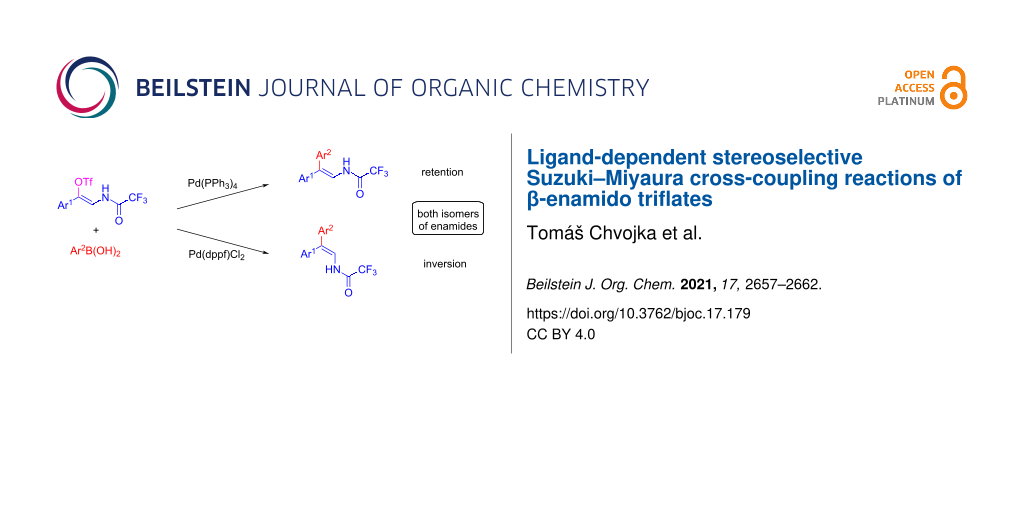
Abstract
The stereoselective Suzuki–Miyaura cross-coupling of (Z)-β-enamido triflates is demonstrated. Depending on the nature of the ligand in the palladium catalyst, either retention or inversion of the configuration during the synthesis of β,β-diaryl-substituted enamides is observed. Thus, the method provides synthetic access to both isomers of the target enamides from (Z)-β-enamido triflates.

Graphical Abstract
Introduction
Enamides are substrates of high value in organic synthesis [1,2]. Their multifacial reactivity has been explored in asymmetric alkylations [3], hydroalkynylations [4], trifluoromethylcyanations [5], heterocycle synthesis [6], asymmetric acylations [7], hydroborations [8], hydrogenations [9], etc. They are also important pharmacophores, which display a range of cytotoxic, antifungal, or antibiotic properties [10-12]. Modern stereoselective syntheses leading to highly substituted enamides include cross-coupling of vinyl (pseudo)halides or organoboron compounds [13], hydroamidation of alkynes [14-16], ynamide functionalization [17-19], or isomerization of N-allyl amides [20], but still possess drawbacks, especially for stereoselective synthesis of tri- and tetrasubstituted enamides.
Recently, we have reported a triflic acid-mediated reaction of N-fluoroalkyl-1,2,3-triazoles leading to (Z)-β-enamido triflates [21] and Lewis acid-mediated reaction to (Z)-β-enamido fluorides [22] and halovinyl imidoyl halides [23]. In addition, Li and co-workers extended the scope of accessible (Z)-β-enamido triflates by denitrogenative reaction of N1-H-1,2,3-triazoles in the presence of acyl halides and sodium triflate [24]. These enamido triflates and halides were found to undergo cross-coupling reactions with retention of configuration on the double bond and served as valuable starting materials for the synthesis of functionalized β,β-disubstituted enamides (Scheme 1A) [21,23].
![[1860-5397-17-179-i1]](/bjoc/content/inline/1860-5397-17-179-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A: Synthesis of (Z)-β-enamido triflates and subsequent stereoselective cross-coupling reactions. B: Ligand-controlled stereoselective synthesis of β,β-diaryl-substituted enamides.
