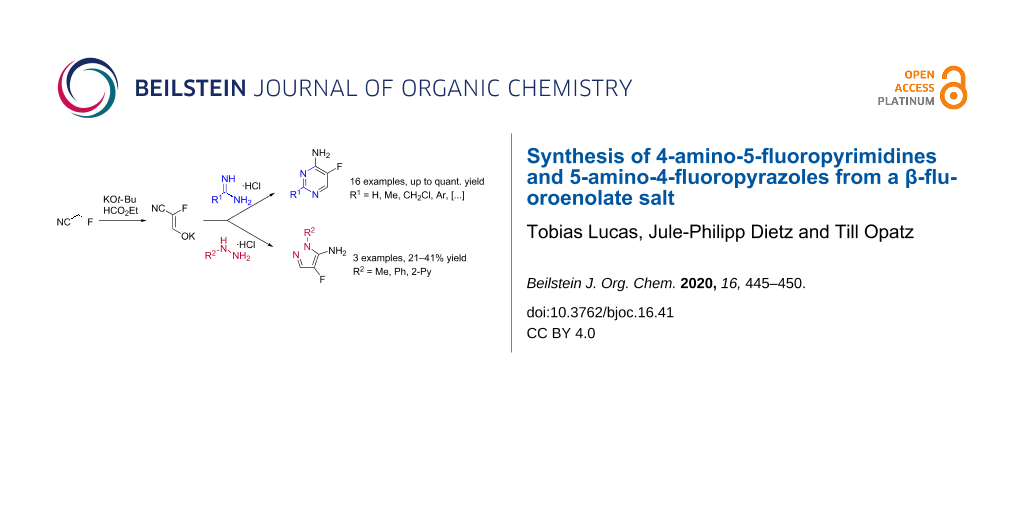
Abstract
A synthesis of fluorinated pyrimidines under mild conditions from amidine hydrochlorides and the recently described potassium 2-cyano-2-fluoroethenolate was developed. A broad substrate scope was tested and mostly excellent yields were obtained. The synthesis of fluorinated aminopyrazoles from the same fluorinated precursor could be demonstrated but proceeded with lower efficiency.

Graphical Abstract
Introduction
Due to its extremely widespread application in virtually all areas of synthetic organic chemistry, fluorine is considered a “magic” element. It confers metabolic stability and other unique properties to organic molecules and a large fraction of modern pharmaceuticals [1-6] and agrochemicals [7] contain at least one fluorine atom [8-15]. One of the approaches for their synthesis is the use of small fluorinated building blocks which avoids the often cumbersome regioselective fluorination at a later synthetic stage [15-19].
Fluorinated pyrimidines, pyrimidine analogues and pyrazoles [20,21] play a prominent role in several clinically important pharmaceuticals [22-25]. These compounds act against HIV or HIV-related diseases [26], such as opportunistic fungal infections, and represent nucleoside analogues which can either act as antimetabolites or as nucleoside reverse transcriptase inhibitors (NRTIs). Two of the most common 5-fluoropyrimidines are the cytostatic 5-fluorouracil (1) [27] and the antimycotic prodrug 5-fluorocytosine (2) [28,29], which is also part of the anti-HIV agents emtricitabine [30] (ingredient of Truvada®) and capecitabine [31] (Xeloda®) (see Figure 1) [32,33].
![[1860-5397-16-41-1]](/bjoc/content/figures/1860-5397-16-41-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The structures of 5-fluorouracil (1), 5-fluorocytosine (2), emtricitabine (3) and capecitabine (4).
Figure 1: The structures of 5-fluorouracil (1), 5-fluorocytosine (2), emtricitabine (3) and capecitabine (4).
