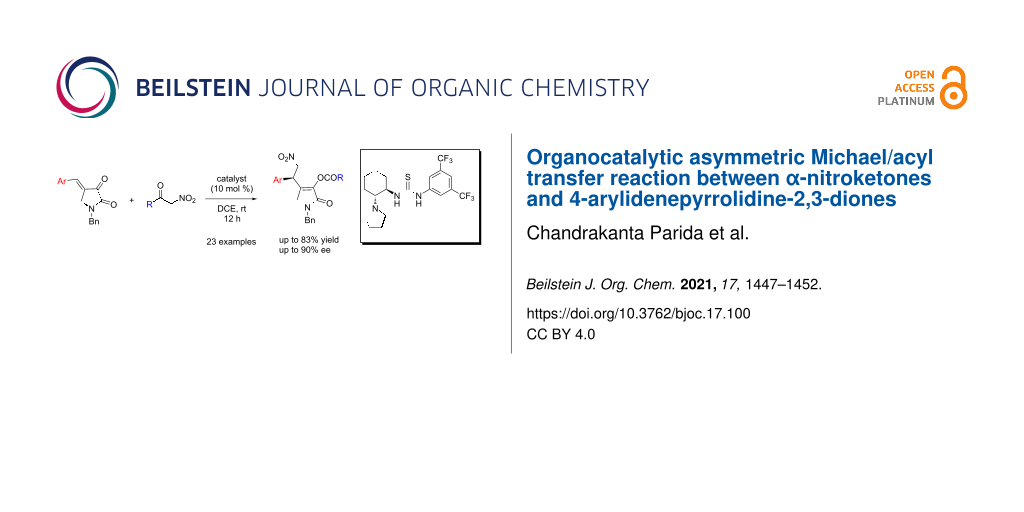
Abstract
An organocatalytic asymmetric Michael/acyl transfer reaction between α-nitroketones and 4-arylidenepyrrolidine-2,3-diones is reported. A bifunctional thiourea catalyst was found to be effective for this reaction. With 10 mol % of the catalyst, good results were attained for a variety of 1,5-dihydro-2H-pyrrol-2-ones under mild reaction conditions.

Graphical Abstract
Introduction
The Michael reaction is a powerful reaction that has been so far applied for the formation of carbon–carbon and carbon–heteroatom bonds in organic synthesis [1,2]. After the renaissance of organocatalysis in the year 2000, this field has been applied tremendously for the development of catalytic asymmetric conjugate addition reactions [3-5]. In particular, the conjugate addition of nitroalkanes and their derivatives to enones has drawn the attention of organic chemists as the corresponding products can be chemoselectively converted to a variety of useful structures [6]. Thus a variety of methods has been developed with a range of different catalysts [7-9]. One of the challenges is to employ highly substituted enones in the reaction. Indeed, additional substituents, especially at the α-position of enones/activated olefins, decreases the reactivity significantly because of unfavorable steric interactions. To overcome this problem, reactive Michael donors must be used to achieve a good conversion in the reaction. In recent years, α-nitroketones have emerged as active nucleophiles in Michael reactions and a range of substrates have been explored [10]. Also, α-nitroketones have been found to be a popular nucleophilic acyl transfer reagent. In 2011, three research groups namely Wang, Yan and Kwong independently revealed the organocatalytic asymmetric conjugate addition of α-nitroketones to β,γ-unsaturated α-keto esters with the concomitant acyl transfer reaction to the keto group [11-13]. Consequently, our group developed an organocatalytic asymmetric Michael–acyl transfer reaction of α-nitroketones with unsaturated pyrazolones, 2-hydroxycinnamaldehydes, γ/δ-hydroxyenones, o-quinone methides, etc. [14-18]. Other groups also contributed contemporarily [19-21].
In recent years 4-arylidenepyrrolidine-2,3-diones have been explored mainly for the preparation of bicyclic dihydropyran derivatives through the catalytic inverse-electron-demand hetero-Diels–Alder reaction [22-24]. We postulated that 4-arylidenepyrrolidine-2,3-diones could also be suitable reaction partners of α-nitroketones. However, during the progress of our work, Bonne, Bugaut and co-workers have shown one example for the reaction of 2-nitroacetophenone with 4-benzylidenepyrrolidine-2,3-dione and only moderate enantioselectivity (50% ee) was achieved (Scheme 1) [25]. Herein, we report a better enantioselective version of the reaction between α-nitroketones and 4-arylidenepyrrolidine-2,3-diones (Scheme 1).