Scheme 1: A: Synthesis of (Z)-β-enamido triflates and subsequent stereoselective cross-coupling reactions. B:...
In the last decade, only a few reports describing isomerization of the double bond of vinyl (pseudo)halides during the Suzuki coupling have been published [25-29]. Typically, inversion of configuration occurs on substrates containing a double bond in conjugation with an electron-withdrawing group, such as the carbonyl group in enones [27,30]. We hypothesized that (Z)-β-enamido triflates could, during the Suzuki cross-coupling, undergo isomerization of the double bond, similarly to enones, and thus serve as starting materials to either (E) or (Z)-isomers of enamides depending on the conditions used. Here we present a study of the effect of ligand on the stereoselective outcome of the Suzuki cross-coupling reaction of various (Z)-β-enamido triflates (Scheme 1B).
Results and Discussion
We initiated our study by examining the Suzuki cross-coupling of vinyl triflate 1a and 3-nitrophenylboronic acid. First, the influence of the catalyst on the stereoselective outcome of the reaction was studied (Table 1). The use of Pd(PPh3)4 resulted in the formation of isomeric products 2aa and 3aa in a 93:7 ratio (Table 1, entry 1). Other catalysts led to significant loss of stereoselectivity on the double bond (Table 1, entries 2–9). When Pd(dppf)Cl2 was employed, isomeric product 3aa was preferentially formed (2aa/3aa, 29:71, Table 1, entry 6). Although other catalysts, such as Pd(acac)2, Pd(dba)2, and Pd(OAc)2(PPh3)2 showed good selectivity to inversion product 3aa, the products were formed in low yields (Table 1, entries 7–9). Further screening of the solvent and the base led to the identification of Pd(PPh3)4 (10 mol %), arylboronic acid (1 equiv), K3PO4 (2 equiv) in THF/H2O 1:1 as ideal conditions providing enamides 2aa and 3aa in a 93:7 ratio in 90% 19F NMR yield (Table S1 in Supporting Information File 1).
Table 1: The effect of different catalysts on product yields and ratios.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-179-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst (10 mol %) | Yield (%)a | 2aa/3aa |
| 1 | Pd(PPh3)4 | 70 | 93:7 |
| 2 | PdCl2(PPh3)2 | 80 | 81:19 |
| 3 | 2-(2'-di-tert-butylphosphine)biphenylpalladium(II) acetate | 42 | 83:17 |
| 4 | Pd(t-Bu3P)2 | 46 | 48:52 |
| 5 | Pd(dtbpf)Cl2 | 66 | 33:67 |
| 6 | Pd(dppf)Cl2 | 70 | 29:71 |
| 7 | Pd(acac)2 | 34 | 24:76 |
| 8 | Pd(dba)2 | 23 | 22:78 |
| 9 | Pd(OAc)2(PPh3)2 | 11 | 18:82 |
aCombined 19F NMR yield of 2aa and 3aa using PhCF3 as an internal standard.
The optimized conditions were applied to the scope study. Substituted phenylboronic acids and vinyl triflates led to the formation of enamides 2 with high stereoselectivity and in good to high NMR yields (Scheme 2). However, isolated yields were found to be lower due to the decomposition of the formed enamides 2 during column chromatography on silica gel. A moderate loss of stereochemistry was observed only in cases of bulky ortho-substitution of either arylboronic acid or vinyl triflate (2ad, 2da, and 2dd). Alkylboronic acids were found to be unreactive even after prolonged reaction time (1a with n-hexylboronic acid, 60 h, rt).
![[1860-5397-17-179-i2]](/bjoc/content/inline/1860-5397-17-179-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope of the Suzuki coupling leading to enamides 2 and 3. aRatio determined by 19F NMR; byield determined by 19F NMR using PhCF3 as an internal standard; cisolated yield; disolated as a mixture of isomers E/Z = 64:36.