While the introduction of fluorine into preexisting heterocycles can require difficult-to-handle, expensive and highly corrosive electrophilic fluorine species [16,34,35], the present approach employs a fluorinated C3 building block recently described and used by our laboratory for the synthesis of 2 [36]. Herein, we present the synthesis of fluorinated pyrimidines and pyrazoles from the same precursor. Potassium (Z)-2-cyano-2-fluoroethenolate (8) was synthesized in three steps starting from chloroacetamide (5). First, a Finkelstein halogen exchange reaction and a dehydration reaction were combined to obtain fluoroacetonitrile (7) in 82% yield [36]. The latter then underwent a Claisen condensation with ethyl formate to obtain 8 in 77% yield with a purity of 90%. The product contained about 10% (1H NMR) of potassium formate, which could not easily be removed without risking the decomposition of the enolate salt (Scheme 1).
![[1860-5397-16-41-i1]](/bjoc/content/inline/1860-5397-16-41-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of potassium (Z)-2-cyano-2-fluoroethenolate (8) by Dietz et al. [36].
Scheme 1: Synthesis of potassium (Z)-2-cyano-2-fluoroethenolate (8) by Dietz et al. [36].
As the fluorine atom is part of a building block, harsh conditions for a late-stage fluorination can be avoided, and even products with sensitive functionalities are accessible.
Results and Discussion
Synthesis of fluorinated pyrimidines
To synthesize 2-alkyl- and 2-aryl-substituted 5-fluoro-4-aminopyrimidines, the reaction of 8 with various amidines was investigated. Different formamidinium and guanidinium salts were tested (Table 1) to evaluate the effect of the counter ion. The hydrochlorides furnished the highest yields and no undesired byproducts were formed. In contrast to the related reaction with guanidinium salts [36], the reactions with amidine hydrochlorides did not require any basic additives.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-41-i4.svg?max-width=637&scale=1.0)
Under these improved conditions, the scope of the reaction for producing alkyl-substituted pyrimidine derivatives was investigated (Scheme 2). Starting from formamidine hydrochloride, compound 10a was synthesized in 85% yield. A variety of amidines with different steric hindrance such as methyl, cyclopropyl and tert-butyl substitution was used, and all substituents were well tolerated, providing the corresponding fluorinated aminoalkylpyrimidines (10d–f) in yields of 81–93%.
![[1860-5397-16-41-i2]](/bjoc/content/inline/1860-5397-16-41-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the cyclization reaction. All yields are those of the purified products. aNo further purification was required to obtain analytically pure material. bUse of sodium methoxide. cUsing O-methylisourea hemisulfate. dReaction time of 24 h. eProduct detected by LC–MS.
Scheme 2: Scope of the cyclization reaction. All yields are those of the purified products. aNo further purif...
In spite of its potential electrophilic reactivity, the chloromethyl derivative 10g was obtained in excellent yield (90%) and could be used for further modifications to construct more complex products. Subsequently, benzamidine derivatives with a broad variety of substituents on the benzenoid core were evaluated as the nucleophiles. Benzamidine hydrochloride itself yielded compound 10h with an excellent yield of 93%. A variety of substituted products including electron-donating (compounds 10i–k) and electron-withdrawing groups (compound 10l) were formed in nearly quantitative yields (95–99%). Only the 4-hydroxyphenyl derivative 10j was obtained in a moderate yield (69%) presumably due to competing polymerization of the enolate.
The 4-chloro and 4-bromo derivatives (10m and 10n) were also obtained in excellent yields of 93% and 97%, respectively, and represent potential substrates for subsequent cross-coupling reactions, which would provide structurally more complex products.
Amidines with heterocyclic substituents were also evaluated. While the pyridine-substituted derivative 10o furnished a yield of 86%, the pyrazolo derivative 10p could only be obtained in a moderate yield of 38%. This might be explained by the tendency of the pyrazolo moiety to act as a leaving group and a following addition–elimination reaction with methanol can take place (the corresponding 2-methoxy-substituted product was detected by LC–MS). This side reaction could also be observed in the attempted synthesis of the triazolo derivative 10r, where the desired pyrimidine could only be detected in traces by LC–MS. Another limitation of the substrate scope was observed in the cyclization with an Nα-protected arginine, where the desired pyrimidine 10q was also only observed in trace amounts while the majority of the starting material remained unchanged.
