Bioorg. Med. Chem. Lett. 2007, 17, 2643–2648. doi:10.1016/j.bmcl.2007.01.098
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2005, 22, 627–646. doi:10.1039/b413750g
Return to citation in text: [1] -
Strunz, G. M.; Findlay, J. A. Pyridine and Piperidine Alkaloids. In The Alkaloids; Brossi, A., Ed.; Academic Press: New York, NY, USA, 1985; pp 89–183. doi:10.1016/s0099-9598(08)60194-7
Return to citation in text: [1] -
Meth-Cohn, O.; Yau, C. C.; Yu, C.-Y. J. Heterocycl. Chem. 1999, 36, 1549–1553. doi:10.1002/jhet.5570360615
Return to citation in text: [1] -
Houghton, P. J.; Woldemariam, T. Z.; Watanabe, Y.; Yates, M. Planta Med. 1999, 65, 250–254. doi:10.1055/s-1999-13988
Return to citation in text: [1] -
Kumar, S.; Bawa, S.; Gupta, H. Mini-Rev. Med. Chem. 2009, 9, 1648–1654. doi:10.2174/138955709791012247
Return to citation in text: [1] -
Fournet, A.; Barrios, A. A.; Muñoz, V.; Hocquemiller, R.; Roblot, F.; Cavé, A.; Richomme, P.; Bruneton, J. Phytother. Res. 1994, 8, 174–178. doi:10.1002/ptr.2650080312
Return to citation in text: [1] -
Nakamura, I.; Yamamoto, Y. Chem. Rev. 2004, 104, 2127–2198. doi:10.1021/cr020095i
Return to citation in text: [1] -
Campeau, L.-C.; Rousseaux, S.; Fagnou, K. J. Am. Chem. Soc. 2005, 127, 18020–18021. doi:10.1021/ja056800x
Return to citation in text: [1] -
Kanyiva, K. S.; Nakao, Y.; Hiyama, T. Angew. Chem., Int. Ed. 2007, 46, 8872–8874. doi:10.1002/anie.200703758
Return to citation in text: [1] -
Wu, J.; Cui, X.; Chen, L.; Jiang, G.; Wu, Y. J. Am. Chem. Soc. 2009, 131, 13888–13889. doi:10.1021/ja902762a
Return to citation in text: [1] -
Mousseau, J. J.; Bull, J. A.; Charette, A. B. Angew. Chem., Int. Ed. 2010, 49, 1115–1118. doi:10.1002/anie.200906020
Return to citation in text: [1] -
Deng, G.; Ueda, K.; Yanagisawa, S.; Itami, K.; Li, C.-J. Chem. – Eur. J. 2009, 15, 333–337. doi:10.1002/chem.200801893
Return to citation in text: [1] -
Tobisu, M.; Hyodo, I.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 12070–12071. doi:10.1021/ja9053509
Return to citation in text: [1] -
Qian, B.; Guo, S.; Shao, J.; Zhu, Q.; Yang, L.; Xia, C.; Huang, H. J. Am. Chem. Soc. 2010, 132, 3650–3651. doi:10.1021/ja910104n
Return to citation in text: [1] -
Qian, B.; Xie, P.; Xie, Y.; Huang, H. Org. Lett. 2011, 13, 2580–2583. doi:10.1021/ol200684b
Return to citation in text: [1] [2] -
Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. – Eur. J. 2010, 16, 2654–2672. doi:10.1002/chem.200902374
Return to citation in text: [1] -
Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e
Return to citation in text: [1] -
Kalyani, D.; Deprez, N. R.; Desai, L. V.; Sanford, M. S. J. Am. Chem. Soc. 2005, 127, 7330–7331. doi:10.1021/ja051402f
Return to citation in text: [1] -
Shabashov, D.; Daugulis, O. Org. Lett. 2005, 7, 3657–3659. doi:10.1021/ol051255q
Return to citation in text: [1] -
Mao, D.; Hong, G.; Wu, S.; Liu, X.; Yu, J.; Wang, L. Eur. J. Org. Chem. 2014, 3009–3019. doi:10.1002/ejoc.201400073
Return to citation in text: [1] -