![[1860-5397-17-100-i1]](/bjoc/content/inline/1860-5397-17-100-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of α-nitroketones with unsaturated pyrazolone and with 4-benzylidenepyrrolidine-2,3-dione.
Scheme 1: Reactions of α-nitroketones with unsaturated pyrazolone and with 4-benzylidenepyrrolidine-2,3-dione....
Results and Discussion
Initially a model reaction was examined between N-benzyl-4-benzylidenepyrrolidine-2,3-dione (1a) and 2-nitro-1-phenylethanone (2a) in the presence of the quinine-derived bifunctional squaramide catalyst I in dichloromethane at room temperature (Table 1). Delightfully, after stirring for 12 hours, a product was isolated in 70% yield that was characterized as compound 3a and was supposed to be formed through conjugate addition followed by benzoyl-transfer reaction. However, only 20% enantiomeric excess was achieved. Then, the tert-leucine-derived squaramide catalyst II was employed and here both yield and ee slightly improved. Next, we turned our attention to bifunctional thiourea catalysts [26,27] that proved to be fruitful. Thus, the quinine and cinchonidine-derived bifunctional thiourea catalysts III and IV were employed in the reaction and moderate enantiomeric excesses were achieved. The yield and enantioselectivity further improved when using the tert-leucine-derived thiourea catalyst V. Also, Takemoto’s catalyst VI [28] was suitable for the reaction though a moderate enantiomeric excess was detected. Finally, the best catalyst turned out to be the pyrrolidine-containing bifunctional thiourea catalyst VII and the desired product was isolated in 80% yield with 80% ee. Then, solvent optimization was carried out to obtain better enantioselectivities. A similar enantioselectivity was attained in α,α,α-trifluorotoluene and tetrahydrofuran as the solvent, whereas in chloroform a slightly improved enantioselectivity of 86% ee was observed. Finally, the best solvent was found to be 1,2-dichloroethane and the product 3a was obtained in 82% yield with 90% ee.
Table 1: Catalyst screening and optimization of the reaction conditions.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-100-i3.svg?max-width=637&scale=1.0)
|
||||
| entrya | catalyst | solvent | yieldb | eec |
| 1 | I | CH2Cl2 | 70 | 20 |
| 2 | II | CH2Cl2 | 73 | 34 |
| 3 | III | CH2Cl2 | 76 | 55 |
| 4 | IV | CH2Cl2 | 78 | 52 |
| 5 | V | CH2Cl2 | 80 | 74 |
| 6 | VI | CH2Cl2 | 75 | 50 |
| 7 | VII | CH2Cl2 | 80 | 80 |
| 8 | VII | PhCF3 | 78 | 78 |
| 9 | VII | THF | 80 | 80 |
| 10 | VII | CHCl3 | 80 | 86 |
| 11 | VII | (CH2Cl)2 | 82 | 90 |
aReactions were carried out with 0.1 mmol of 1a and 0.1 mmol of 2a in 0.6 mL solvent at 25 °C for 12 hours; bisolated yield after silica gel column chromatography; cdetermined by chiral HPLC.
After having identified the optimized conditions we ventured in the scope and generality of the reaction. Initially a variety of α-nitroketones 1 having different aryl substituents were tested (Table 2). In fact, different ortho-, meta-, and para-substitutions on the phenyl group were compatible with the reaction conditions and satisfactory results were obtained (Table 2, entries 2–11). For example, p-tolyl-containing nitroketone 2b delivered the product 3b in 80% yield with 88% ee (Table 2, entry 2). A similar enantioselectivity was obtained for product 3c with a p-anisyl group (Table 2, entry 3). Interestingly, the enantioselectivity dropped slightly when replacing a p-methoxy substituent with a p-ethoxy group and product 3d was isolated in 78% yield with 80% ee (Table 2, entry 4). Also, a biphenyl group was tolerated and a good result was achieved (Table 2, entry 5). Then, 4-fluoro and 4-bromo-containing nitroketones 2f and 2g were employed in the reaction and gratifyingly the same 90% ee were obtained for both products 3f and 3g (Table 2, entries 6 and 7). meta-Substitutions were also tolerated in the reaction although decreased enantioselectivities were detected for the products 3h and 3i, respectively (Table 2, entries 8 and 9). Then, o-methyl- and o-methoxyphenyl-substituted nitroketones 2j and 2k were employed in the reaction. Here also, the reactions progressed well to provide products 3j and 3k in moderate yields and enantioselectivities (Table 2, entries 10 and 11). The 2-naphthyl-substituted nitroketone 2l also participated in the reaction to deliver 3l in 80% ee (Table 2, entry 12). Moreover, the hydrocinnamyl group containing nitroketone 2m also took part in the reaction and the corresponding product 3m was isolated in 65% yield with 64% ee (Table 2, entry 13). Finally, nitroketone 2n with a cyclohexyl group was engaged in the reaction and a moderate enantioselectivity was detected for product 3n (Table 2, entry 14).