Synthesis of fluorinated aminopyrazoles
As in the cyclocondensation with amidines, the reaction of 8 with other bifunctional nucleophiles could also result in the formation of fluorinated heterocycles. While attempts to obtain an aminooxazole upon reaction with hydroxylamine were unsuccessful, substituted hydrazines turned out to provide moderate yields of fluorinated pyrazoles. Phenylhydrazine (12a) was chosen as the model substrate for optimization (Table 2).
Table 2: Synthesis of fluorinated pyrazoles.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-41-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | R | Solvent | T [°C] | Additive | Results |
| 1 | Ph | MeOH | rt | – | traces of 13a and 13ba |
| 2 | Ph | MeOH | 5–10 | – | polymerization |
| 3 | Ph | MeOH | −10 | – | polymerization |
| 4 | Phb | MeOH | −10 | – | polymerization |
| 5 | Ph | MeOH | 5–10 | AcOH | polymerization |
| 6 | Ph | MeOH | 0–5 | NaOMe | 13a–ca |
| 7 | Ph | MeOH | 0–5 | DBU | polymerization |
| 8 | Ph | MeOH | 0–5 | NaOHaq | 13a (19%)c |
| 9 | Ph | H2O | rt | – | traces of 13aa |
| 10 | Ph | H2O | rt | AcOH | polymerization |
| 11 | Phb | H2O | rt | NaOHaq | 13a (36%)c |
| 12 | Me | H2O | rt | – | 13d (41%)c |
| 13 | 2-Py | H2O | rt | – | 13e (20%)c |
aDetected by LC–MS. bThe corresponding hydrochloride was used. cIsolated yield.
In the first reactions, only traces of the desired compound 13a were observed. Changing the temperature did not increase the yield. Acidic conditions were tested next but phenylhydrazine hydrochloride and acetic acid again gave only traces of 13a and polymerization of the enolate took place. To stabilize the starting material, basic conditions were tested as well and various bases were used. With sodium methoxide, only traces of compound 13a could be detected and in addition, the dehalogenated- and methoxy-substituted derivatives (13b and 13c) were observed (Scheme 3). This prompted us to investigate the use of non-nucleophilic bases, but with DBU, polymerization of the fluoroenolate took place.
![[1860-5397-16-41-i3]](/bjoc/content/inline/1860-5397-16-41-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Cyclization with phenylhydrazine (12a) to obtain the desired pyrazole 13a and the byproducts 13b and 13c (detected via LC–MS).
Scheme 3: Cyclization with phenylhydrazine (12a) to obtain the desired pyrazole 13a and the byproducts 13b an...
When using aqueous sodium hydroxide as the base, a 19% yield could be achieved. Switching the solvent to water led to an improvement of the yield to 36% (Table 2). In water, but without using a base, the N-methylpyrazole 13d and the N-(2-pyridyl)-substituted derivative 13e could be obtained in 41% and 21% yield, respectively.
The formation of the other possible aminopyrazole regioisomer was not observed, which could be explained by the preferential attack of the less hindered nitrogen atom of the hydrazine on the more reactive carbon atom of the fluoroenolate in a Michael-type addition. The cyclization did not require a deprotonation of the RNH moiety.
Conclusion
In summary, a synthesis of fluorinated pyrimidines under mild conditions using fluoroenolate 8 and amidines in a cyclocondensation was developed. A series of substrates carrying aliphatic and aromatic substituents was successfully tested and various functional groups were tolerated. The products could be mostly isolated in excellent yields. Furthermore, the synthesis of fluorinated pyrazoles from the same fluorinated enolate was demonstrated, albeit in only low to moderate yields. Nevertheless, the presented method provides a simple and short route to hitherto unknown compounds of this type.