Yaragorla, S.; Dada, R.; Singh, G. Synlett 2016, 27, 912–918. doi:10.1055/s-0035-1560385
Return to citation in text: [1] -
Yaragorla, S.; Singh, G.; Dada, R. Tetrahedron Lett. 2016, 57, 591–594. doi:10.1016/j.tetlet.2015.12.096
Return to citation in text: [1] -
Wang, F.-F.; Luo, C.-P.; Wang, Y.; Deng, G.; Yang, L. Org. Biomol. Chem. 2012, 10, 8605–8608. doi:10.1039/c2ob26604k
Return to citation in text: [1] -
Nageswara Rao, N.; Meshram, H. M. Tetrahedron Lett. 2013, 54, 5087–5090. doi:10.1016/j.tetlet.2013.07.053
Return to citation in text: [1] -
Percino, M. J.; Chapela, V. M.; Cerón, M.; Soriano-Moro, G.; Castro, M. E. Res. Chem. Intermed. 2015, 41, 3563–3572. doi:10.1007/s11164-013-1471-y
Return to citation in text: [1] -
Zhang, X.-Y.; Dong, D.-Q.; Yue, T.; Hao, S.-H.; Wang, Z.-L. Tetrahedron Lett. 2014, 55, 5462–5464. doi:10.1016/j.tetlet.2014.08.034
Return to citation in text: [1] -
Wang, Z.-L. RSC Adv. 2015, 5, 5563–5566. doi:10.1039/c4ra14486d
Return to citation in text: [1] -
Yaragorla, S.; Singh, G.; Dada, R. Tetrahedron Lett. 2015, 56, 5924–5929. doi:10.1016/j.tetlet.2015.09.035
Return to citation in text: [1] -
Chen, Z.; Luo, M.; Wen, Y.; Luo, G.; Liu, L. Org. Lett. 2014, 16, 3020–3023. doi:10.1021/ol501137x
Return to citation in text: [1] -
Nallasivam, J. L.; Fernandes, R. A. Eur. J. Org. Chem. 2015, 3558–3567. doi:10.1002/ejoc.201500353
Return to citation in text: [1] -
Sharma, R.; Abdullaha, M.; Bharate, S. B. J. Org. Chem. 2017, 82, 9786–9793. doi:10.1021/acs.joc.7b00856
Return to citation in text: [1] -
Latha, D. S.; Yaragorla, S. Eur. J. Org. Chem. 2020, 2155–2179. doi:10.1002/ejoc.201901899
Return to citation in text: [1] -
Anastas, P. T.; Heine, L. G.; Williamson, T. C. Green Chemical Syntheses and Processes; Oxford University Press: Oxford, U.K., 2000.
Return to citation in text: [1] -
Anastas, P. T.; Warner, J. C. Green chemistry: theory and practice; Oxford University Press: Oxford, U.K., 1998.
Return to citation in text: [1] -
Varma, R. S. In Green chemistry: challenging perspectives; Tundo, P.; Anastas, P. T., Eds.; Oxford University Press: Oxford, U.K., 2000; pp 221–244.
Return to citation in text: [1] -
Xu, L.; Shao, Z.; Wang, L.; Zhao, H.; Xiao, J. Tetrahedron Lett. 2014, 55, 6856–6860. doi:10.1016/j.tetlet.2014.10.079
Return to citation in text: [1] -
Malviya, B. K.; Singh, K.; Jaiswal, P. K.; Shukla, M.; Verma, V. P.; Vanangamudi, M.; Jassal, A. K.; Punjabi, P. B.; Sharma, S. ACS Omega 2019, 4, 12146–12155. doi:10.1021/acsomega.9b01514
Return to citation in text: [1] -
Goutam, B. Catalyst-free Organic Synthesis; Royal Society of Chemistry: Cambridge. U.K., 2017. doi:10.1039/9781788012782
Return to citation in text: [1] -
Rao, Y. S.; Latha, D. S.; Devunuri, N.; Almansour, A. I.; Arumugam, N.; Yaragorla, S. Eur. J. Org. Chem. 2020, 4134–4145. doi:10.1002/ejoc.202000511
Return to citation in text: [1]
| 1. | Chelucci, G.; Orrù, G.; Pinna, G. A. Tetrahedron 2003, 59, 9471–9515. doi:10.1016/j.tet.2003.09.066 |