Table 2: Scope of α-nitroketones 2 in the reaction with 1a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-100-i4.svg?max-width=637&scale=1.0)
|
||||
| entrya | R | 3 | yieldb | eec |
| 1 | Ph | 3a | 80 | 90 |
| 2 | 4-MeC6H4 | 3b | 80 | 88 |
| 3 | 4-MeOC6H4 | 3c | 82 | 88 |
| 4 | 4-EtOC6H4 | 3d | 78 | 80 |
| 5 | 4-PhC6H4 | 3e | 82 | 82 |
| 6 | 4-FC6H4 | 3f | 79 | 90 |
| 7 | 4-BrC6H4 | 3g | 78 | 90 |
| 8 | 3-MeC6H4 | 3h | 70 | 72 |
| 9 | 3-MeOC6H4 | 3i | 72 | 66 |
| 10 | 2-MeC6H4 | 3j | 65 | 68 |
| 11 | 2-MeOC6H4 | 3k | 68 | 70 |
| 12 | 2-naphthyl | 3l | 75 | 80 |
| 13 | PhCH2CH2 | 3m | 65 | 64 |
| 14 | cyclohexyl | 3n | 70 | 72 |
aThe reactions were carried out with 0.1 mmol of 1a and 0.1 mmol of 2 in 0.6 mL 1,2-dichloroethane at 25 °C for 12 hours; bisolated yield after silica gel column chromatography; cdetermined by chiral HPLC.
In the next step, we investigated the scope of the reaction of substrate 2a with a variety of pyrrolidine-2,3-diones 1 having different benzylidene substituents under the optimized conditions (Table 3). It turned out that a range of substitutions was tolerated and good results were attained. Initially, different para-substituted arylidene substrates were screened that smoothly afforded products 3o–s (Table 3, entries 1–5). For example, the pyrrolidine-2,3-dione 1b with a 4-methylbenzylidene-substituent provided the product 3o in 83% yield and 72% ee (Table 3, entry 1). A similar enantioselectivity was obtained with the 4-tert-butylenzylidene-substituted pyrrolidine-2,3-dione 1c (Table 3, entry 2). Then, different 4-halobenzylidene-substituted pyrrolidine-2,3-diones 1d–f were employed in the reaction and mixed results were obtained. Although product 3q having a 4-fluorophenyl-substitution was isolated in 80% yield and 84% ee, slightly decreased enantioselectivities were obtained for the corresponding 4-chloro- (3r, 70% ee) and 4-bromophenyl (3s, 76% ee) derivatives (Table 3, entries 3–5). These products could be particularly useful for further transformations via cross-coupling reactions. The ortho-fluoroarylidene-substituted pyrrolidine-2,3-dione 1g also participated in the reaction to deliver product 3t in 86% ee (Table 3, entry 6). 2,4-Disubstitution at the aromatic ring was also tolerated in the reaction and a moderate enantioselectivity was observed for the 2,4-difluorophenyl-substituted product 3u (Table 3, entry 7). The 3,5-dimethoxybenzylidene-containing pyrrolidine-2,3-dione 1i was prepared and also engaged in the reaction. Here also, a smooth conversion was detected and the product 3v was isolated in 80% yield with 72% ee (Table 3, entry 8). Finally, pyrrolidine-2,3-dione 1j containing a heteroaromatic group was also screened and an acceptable enantioselectivity for the 2-thienyl-substituted product 3w was witnessed (Table 3, entry 9).