Supporting Information
| Supporting Information File 1: Chemical procedures and analytical data, including copies of 1H NMR and 13C NMR spectra. | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992–1012. doi:10.1016/j.jfluchem.2006.05.006
Return to citation in text: [1] -
Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943
Return to citation in text: [1] -
Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; Gershonov, E.; Saphier, S. J. Med. Chem. 2019, 62, 5628–5637. doi:10.1021/acs.jmedchem.9b00604
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. J. Med. Chem. 2017, 60, 797–804. doi:10.1021/acs.jmedchem.6b01691
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788
Return to citation in text: [1] -
Maienfisch, P.; Hall, R. G. Chimia 2004, 58, 93–99. doi:10.2533/000942904777678091
Return to citation in text: [1] -
Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833
Return to citation in text: [1] -
Dolbier, W. R. Guide to fluorine NMR for organic chemists; Wiley-VCH: Weinheim, Germany, 2016. doi:10.1002/9781118831106
Return to citation in text: [1] -
Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023
Return to citation in text: [1] -
Ismail, F. M. D. J. Fluorine Chem. 2002, 118, 27–33. doi:10.1016/s0022-1139(02)00201-4
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011
Return to citation in text: [1] -
Politanskaya, L. V.; Selivanova, G. A.; Panteleeva, E. V.; Tretyakov, E. V.; Platonov, V. E.; Nikul'shin, P. V.; Vinogradov, A. S.; Zonov, Y. V.; Karpov, V. M.; Mezhenkova, T. V.; Vasilyev, A. V.; Koldobskii, A. B.; Shilova, O. S.; Morozova, S. M.; Burgart, Y. V.; Shchegolkov, E. V.; Saloutin, V. I.; Sokolov, V. B.; Aksinenko, A. Yu.; Nenajdenko, V. G.; Moskalik, M. Yu.; Astakhova, V. V.; Shainyan, B. A.; Tabolin, A. A.; Ioffe, S. L.; Muzalevskiy, V. M.; Balenkova, E. S.; Shastin, A. V.; Tyutyunov, A. A.; Boiko, V. E.; Igumnov, S. M.; Dilman, A. D.; Adonin, N. Yu.; Bardin, V. V.; Masoud, S. M.; Vorobyeva, D. V.; Osipov, S. N.; Nosova, E. V.; Lipunova, G. N.; Charushin, V. N.; Prima, D. O.; Makarov, A. G.; Zibarev, A. V.; Trofimov, B. A.; Sobenina, L. N.; Belyaeva, K. V.; Sosnovskikh, V. Ya.; Obydennov, D. L.; Usachev, S. A. Russ. Chem. Rev. 2019, 88, 425–569. doi:10.1070/rcr4871
Return to citation in text: [1] -
Muzalevskiy, V. M.; Nenajdenko, V. G. Org. Biomol. Chem. 2018, 16, 7935–7946. doi:10.1039/c8ob02247j
Return to citation in text: [1] [2] -
Kirk, K. L. Org. Process Res. Dev. 2008, 12, 305–321. doi:10.1021/op700134j
Return to citation in text: [1] [2] -
Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R. D., Ed.; Springer: Heidelberg, Germany, 1997; pp 131–195. doi:10.1007/3-540-69197-9_4
Return to citation in text: [1] -
Nenajdenko, V. G.; Sanin, A. V.; Balenkova, E. S. Molecules 1997, 2, 186–232. doi:10.3390/21200186
Return to citation in text: [1] -