Scheme 2: Substrate scope of the Suzuki coupling leading to enamides 2 and 3. aRatio determined by 19F NMR; b...
Next, conditions for the formation of isomeric products were optimized. Screening the reaction conditions identified Pd(dppf)Cl2 (10 mol %), arylboronic acid (1.2 equiv), KF (2 equiv) in THF/H2O 15:1 at 50 °C as a system affording the best obtained ratio and yield in favor to product 3aa (Table S2 in Supporting Information File 1). Subsequently the optimized conditions for preferential formation of 3 were used in the scope study and in all cases, isomeric products 3 formed preferentially (Scheme 2). The highest selectivity in favor to enamide 3 was noted in the Suzuki coupling of vinyl triflate 1d bearing a 2-trifluoromethylphenyl group which gave enamides 3da (2da/3da, 20:80) and 3dd (2dd/3dd, <1:99). It is worth mentioning that the reaction of vinyl triflate 1c with 3-methoxyphenylboronic acid led to full decomposition of the starting material. The stereochemistry of the double bond in compounds 2 and 3 was determined by 2D ROESY NMR analysis showing interaction between the alkenyl hydrogen and ortho-hydrogens on the aryl rings.
Based on previously proposed mechanisms of isomerization in Suzuki cross-coupling reactions, we suggest the following explanation for the observed isomerization [25,29] (Scheme 3). In the first step, vinyl triflate undergoes oxidative addition to give complex 4, which subsequently transmetalates with arylboronic acid to form palladium complex 5. In the case of Pd(PPh3)4, reductive elimination occurs to give enamide 2. However, using catalysts with very bulky ligands, such as Pd(dppf)Cl2 causes the tautomerization of complex 5 [30] to zwitterionic carbene 6 which can now isomerize through the C–C bond rotation to the thermodynamically more stable palladium complex 7, followed by reductive elimination to enamide 3. A possible isomerization of enamides 2 or 3 in the presence of a catalyst was ruled out because the treatment of 2ca under conditions leading to inversion of the configuration did not affect the ratio between the resulting enamides.
![[1860-5397-17-179-i3]](/bjoc/content/inline/1860-5397-17-179-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanisms for the formed Suzuki coupling retention products 2 and inversion products 3.
Scheme 3: Proposed mechanisms for the formed Suzuki coupling retention products 2 and inversion products 3.
Conclusion
In conclusion, the stereoselective outcome of the Suzuki cross-coupling of vinyl triflates 1 with arylboronic acids was found to be catalyst dependent. The use of Pd(PPh3)4 led to the selective formation of enamides 2 with retention of configuration of the double bond. Reactions with other catalysts provided significant losses of stereoselectivity on the double bond. When Pd(dppf)Cl2 was used, enamides 3 with inversion of the configuration of the double bond were formed preferably. Both conditions were applied to a range of arylboronic acids and (Z)-β-enamido triflates.