Table 3: Scope of pyrrolidine-2,3-diones 1 in the reaction with 2a.
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-100-i5.svg?max-width=637&scale=1.0)
|
|||||
| entrya | R1 | 1 | 3 | yieldb | eec |
| 1 | 4-MeC6H4 | 1b | 3o | 83 | 72 |
| 2 | 4-t-BuC6H4 | 1c | 3p | 80 | 72 |
| 3 | 4-FC6H4 | 1d | 3q | 80 | 84 |
| 4 | 4-ClC6H4 | 1e | 3r | 79 | 70 |
| 5 | 4-BrC6H4 | 1f | 3s | 82 | 76 |
| 6 | 2-FC6H4 | 1g | 3t | 79 | 86 |
| 7 | 2,4-F2C6H3 | 1h | 3u | 78 | 72 |
| 8 | 3,5-(MeO)2C6H3 | 1i | 3v | 80 | 72 |
| 9 | 2-thienyl | 1j | 3w | 81 | 82 |
aReactions were carried out with 0.1 mmol of 1 and 0.1 mmol of 2a in 0.6 mL 1,2-dichloroethane at 25 °C for 12 hours; bisolated yield after silica gel column chromatography; cdetermined by chiral HPLC.
To further expand the scope of the reaction, 4-benzylidenedihydrofuran-2,3-dione (4) was prepared and reacted with nitroketones 2b and 2c, respectively. To our delight, the reactions proceeded smoothly at room temperature providing the desired products 5a and 5b in good yields and enantioselectivities (Scheme 2).
![[1860-5397-17-100-i2]](/bjoc/content/inline/1860-5397-17-100-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of 4-benzylidenedihydrofuran-2,3-dione (4) with α-nitroketones 2b,c. Reaction conditions: furan 4 (0.1 mmol), α-nitroketone 2 (0.1 mmol), 10 mol % VII in 0.6 mL 1,2-dichloroethane were reacted at 25 °C for 12 hours. Yields correspond to isolated yields after silica gel column chromatography and ees were determined by chiral HPLC.
Scheme 2: Reaction of 4-benzylidenedihydrofuran-2,3-dione (4) with α-nitroketones 2b,c. Reaction conditions: ...
Conclusion
In summary, in this paper we reported an organocatalytic asymmetric Michael/acyl transfer reaction between α-nitroketones and 4-arylidenepyrrolidine-2,3-diones/4-benzylidenedihydrofuran-2,3-dione. The products were obtained in good yields with moderate to high enantioselectivities. An easily available bifunctional thiourea catalyst was employed in the methodology.
Supporting Information
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 10.9 MB | Download |
References
-
Krause, N.; Hoffmann-Röder, A. Synthesis 2001, 171–196. doi:10.1055/s-2001-10803
Return to citation in text: [1] -
Sibi, M. P.; Manyem, S. Tetrahedron 2000, 56, 8033–8061. doi:10.1016/s0040-4020(00)00618-9
Return to citation in text: [1] -
Alonso, D. A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Molecules 2017, 22, 895. doi:10.3390/molecules22060895
Return to citation in text: [1] -
Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653
Return to citation in text: [1] -
Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065–2092. doi:10.1055/s-2007-983747
Return to citation in text: [1] -
Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Chem. Rev. 2005, 105, 933–972. doi:10.1021/cr040602r
Return to citation in text: [1] -
Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g
Return to citation in text: [1] -
Kwiatkowski, P.; Cholewiak, A.; Kasztelan, A. Org. Lett. 2014, 16, 5930–5933. doi:10.1021/ol502941d