Commare, B.; Schmitt, E.; Aribi, F.; Panossian, A.; Vors, J.-P.; Pazenok, S.; Leroux, F. R. Molecules 2017, 22, 977. doi:10.3390/molecules22060977
Return to citation in text: [1] -
Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459
Return to citation in text: [1] -
Schmitt, E.; Panossian, A.; Vors, J.-P.; Funke, C.; Lui, N.; Pazenok, S.; Leroux, F. R. Chem. – Eur. J. 2016, 22, 11239–11244. doi:10.1002/chem.201601621
Return to citation in text: [1] -
Muzalevskiy, V. M.; Rulev, A. Y.; Romanov, A. R.; Kondrashov, E. V.; Ushakov, I. A.; Chertkov, V. A.; Nenajdenko, V. G. J. Org. Chem. 2017, 82, 7200–7214. doi:10.1021/acs.joc.7b00774
Return to citation in text: [1] -
Pankiewicz, K. W. Carbohydr. Res. 2000, 327, 87–105. doi:10.1016/s0008-6215(00)00089-6
Return to citation in text: [1] -
Wójtowicz-Rajchel, H. J. Fluorine Chem. 2012, 143, 11–48. doi:10.1016/j.jfluchem.2012.06.026
Return to citation in text: [1] -
Fustero, S.; Simón-Fuentes, A.; Delgado, O.; Román, R. Fluorinated Pyrazoles and Indazoles. In 5-Membered Heterocycles and Macrocycles; Nenajdenko, V. G., Ed.; Fluorine in Heterocyclic Chemistry, Vol. 1; Springer International Publishing: Cham, Switzerland, 2014; pp 279–321. doi:10.1007/978-3-319-04346-3_7
Return to citation in text: [1] -
Meanwell, N. A. J. Med. Chem. 2011, 54, 2529–2591. doi:10.1021/jm1013693
Return to citation in text: [1] -
Longley, D. B.; Harkin, D. P.; Johnston, P. G. Nat. Rev. Cancer 2003, 3, 330–338. doi:10.1038/nrc1074
Return to citation in text: [1] -
Vermes, A.; Guchelaar, H.-J.; Dankert, J. J. Antimicrob. Chemother. 2000, 46, 171–179. doi:10.1093/jac/46.2.171
Return to citation in text: [1] -
Bennett, J. E. Ann. Intern. Med. 1977, 86, 319–322. doi:10.7326/0003-4819-86-3-319
Return to citation in text: [1] -
Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Murphey-Corb, M.; Rajakumar, P.; Michael, H.; Nyaundi, J.; Didier, P. J.; Reeve, A. B.; Mitsuya, H.; Sarafianos, S. G.; Parniak, M. A. Antimicrob. Agents Chemother. 2012, 56, 4707–4712. doi:10.1128/aac.00723-12
Return to citation in text: [1] -
Harsanyi, A.; Conte, A.; Pichon, L.; Rabion, A.; Grenier, S.; Sandford, G. Org. Process Res. Dev. 2017, 21, 273–276. doi:10.1021/acs.oprd.6b00420
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Shimizu, M.; Hiyama, T. Angew. Chem., Int. Ed. 2005, 44, 214–231. doi:10.1002/anie.200460441
Return to citation in text: [1] -
Dietz, J.-P.; Derstine, B. P.; Ferenc, D.; Crawford, E. T.; Arduengo, A. J., III; Gupton, B. F.; McQuade, D. T.; Opatz, T. Eur. J. Org. Chem. 2019, 5519–5526. doi:10.1002/ejoc.201900629
Return to citation in text: [1] [2] [3] [4]
| 36. | Dietz, J.-P.; Derstine, B. P.; Ferenc, D.; Crawford, E. T.; Arduengo, A. J., III; Gupton, B. F.; McQuade, D. T.; Opatz, T. Eur. J. Org. Chem. 2019, 5519–5526. doi:10.1002/ejoc.201900629 |
| 1. | Bégué, J.-P.; Bonnet-Delpon, D. J. Fluorine Chem. 2006, 127, 992–1012. doi:10.1016/j.jfluchem.2006.05.006 |