Supporting Information
| Supporting Information File 1: Experimental part, optimization, compound characterization, and copies of NMR spectra. | ||
| Format: PDF | Size: 6.7 MB | Download |
References
-
Gopalaiah, K.; Kagan, H. B. Chem. Rev. 2011, 111, 4599–4657. doi:10.1021/cr100031f
Return to citation in text: [1] -
Beltran, F.; Miesch, L. Synthesis 2020, 52, 2497–2511. doi:10.1055/s-0040-1707403
Return to citation in text: [1] -
Zhao, B.; Shang, R.; Wang, G.-Z.; Wang, S.; Chen, H.; Fu, Y. ACS Catal. 2020, 10, 1334–1343. doi:10.1021/acscatal.9b04699
Return to citation in text: [1] -
Zhang, W.-W.; Zhang, S.-L.; Li, B.-J. Angew. Chem., Int. Ed. 2020, 59, 6874–6880. doi:10.1002/anie.201916088
Return to citation in text: [1] -
Zhang, G.; Zhou, S.; Fu, L.; Chen, P.; Li, Y.; Zou, J.; Liu, G. Angew. Chem., Int. Ed. 2020, 59, 20439–20444. doi:10.1002/anie.202008338
Return to citation in text: [1] -
Zhen, L.; Tong, S.; Zhu, J.; Wang, M.-X. Chem. – Eur. J. 2020, 26, 401–405. doi:10.1002/chem.201904596
Return to citation in text: [1] -
Liu, R.-H.; Shen, Z.-Y.; Wang, C.; Loh, T.-P.; Hu, X.-H. Org. Lett. 2020, 22, 944–949. doi:10.1021/acs.orglett.9b04495
Return to citation in text: [1] -
Bai, X.-Y.; Zhao, W.; Sun, X.; Li, B.-J. J. Am. Chem. Soc. 2019, 141, 19870–19878. doi:10.1021/jacs.9b10578
Return to citation in text: [1] -
Friedfeld, M. R.; Zhong, H.; Ruck, R. T.; Shevlin, M.; Chirik, P. J. Science 2018, 360, 888–893. doi:10.1126/science.aar6117
Return to citation in text: [1] -
Martín, M. J.; Coello, L.; Fernández, R.; Reyes, F.; Rodríguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sánchez-Sancho, F.; Bueno, S.; de Eguilior, C.; Francesch, A.; Munt, S.; Cuevas, C. J. Am. Chem. Soc. 2013, 135, 10164–10171. doi:10.1021/ja404578u
Return to citation in text: [1] -
Lin, S.; Yang, Z.-Q.; Kwok, B. H. B.; Koldobskiy, M.; Crews, C. M.; Danishefsky, S. J. J. Am. Chem. Soc. 2004, 126, 6347–6355. doi:10.1021/ja049821k
Return to citation in text: [1] -
Shen, R.; Lin, C. T.; Bowman, E. J.; Bowman, B. J.; Porco, J. A. J. Am. Chem. Soc. 2003, 125, 7889–7901. doi:10.1021/ja0352350
Return to citation in text: [1] -
Correa, A.; Bolm, C. Metal-Catalyzed C(sp2)–N Bond Formation. In Amination and Formation of sp2 C-N Bonds; Taillefer, M.; Ma, D., Eds.; Topics in Organometallic Chemistry, Vol. 46; Springer: Berlin, Heidelberg, 2013; pp 55–85. doi:10.1007/3418_2012_57
Return to citation in text: [1] -
Arndt, M.; Salih, K. S. M.; Fromm, A.; Goossen, L. J.; Menges, F.; Niedner-Schatteburg, G. J. Am. Chem. Soc. 2011, 133, 7428–7449. doi:10.1021/ja111389r
Return to citation in text: [1] -
Gooßen, L. J.; Salih, K. S. M.; Blanchot, M. Angew. Chem., Int. Ed. 2008, 47, 8492–8495. doi:10.1002/anie.200803068
Return to citation in text: [1] -
