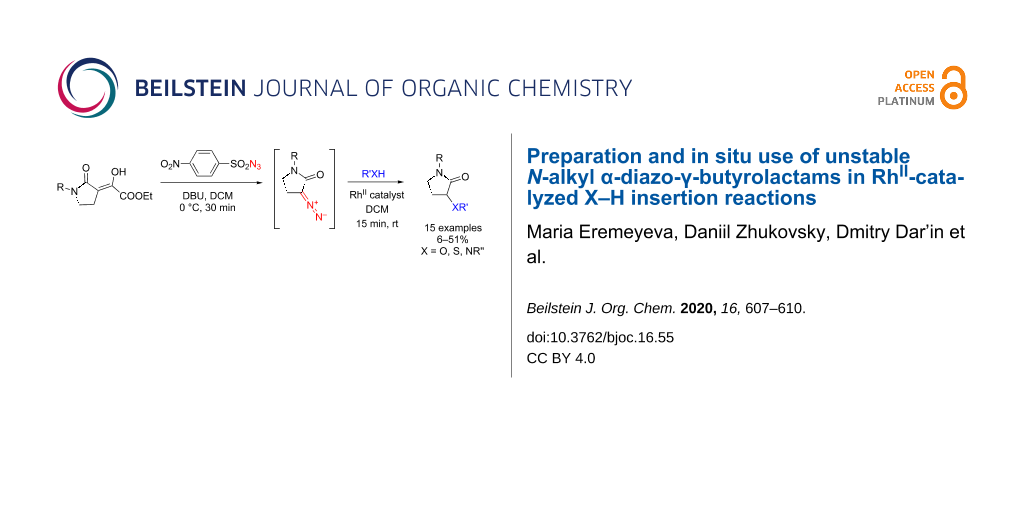
Abstract
N-Alkyl α-diazo-γ-butyrolactams, previously found to be unstable and to undergo unproductive dimerization to bishydrazones, were successfully converted immediately to various X–H insertion products with alcohols, aromatic amines and thiols via an in situ RhII-catalyzed reaction. With aliphatic amines or unreactive, sterically hindered anilines, the reactions tended to yield enamine adducts.

Graphical Abstract
Introduction
Recently, we described the first synthesis and subsequent transformations of a rare type of cyclic α-diazocarbonyl compounds, namely, α-diazo-γ-butyrolactams [1]. In particular, N-aryl-α-diazo-γ-butyrolactams 1 were efficiently transformed into pyrrolinones 2 upon the treatment with AgOTf (1 mol %) and into α-alkoxy derivatives 3 via Rh2(OAc)4-catalyzed O–H insertion reactions with various alcohols. In contrast, N-alkyl-α-diazo-γ-butyrolactams 4 did not undergo these reactions typical of α-diazocarbonyl compounds, as they rapidly dimerized to give bishydrazones 5 (Figure 1). The instability of N-alkyl-α-diazo-γ-butyrolactams 4 compared to the N-aryl counterparts 1, was most likely related to the reduced electron-withdrawing character of the lactam carbonyl group in the former compared to the latter. This assumption is further supported by the fact that ortho-substituted N-aryl derivatives 1 (in which the conjugation of the aromatic ring with the lone pair of the lactam nitrogen atom is reduced due to the sterically forced loss of the coplanarity between the aromatic ring and the aminocarbonyl moiety) are as unstable as the N-alkyl derivatives 4 [1].
![[1860-5397-16-55-1]](/bjoc/content/figures/1860-5397-16-55-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Previously reported uses of α-diazo-γ-butyrolactams 1 and 4.
Figure 1: Previously reported uses of α-diazo-γ-butyrolactams 1 and 4.
Faced with this serious limitation of the reactivity scope, we set off to investigate the possibility of using unstable compounds 4 in situ, promptly after their formation, in various RhII-catalyzed X–H insertion reactions, particularly the recently described rhodium carbene insertion into O–H [1], N–H [2] and S–H [3] bonds of alcohols, aromatic amines, and thiols, respectively. Herein, we report the results of these studies.
Results and Discussion
Three N-alkyl-α-ethoxalyl-γ-lactams 6a–c, prepared by oxalylation of the respective γ-lactams as decribed previously [1], underwent a rapid diazo transfer reaction via the conventional protocol [4,5] employing 4-nitrobenzenesulfonyl azide and DBU. A quick filtration through a plug of alumina (in lieu of silica gel, which led to decomposition of the diazo compounds 4a–c), and addition of an alcohol, a thiol, or an aromatic amine along with a RhII catalyst resulted in a rapid insertion reaction and the isolation of the desired α-substituted γ-lactams 7a–o in modest yields (Scheme 1). It should be noted that, after some experimentation, the reactions with alcohols and thiols were found to be efficiently catalyzed by 1 mol % of Rh2(OAc)4 and completed within 30 min. For aromatic amines, this catalyst proved inefficient and was replaced by 0.5 mol % of Rh2(esp)2. The attempted change of the catalyst to Rh2(esp)2 in the reactions with alcohols and thiols (which earlier gave a remarkable improvement of the product yields of NH-insertion reactions [2]) resulted in no notable improvement in this case.