| 2. | Müller, K.; Faeh, C.; Diederich, F. Science 2007, 317, 1881–1886. doi:10.1126/science.1131943 |
| 3. | Zafrani, Y.; Sod-Moriah, G.; Yeffet, D.; Berliner, A.; Amir, D.; Marciano, D.; Elias, S.; Katalan, S.; Ashkenazi, N.; Madmon, M.; Gershonov, E.; Saphier, S. J. Med. Chem. 2019, 62, 5628–5637. doi:10.1021/acs.jmedchem.9b00604 |
| 4. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 5. | Zafrani, Y.; Yeffet, D.; Sod-Moriah, G.; Berliner, A.; Amir, D.; Marciano, D.; Gershonov, E.; Saphier, S. J. Med. Chem. 2017, 60, 797–804. doi:10.1021/acs.jmedchem.6b01691 |
| 6. | Meanwell, N. A. J. Med. Chem. 2018, 61, 5822–5880. doi:10.1021/acs.jmedchem.7b01788 |
| 20. | Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. Chem. Rev. 2011, 111, 6984–7034. doi:10.1021/cr2000459 |
| 21. | Schmitt, E.; Panossian, A.; Vors, J.-P.; Funke, C.; Lui, N.; Pazenok, S.; Leroux, F. R. Chem. – Eur. J. 2016, 22, 11239–11244. doi:10.1002/chem.201601621 |
| 36. | Dietz, J.-P.; Derstine, B. P.; Ferenc, D.; Crawford, E. T.; Arduengo, A. J., III; Gupton, B. F.; McQuade, D. T.; Opatz, T. Eur. J. Org. Chem. 2019, 5519–5526. doi:10.1002/ejoc.201900629 |
| 15. | Muzalevskiy, V. M.; Nenajdenko, V. G. Org. Biomol. Chem. 2018, 16, 7935–7946. doi:10.1039/c8ob02247j |
| 16. | Kirk, K. L. Org. Process Res. Dev. 2008, 12, 305–321. doi:10.1021/op700134j |
| 17. | Percy, J. M. Building Block Approaches to Aliphatic Organofluorine Compounds. In Organofluorine Chemistry: Techniques and Synthons; Chambers, R. D., Ed.; Springer: Heidelberg, Germany, 1997; pp 131–195. doi:10.1007/3-540-69197-9_4 |
| 18. | Nenajdenko, V. G.; Sanin, A. V.; Balenkova, E. S. Molecules 1997, 2, 186–232. doi:10.3390/21200186 |
| 19. | Commare, B.; Schmitt, E.; Aribi, F.; Panossian, A.; Vors, J.-P.; Pazenok, S.; Leroux, F. R. Molecules 2017, 22, 977. doi:10.3390/molecules22060977 |
| 36. | Dietz, J.-P.; Derstine, B. P.; Ferenc, D.; Crawford, E. T.; Arduengo, A. J., III; Gupton, B. F.; McQuade, D. T.; Opatz, T. Eur. J. Org. Chem. 2019, 5519–5526. doi:10.1002/ejoc.201900629 |
| 8. | Jeschke, P. ChemBioChem 2004, 5, 570–589. doi:10.1002/cbic.200300833 |
| 9. | Dolbier, W. R. Guide to fluorine NMR for organic chemists; Wiley-VCH: Weinheim, Germany, 2016. doi:10.1002/9781118831106 |
| 10. | Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023 |
| 11. | Ismail, F. M. D. J. Fluorine Chem. 2002, 118, 27–33. doi:10.1016/s0022-1139(02)00201-4 |
| 12. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 13. | Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011 |