| 2. | Prehn, K.; Warburg, A.; Schilling, T.; Bron, M.; Schulte, K. Compos. Sci. Technol. 2009, 69, 1570–1579. doi:10.1016/j.compscitech.2008.09.006 |
| 3. | Kem, W. R.; Soti, F.; Rittschof, D. Biomol. Eng. 2003, 20, 355–361. doi:10.1016/s1389-0344(03)00049-2 |
| 4. | Friesen, R. W.; Brideau, C.; Chan, C. C.; Charleson, S.; Deschênes, D.; Dubé, D.; Ethier, D.; Fortin, R.; Gauthier, J. Y.; Girard, Y.; Gordon, R.; Greig, G. M.; Riendeau, D.; Savoie, C.; Wang, Z.; Wong, E.; Visco, D.; Xu, L. J.; Young, R. N. Bioorg. Med. Chem. Lett. 1998, 8, 2777–2782. doi:10.1016/s0960-894x(98)00499-5 |
| 12. | Kumar, S.; Bawa, S.; Gupta, H. Mini-Rev. Med. Chem. 2009, 9, 1648–1654. doi:10.2174/138955709791012247 |
| 13. | Fournet, A.; Barrios, A. A.; Muñoz, V.; Hocquemiller, R.; Roblot, F.; Cavé, A.; Richomme, P.; Bruneton, J. Phytother. Res. 1994, 8, 174–178. doi:10.1002/ptr.2650080312 |
| 33. | Zhang, X.-Y.; Dong, D.-Q.; Yue, T.; Hao, S.-H.; Wang, Z.-L. Tetrahedron Lett. 2014, 55, 5462–5464. doi:10.1016/j.tetlet.2014.08.034 |
| 9. | Strunz, G. M.; Findlay, J. A. Pyridine and Piperidine Alkaloids. In The Alkaloids; Brossi, A., Ed.; Academic Press: New York, NY, USA, 1985; pp 89–183. doi:10.1016/s0099-9598(08)60194-7 |
| 10. | Meth-Cohn, O.; Yau, C. C.; Yu, C.-Y. J. Heterocycl. Chem. 1999, 36, 1549–1553. doi:10.1002/jhet.5570360615 |
| 11. | Houghton, P. J.; Woldemariam, T. Z.; Watanabe, Y.; Yates, M. Planta Med. 1999, 65, 250–254. doi:10.1055/s-1999-13988 |
| 6. | Solomon, V. R.; Lee, H. Curr. Med. Chem. 2011, 18, 1488–1508. doi:10.2174/092986711795328382 |
| 7. | Wilson, K. J.; van Niel, M. B.; Cooper, L.; Bloomfield, D.; O’Connor, D.; Fish, L. R.; MacLeod, A. M. Bioorg. Med. Chem. Lett. 2007, 17, 2643–2648. doi:10.1016/j.bmcl.2007.01.098 |
| 8. | Michael, J. P. Nat. Prod. Rep. 2005, 22, 627–646. doi:10.1039/b413750g |
| 31. | Nageswara Rao, N.; Meshram, H. M. Tetrahedron Lett. 2013, 54, 5087–5090. doi:10.1016/j.tetlet.2013.07.053 |
| 5. | Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Nat. Rev. Drug Discovery 2002, 1, 493–502. doi:10.1038/nrd839 |
| 32. | Percino, M. J.; Chapela, V. M.; Cerón, M.; Soriano-Moro, G.; Castro, M. E. Res. Chem. Intermed. 2015, 41, 3563–3572. doi:10.1007/s11164-013-1471-y |
| 21. | Qian, B.; Guo, S.; Shao, J.; Zhu, Q.; Yang, L.; Xia, C.; Huang, H. J. Am. Chem. Soc. 2010, 132, 3650–3651. doi:10.1021/ja910104n |
| 22. | Qian, B.; Xie, P.; Xie, Y.; Huang, H. Org. Lett. 2011, 13, 2580–2583. doi:10.1021/ol200684b |
| 29. | Yaragorla, S.; Singh, G.; Dada, R. Tetrahedron Lett. 2016, 57, 591–594. doi:10.1016/j.tetlet.2015.12.096 |
| 16. | Kanyiva, K. S.; Nakao, Y.; Hiyama, T. Angew. Chem., Int. Ed. 2007, 46, 8872–8874. doi:10.1002/anie.200703758 |
| 17. | Wu, J.; Cui, X.; Chen, L.; Jiang, G.; Wu, Y. J. Am. Chem. Soc. 2009, 131, 13888–13889. doi:10.1021/ja902762a |
| 18. | Mousseau, J. J.; Bull, J. A.; Charette, A. B. Angew. Chem., Int. Ed. 2010, 49, 1115–1118. doi:10.1002/anie.200906020 |