Return to citation in text: [1] -
Bera, K.; Namboothiri, I. N. N. J. Org. Chem. 2015, 80, 1402–1413. doi:10.1021/jo502332r
Return to citation in text: [1] -
Gharui, C.; Pan, S. C. Org. Biomol. Chem. 2019, 17, 5190–5211. doi:10.1039/c9ob00828d
Return to citation in text: [1] -
Gao, Y.; Ren, Q.; Siau, W.-Y.; Wang, J. Chem. Commun. 2011, 47, 5819–5821. doi:10.1039/c1cc11124h
Return to citation in text: [1] -
Lu, R.-j.; Yan, Y.-y.; Wang, J.-j.; Du, Q.-s.; Nie, S.-z.; Yan, M. J. Org. Chem. 2011, 76, 6230–6239. doi:10.1021/jo2009752
Return to citation in text: [1] -
Li, P.; Chan, S. H.; Chan, A. S. C.; Kwong, F. Y. Org. Biomol. Chem. 2011, 9, 7997–7999. doi:10.1039/c1ob06191g
Return to citation in text: [1] -
Maity, R.; Gharui, C.; Sil, A. K.; Pan, S. C. Org. Lett. 2017, 19, 662–665. doi:10.1021/acs.orglett.6b03823
Return to citation in text: [1] -
Maity, R.; Pan, S. C. Org. Biomol. Chem. 2018, 16, 1598–1608. doi:10.1039/c8ob00078f
Return to citation in text: [1] -
Mondal, K.; Pan, S. C. J. Org. Chem. 2018, 83, 5301–5312. doi:10.1021/acs.joc.8b00436
Return to citation in text: [1] -
Gharui, C.; Behera, D.; Pan, S. C. Adv. Synth. Catal. 2018, 360, 4502–4508. doi:10.1002/adsc.201801015
Return to citation in text: [1] -
Maity, R.; Sahoo, S. C.; Pan, S. C. Eur. J. Org. Chem. 2019, 2297–2304. doi:10.1002/ejoc.201900069
Return to citation in text: [1] -
Liu, Y.; Mo, Y.; Dong, X.; Chen, L.; Ye, L.; Li, X.; Zhao, Z.; Li, X. Tetrahedron 2019, 75, 2466–2471. doi:10.1016/j.tet.2019.03.021
Return to citation in text: [1] -
Song, Y.-X.; Du, D.-M. Adv. Synth. Catal. 2019, 361, 5042–5049. doi:10.1002/adsc.201900901
Return to citation in text: [1] -
Zhou, J.; Bai, L.-J.; Liang, G.-J.; Xu, Q.-G.; Zhou, L.-P.; Zhou, H. Org. Biomol. Chem. 2020, 18, 2641–2645. doi:10.1039/d0ob00397b
Return to citation in text: [1] -
Li, J.-L.; Fu, L.; Wu, J.; Yang, K.-C.; Li, Q.-Z.; Gou, X.-J.; Peng, C.; Han, B.; Shen, X.-D. Chem. Commun. 2017, 53, 6875–6878. doi:10.1039/c7cc02921g
Return to citation in text: [1] -
Hu, X.; Zhou, Y.; Lu, Y.; Zou, S.; Lin, L.; Liu, X.; Feng, X. J. Org. Chem. 2018, 83, 8679–8687. doi:10.1021/acs.joc.8b00839
Return to citation in text: [1] -
Wang, Y.; Chen, Y.; Li, X.; Mao, Y.; Chen, W.; Zhan, R.; Huang, H. Org. Biomol. Chem. 2019, 17, 3945–3950. doi:10.1039/c9ob00419j
Return to citation in text: [1] -
Fofana, M.; Dudognon, Y.; Bertrand, L.; Constantieux, T.; Rodriguez, J.; Ndiaye, I.; Bonne, D.; Bugaut, X. Eur. J. Org. Chem. 2020, 3486–3490. doi:10.1002/ejoc.202000460
Return to citation in text: [1] -
Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/b719249e
Return to citation in text: [1] -
Siau, W.-Y.; Wang, J. Catal. Sci. Technol. 2011, 1, 1298–1310. doi:10.1039/c1cy00271f
Return to citation in text: [1] -
Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672–12673. doi:10.1021/ja036972z
Return to citation in text: [1]
| 1. | Krause, N.; Hoffmann-Röder, A. Synthesis 2001, 171–196. doi:10.1055/s-2001-10803 |
| 2. | Sibi, M. P.; Manyem, S. Tetrahedron 2000, 56, 8033–8061. doi:10.1016/s0040-4020(00)00618-9 |