| 14. | Politanskaya, L. V.; Selivanova, G. A.; Panteleeva, E. V.; Tretyakov, E. V.; Platonov, V. E.; Nikul'shin, P. V.; Vinogradov, A. S.; Zonov, Y. V.; Karpov, V. M.; Mezhenkova, T. V.; Vasilyev, A. V.; Koldobskii, A. B.; Shilova, O. S.; Morozova, S. M.; Burgart, Y. V.; Shchegolkov, E. V.; Saloutin, V. I.; Sokolov, V. B.; Aksinenko, A. Yu.; Nenajdenko, V. G.; Moskalik, M. Yu.; Astakhova, V. V.; Shainyan, B. A.; Tabolin, A. A.; Ioffe, S. L.; Muzalevskiy, V. M.; Balenkova, E. S.; Shastin, A. V.; Tyutyunov, A. A.; Boiko, V. E.; Igumnov, S. M.; Dilman, A. D.; Adonin, N. Yu.; Bardin, V. V.; Masoud, S. M.; Vorobyeva, D. V.; Osipov, S. N.; Nosova, E. V.; Lipunova, G. N.; Charushin, V. N.; Prima, D. O.; Makarov, A. G.; Zibarev, A. V.; Trofimov, B. A.; Sobenina, L. N.; Belyaeva, K. V.; Sosnovskikh, V. Ya.; Obydennov, D. L.; Usachev, S. A. Russ. Chem. Rev. 2019, 88, 425–569. doi:10.1070/rcr4871 |
| 15. | Muzalevskiy, V. M.; Nenajdenko, V. G. Org. Biomol. Chem. 2018, 16, 7935–7946. doi:10.1039/c8ob02247j |
| 16. | Kirk, K. L. Org. Process Res. Dev. 2008, 12, 305–321. doi:10.1021/op700134j |
| 34. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 35. | Shimizu, M.; Hiyama, T. Angew. Chem., Int. Ed. 2005, 44, 214–231. doi:10.1002/anie.200460441 |
| 7. | Maienfisch, P.; Hall, R. G. Chimia 2004, 58, 93–99. doi:10.2533/000942904777678091 |
| 36. | Dietz, J.-P.; Derstine, B. P.; Ferenc, D.; Crawford, E. T.; Arduengo, A. J., III; Gupton, B. F.; McQuade, D. T.; Opatz, T. Eur. J. Org. Chem. 2019, 5519–5526. doi:10.1002/ejoc.201900629 |
| 28. | Vermes, A.; Guchelaar, H.-J.; Dankert, J. J. Antimicrob. Chemother. 2000, 46, 171–179. doi:10.1093/jac/46.2.171 |
| 29. | Bennett, J. E. Ann. Intern. Med. 1977, 86, 319–322. doi:10.7326/0003-4819-86-3-319 |
| 31. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 27. | Longley, D. B.; Harkin, D. P.; Johnston, P. G. Nat. Rev. Cancer 2003, 3, 330–338. doi:10.1038/nrc1074 |
| 32. | Murphey-Corb, M.; Rajakumar, P.; Michael, H.; Nyaundi, J.; Didier, P. J.; Reeve, A. B.; Mitsuya, H.; Sarafianos, S. G.; Parniak, M. A. Antimicrob. Agents Chemother. 2012, 56, 4707–4712. doi:10.1128/aac.00723-12 |
| 33. | Harsanyi, A.; Conte, A.; Pichon, L.; Rabion, A.; Grenier, S.; Sandford, G. Org. Process Res. Dev. 2017, 21, 273–276. doi:10.1021/acs.oprd.6b00420 |
| 22. | Muzalevskiy, V. M.; Rulev, A. Y.; Romanov, A. R.; Kondrashov, E. V.; Ushakov, I. A.; Chertkov, V. A.; Nenajdenko, V. G. J. Org. Chem. 2017, 82, 7200–7214. doi:10.1021/acs.joc.7b00774 |
| 23. | Pankiewicz, K. W. Carbohydr. Res. 2000, 327, 87–105. doi:10.1016/s0008-6215(00)00089-6 |
| 24. | Wójtowicz-Rajchel, H. J. Fluorine Chem. 2012, 143, 11–48. doi:10.1016/j.jfluchem.2012.06.026 |
| 25. | Fustero, S.; Simón-Fuentes, A.; Delgado, O.; Román, R. Fluorinated Pyrazoles and Indazoles. In 5-Membered Heterocycles and Macrocycles; Nenajdenko, V. G., Ed.; Fluorine in Heterocyclic Chemistry, Vol. 1; Springer International Publishing: Cham, Switzerland, 2014; pp 279–321. doi:10.1007/978-3-319-04346-3_7 |
| 30. | Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840 |
© 2020 Lucas et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)