Herrero, M. T.; de Sarralde, J. D.; SanMartin, R.; Bravo, L.; Domínguez, E. Adv. Synth. Catal. 2012, 354, 3054–3064. doi:10.1002/adsc.201200430
Return to citation in text: [1] -
Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569
Return to citation in text: [1] -
Mallick, R. K.; Prabagar, B.; Sahoo, A. K. J. Org. Chem. 2017, 82, 10583–10594. doi:10.1021/acs.joc.7b02124
Return to citation in text: [1] -
Baldassari, L. L.; de la Torre, A.; Li, J.; Lüdtke, D. S.; Maulide, N. Angew. Chem., Int. Ed. 2017, 56, 15723–15727. doi:10.1002/anie.201709128
Return to citation in text: [1] -
Trost, B. M.; Cregg, J. J.; Quach, N. J. Am. Chem. Soc. 2017, 139, 5133–5139. doi:10.1021/jacs.7b00564
Return to citation in text: [1] -
Markos, A.; Voltrová, S.; Motornov, V.; Tichý, D.; Klepetářová, B.; Beier, P. Chem. – Eur. J. 2019, 25, 7640–7644. doi:10.1002/chem.201901632
Return to citation in text: [1] [2] -
Markos, A.; Janecký, L.; Klepetářová, B.; Pohl, R.; Beier, P. Org. Lett. 2021, 23, 4224–4227. doi:10.1021/acs.orglett.1c01183
Return to citation in text: [1] -
Markos, A.; Janecký, L.; Chvojka, T.; Martinek, T.; MartinezSeara, H.; Klepetářová, B.; Beier, P. Adv. Synth. Catal. 2021, 363, 3258–3266. doi:10.1002/adsc.202100009
Return to citation in text: [1] [2] -
Wang, T.; Tang, Z.; Luo, H.; Tian, Y.; Xu, M.; Lu, Q.; Li, B. Org. Lett. 2021, 23, 6293–6298. doi:10.1021/acs.orglett.1c02087
Return to citation in text: [1] -
Lu, G.-P.; Voigtritter, K. R.; Cai, C.; Lipshutz, B. H. J. Org. Chem. 2012, 77, 3700–3703. doi:10.1021/jo300437t
Return to citation in text: [1] [2] -
Feceu, A.; Sangster, L. E.; Martin, D. B. C. Org. Lett. 2018, 20, 3151–3155. doi:10.1021/acs.orglett.8b00809
Return to citation in text: [1] -
Chehal, N. K.; Budzelaar, P. H. M.; Hultin, P. G. Org. Biomol. Chem. 2018, 16, 1134–1143. doi:10.1039/c7ob02925j
Return to citation in text: [1] [2] -
Molinaro, C.; Scott, J. P.; Shevlin, M.; Wise, C.; Ménard, A.; Gibb, A.; Junker, E. M.; Lieberman, D. J. Am. Chem. Soc. 2015, 137, 999–1006. doi:10.1021/ja511872a
Return to citation in text: [1] -
Li, B. X.; Le, D. N.; Mack, K. A.; McClory, A.; Lim, N.-K.; Cravillion, T.; Savage, S.; Han, C.; Collum, D. B.; Zhang, H.; Gosselin, F. J. Am. Chem. Soc. 2017, 139, 10777–10783. doi:10.1021/jacs.7b05071
Return to citation in text: [1] [2] -
Trost, B. M.; Tracy, J. S. ACS Catal. 2019, 9, 1584–1594. doi:10.1021/acscatal.8b04567
Return to citation in text: [1] [2]
| 25. | Lu, G.-P.; Voigtritter, K. R.; Cai, C.; Lipshutz, B. H. J. Org. Chem. 2012, 77, 3700–3703. doi:10.1021/jo300437t |
| 26. | Feceu, A.; Sangster, L. E.; Martin, D. B. C. Org. Lett. 2018, 20, 3151–3155. doi:10.1021/acs.orglett.8b00809 |
| 27. | Chehal, N. K.; Budzelaar, P. H. M.; Hultin, P. G. Org. Biomol. Chem. 2018, 16, 1134–1143. doi:10.1039/c7ob02925j |