| 19. | Deng, G.; Ueda, K.; Yanagisawa, S.; Itami, K.; Li, C.-J. Chem. – Eur. J. 2009, 15, 333–337. doi:10.1002/chem.200801893 |
| 20. | Tobisu, M.; Hyodo, I.; Chatani, N. J. Am. Chem. Soc. 2009, 131, 12070–12071. doi:10.1021/ja9053509 |
| 30. | Wang, F.-F.; Luo, C.-P.; Wang, Y.; Deng, G.; Yang, L. Org. Biomol. Chem. 2012, 10, 8605–8608. doi:10.1039/c2ob26604k |
| 15. | Campeau, L.-C.; Rousseaux, S.; Fagnou, K. J. Am. Chem. Soc. 2005, 127, 18020–18021. doi:10.1021/ja056800x |
| 14. | Nakamura, I.; Yamamoto, Y. Chem. Rev. 2004, 104, 2127–2198. doi:10.1021/cr020095i |
| 23. | Jazzar, R.; Hitce, J.; Renaudat, A.; Sofack-Kreutzer, J.; Baudoin, O. Chem. – Eur. J. 2010, 16, 2654–2672. doi:10.1002/chem.200902374 |
| 24. | Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147–1169. doi:10.1021/cr900184e |
| 25. | Kalyani, D.; Deprez, N. R.; Desai, L. V.; Sanford, M. S. J. Am. Chem. Soc. 2005, 127, 7330–7331. doi:10.1021/ja051402f |
| 26. | Shabashov, D.; Daugulis, O. Org. Lett. 2005, 7, 3657–3659. doi:10.1021/ol051255q |
| 27. | Mao, D.; Hong, G.; Wu, S.; Liu, X.; Yu, J.; Wang, L. Eur. J. Org. Chem. 2014, 3009–3019. doi:10.1002/ejoc.201400073 |
| 28. | Yaragorla, S.; Dada, R.; Singh, G. Synlett 2016, 27, 912–918. doi:10.1055/s-0035-1560385 |
| 22. | Qian, B.; Xie, P.; Xie, Y.; Huang, H. Org. Lett. 2011, 13, 2580–2583. doi:10.1021/ol200684b |
| 35. | Yaragorla, S.; Singh, G.; Dada, R. Tetrahedron Lett. 2015, 56, 5924–5929. doi:10.1016/j.tetlet.2015.09.035 |
| 36. | Chen, Z.; Luo, M.; Wen, Y.; Luo, G.; Liu, L. Org. Lett. 2014, 16, 3020–3023. doi:10.1021/ol501137x |
| 46. | Rao, Y. S.; Latha, D. S.; Devunuri, N.; Almansour, A. I.; Arumugam, N.; Yaragorla, S. Eur. J. Org. Chem. 2020, 4134–4145. doi:10.1002/ejoc.202000511 |
| 41. | Anastas, P. T.; Warner, J. C. Green chemistry: theory and practice; Oxford University Press: Oxford, U.K., 1998. |
| 42. | Varma, R. S. In Green chemistry: challenging perspectives; Tundo, P.; Anastas, P. T., Eds.; Oxford University Press: Oxford, U.K., 2000; pp 221–244. |
| 43. | Xu, L.; Shao, Z.; Wang, L.; Zhao, H.; Xiao, J. Tetrahedron Lett. 2014, 55, 6856–6860. doi:10.1016/j.tetlet.2014.10.079 |
| 44. | Malviya, B. K.; Singh, K.; Jaiswal, P. K.; Shukla, M.; Verma, V. P.; Vanangamudi, M.; Jassal, A. K.; Punjabi, P. B.; Sharma, S. ACS Omega 2019, 4, 12146–12155. doi:10.1021/acsomega.9b01514 |
| 45. | Goutam, B. Catalyst-free Organic Synthesis; Royal Society of Chemistry: Cambridge. U.K., 2017. doi:10.1039/9781788012782 |
| 39. | Latha, D. S.; Yaragorla, S. Eur. J. Org. Chem. 2020, 2155–2179. doi:10.1002/ejoc.201901899 |
| 40. | Anastas, P. T.; Heine, L. G.; Williamson, T. C. Green Chemical Syntheses and Processes; Oxford University Press: Oxford, U.K., 2000. |
| 37. | Nallasivam, J. L.; Fernandes, R. A. Eur. J. Org. Chem. 2015, 3558–3567. doi:10.1002/ejoc.201500353 |
| 38. | Sharma, R.; Abdullaha, M.; Bharate, S. B. J. Org. Chem. 2017, 82, 9786–9793. doi:10.1021/acs.joc.7b00856 |
© 2020 Yennamaneni et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)