| 10. | Gharui, C.; Pan, S. C. Org. Biomol. Chem. 2019, 17, 5190–5211. doi:10.1039/c9ob00828d |
| 7. | Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g |
| 8. | Kwiatkowski, P.; Cholewiak, A.; Kasztelan, A. Org. Lett. 2014, 16, 5930–5933. doi:10.1021/ol502941d |
| 9. | Bera, K.; Namboothiri, I. N. N. J. Org. Chem. 2015, 80, 1402–1413. doi:10.1021/jo502332r |
| 6. | Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Chem. Rev. 2005, 105, 933–972. doi:10.1021/cr040602r |
| 3. | Alonso, D. A.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Molecules 2017, 22, 895. doi:10.3390/molecules22060895 |
| 4. | Tsogoeva, S. B. Eur. J. Org. Chem. 2007, 1701–1716. doi:10.1002/ejoc.200600653 |
| 5. | Vicario, J. L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065–2092. doi:10.1055/s-2007-983747 |
| 22. | Li, J.-L.; Fu, L.; Wu, J.; Yang, K.-C.; Li, Q.-Z.; Gou, X.-J.; Peng, C.; Han, B.; Shen, X.-D. Chem. Commun. 2017, 53, 6875–6878. doi:10.1039/c7cc02921g |
| 23. | Hu, X.; Zhou, Y.; Lu, Y.; Zou, S.; Lin, L.; Liu, X.; Feng, X. J. Org. Chem. 2018, 83, 8679–8687. doi:10.1021/acs.joc.8b00839 |
| 24. | Wang, Y.; Chen, Y.; Li, X.; Mao, Y.; Chen, W.; Zhan, R.; Huang, H. Org. Biomol. Chem. 2019, 17, 3945–3950. doi:10.1039/c9ob00419j |
| 26. | Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/b719249e |
| 27. | Siau, W.-Y.; Wang, J. Catal. Sci. Technol. 2011, 1, 1298–1310. doi:10.1039/c1cy00271f |
| 19. | Liu, Y.; Mo, Y.; Dong, X.; Chen, L.; Ye, L.; Li, X.; Zhao, Z.; Li, X. Tetrahedron 2019, 75, 2466–2471. doi:10.1016/j.tet.2019.03.021 |
| 20. | Song, Y.-X.; Du, D.-M. Adv. Synth. Catal. 2019, 361, 5042–5049. doi:10.1002/adsc.201900901 |
| 21. | Zhou, J.; Bai, L.-J.; Liang, G.-J.; Xu, Q.-G.; Zhou, L.-P.; Zhou, H. Org. Biomol. Chem. 2020, 18, 2641–2645. doi:10.1039/d0ob00397b |
| 28. | Okino, T.; Hoashi, Y.; Takemoto, Y. J. Am. Chem. Soc. 2003, 125, 12672–12673. doi:10.1021/ja036972z |
| 14. | Maity, R.; Gharui, C.; Sil, A. K.; Pan, S. C. Org. Lett. 2017, 19, 662–665. doi:10.1021/acs.orglett.6b03823 |
| 15. | Maity, R.; Pan, S. C. Org. Biomol. Chem. 2018, 16, 1598–1608. doi:10.1039/c8ob00078f |
| 16. | Mondal, K.; Pan, S. C. J. Org. Chem. 2018, 83, 5301–5312. doi:10.1021/acs.joc.8b00436 |
| 17. | Gharui, C.; Behera, D.; Pan, S. C. Adv. Synth. Catal. 2018, 360, 4502–4508. doi:10.1002/adsc.201801015 |
| 18. | Maity, R.; Sahoo, S. C.; Pan, S. C. Eur. J. Org. Chem. 2019, 2297–2304. doi:10.1002/ejoc.201900069 |
| 11. | Gao, Y.; Ren, Q.; Siau, W.-Y.; Wang, J. Chem. Commun. 2011, 47, 5819–5821. doi:10.1039/c1cc11124h |
| 12. | Lu, R.-j.; Yan, Y.-y.; Wang, J.-j.; Du, Q.-s.; Nie, S.-z.; Yan, M. J. Org. Chem. 2011, 76, 6230–6239. doi:10.1021/jo2009752 |
| 13. | Li, P.; Chan, S. H.; Chan, A. S. C.; Kwong, F. Y. Org. Biomol. Chem. 2011, 9, 7997–7999. doi:10.1039/c1ob06191g |
| 25. | Fofana, M.; Dudognon, Y.; Bertrand, L.; Constantieux, T.; Rodriguez, J.; Ndiaye, I.; Bonne, D.; Bugaut, X. Eur. J. Org. Chem. 2020, 3486–3490. doi:10.1002/ejoc.202000460 |
© 2021 Parida and Pan; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)