![[1860-5397-16-55-i1]](/bjoc/content/inline/1860-5397-16-55-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Generation and in situ RhII-catalyzed X–H insertion reactions of the diazo compounds 4a–c. Conditions: aRhII catalyst: 1 mol % Rh2(OAc)4 for X = O or S and 0.5 mol % Rh2(esp)2 for X = NR′′.
Scheme 1: Generation and in situ RhII-catalyzed X–H insertion reactions of the diazo compounds 4a–c. Conditio...
The only attempt to employ an aliphatic amine, cyclopropylamine (which would presumably be less reactive in the RhII-catalyzed insertion reaction [2]) resulted in the formation of a sole identifiable product – enamine 8a, isolated chromatographically from a complex mixture of unidentified byproducts. The formation of 8a (that was also observed previously, along with the expected, saturated coupling product of the RhII-catalyzed reaction of cyclopropylamine with N-phenyl-α-diazo-2-pyrrolidone [2]) can be rationalized, as proposed previously [2], either by the oxidation of diazolactam 6c to a respective ketone (a process described in the literature for other α-diazocarbonyl compounds [6]), followed by a nucleophilic attack of cyclopropylamine. Alternatively, the formation of the enamine product could be envisaged via the reaction of the amine with bishydrazone 5, which would have formed, if the N–H insertion pathway was not sufficiently rapid. Both assumptions are in line with the formation of the similar enamine coupling product 8b, that we observed with 2,6-dimethylaniline. With this unreactive, sterically hindered aromatic amine, 6c is likely to undergo either the unwanted N2→O oxidation or dimerize to bishydrazone 5, whereupon the resulting intermediate would be eventually trapped by the aniline to give 8b (Scheme 2). The viability of either (or both) of these possibilities is currently investigated. It should be noted that a similar Rh2(esp)2-catalyzed reaction of one of the N-aryl-α-diazo-γ-butyrolactams 1 with 2,6-dimethylaniline previously gave an excellent yield of the N–H insertion product [2].
![[1860-5397-16-55-i2]](/bjoc/content/inline/1860-5397-16-55-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of the enamine coupling products 8a and b.
Scheme 2: Formation of the enamine coupling products 8a and b.
Conclusion
We demonstrated that the scope of α-diazo-γ-butyrolactams being capable to undergo RhII-catalyzed X–H insertion reactions with alcohols, thiols, and aromatic amines can be extended to unstable N-alkyl derivatives, for which rapid, unproductive dimerization was previously observed. This was achieved through the immediate addition of the X–H insertion partner and a RhII catalyst to the solution of the diazo compound. The reactions are rapid, albeit moderately yielding. Despite the latter drawback, the range of 1,3-disubstituted 2-pyrrolidones attainable via the intermediate formation of α-diazo-γ-butyrolactams was substantially expanded, thereby making this approach more useful for potential medicinal chemistry exploration of these disubstituted γ-lactams.
Supporting Information
| Supporting Information File 1: General experimental information, synthetic procedures, analytical data and NMR spectra for the reported compounds. | ||
| Format: PDF | Size: 3.7 MB | Download |
References
-
Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133
Return to citation in text: [1] [2] [3] [4] -
Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Barkhatova, D.; Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 5798–5800. doi:10.1002/ejoc.201900944
Return to citation in text: [1] -
Kantin, G.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2018, 4857–4859. doi:10.1002/ejoc.201800955
Return to citation in text: [1] -
Clarke, L. A.; Ring, A.; Ford, A.; Sinha, A. S.; Lawrence, S. E.; Maguire, A. R. Org. Biomol. Chem. 2014, 12, 7612–7628. doi:10.1039/c4ob01430h
Return to citation in text: [1] -
Hansen, S. R.; Spangler, J. E.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2012, 14, 4626–4629. doi:10.1021/ol3020754
Return to citation in text: [1]
| 1. | Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133 |
| 3. | Barkhatova, D.; Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 5798–5800. doi:10.1002/ejoc.201900944 |
| 2. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
| 1. | Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133 |
| 2. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
| 1. | Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133 |
| 2. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
| 2. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
| 2. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
| 6. | Hansen, S. R.; Spangler, J. E.; Hansen, J. H.; Davies, H. M. L. Org. Lett. 2012, 14, 4626–4629. doi:10.1021/ol3020754 |
| 4. | Kantin, G.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2018, 4857–4859. doi:10.1002/ejoc.201800955 |
| 5. | Clarke, L. A.; Ring, A.; Ford, A.; Sinha, A. S.; Lawrence, S. E.; Maguire, A. R. Org. Biomol. Chem. 2014, 12, 7612–7628. doi:10.1039/c4ob01430h |
| 1. | Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133 |
| 2. | Zhukovsky, D.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2019, 4377–4383. doi:10.1002/ejoc.201900565 |
© 2020 Eremeyeva et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)