| 28. | Molinaro, C.; Scott, J. P.; Shevlin, M.; Wise, C.; Ménard, A.; Gibb, A.; Junker, E. M.; Lieberman, D. J. Am. Chem. Soc. 2015, 137, 999–1006. doi:10.1021/ja511872a |
| 29. | Li, B. X.; Le, D. N.; Mack, K. A.; McClory, A.; Lim, N.-K.; Cravillion, T.; Savage, S.; Han, C.; Collum, D. B.; Zhang, H.; Gosselin, F. J. Am. Chem. Soc. 2017, 139, 10777–10783. doi:10.1021/jacs.7b05071 |
| 24. | Wang, T.; Tang, Z.; Luo, H.; Tian, Y.; Xu, M.; Lu, Q.; Li, B. Org. Lett. 2021, 23, 6293–6298. doi:10.1021/acs.orglett.1c02087 |
| 21. | Markos, A.; Voltrová, S.; Motornov, V.; Tichý, D.; Klepetářová, B.; Beier, P. Chem. – Eur. J. 2019, 25, 7640–7644. doi:10.1002/chem.201901632 |
| 23. | Markos, A.; Janecký, L.; Chvojka, T.; Martinek, T.; MartinezSeara, H.; Klepetářová, B.; Beier, P. Adv. Synth. Catal. 2021, 363, 3258–3266. doi:10.1002/adsc.202100009 |
| 1. | Gopalaiah, K.; Kagan, H. B. Chem. Rev. 2011, 111, 4599–4657. doi:10.1021/cr100031f |
| 2. | Beltran, F.; Miesch, L. Synthesis 2020, 52, 2497–2511. doi:10.1055/s-0040-1707403 |
| 6. | Zhen, L.; Tong, S.; Zhu, J.; Wang, M.-X. Chem. – Eur. J. 2020, 26, 401–405. doi:10.1002/chem.201904596 |
| 22. | Markos, A.; Janecký, L.; Klepetářová, B.; Pohl, R.; Beier, P. Org. Lett. 2021, 23, 4224–4227. doi:10.1021/acs.orglett.1c01183 |
| 5. | Zhang, G.; Zhou, S.; Fu, L.; Chen, P.; Li, Y.; Zou, J.; Liu, G. Angew. Chem., Int. Ed. 2020, 59, 20439–20444. doi:10.1002/anie.202008338 |
| 23. | Markos, A.; Janecký, L.; Chvojka, T.; Martinek, T.; MartinezSeara, H.; Klepetářová, B.; Beier, P. Adv. Synth. Catal. 2021, 363, 3258–3266. doi:10.1002/adsc.202100009 |
| 4. | Zhang, W.-W.; Zhang, S.-L.; Li, B.-J. Angew. Chem., Int. Ed. 2020, 59, 6874–6880. doi:10.1002/anie.201916088 |
| 20. | Trost, B. M.; Cregg, J. J.; Quach, N. J. Am. Chem. Soc. 2017, 139, 5133–5139. doi:10.1021/jacs.7b00564 |
| 3. | Zhao, B.; Shang, R.; Wang, G.-Z.; Wang, S.; Chen, H.; Fu, Y. ACS Catal. 2020, 10, 1334–1343. doi:10.1021/acscatal.9b04699 |
| 21. | Markos, A.; Voltrová, S.; Motornov, V.; Tichý, D.; Klepetářová, B.; Beier, P. Chem. – Eur. J. 2019, 25, 7640–7644. doi:10.1002/chem.201901632 |
| 10. | Martín, M. J.; Coello, L.; Fernández, R.; Reyes, F.; Rodríguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sánchez-Sancho, F.; Bueno, S.; de Eguilior, C.; Francesch, A.; Munt, S.; Cuevas, C. J. Am. Chem. Soc. 2013, 135, 10164–10171. doi:10.1021/ja404578u |
| 11. | Lin, S.; Yang, Z.-Q.; Kwok, B. H. B.; Koldobskiy, M.; Crews, C. M.; Danishefsky, S. J. J. Am. Chem. Soc. 2004, 126, 6347–6355. doi:10.1021/ja049821k |
| 12. | Shen, R.; Lin, C. T.; Bowman, E. J.; Bowman, B. J.; Porco, J. A. J. Am. Chem. Soc. 2003, 125, 7889–7901. doi:10.1021/ja0352350 |
| 14. | Arndt, M.; Salih, K. S. M.; Fromm, A.; Goossen, L. J.; Menges, F.; Niedner-Schatteburg, G. J. Am. Chem. Soc. 2011, 133, 7428–7449. doi:10.1021/ja111389r |
| 15. | Gooßen, L. J.; Salih, K. S. M.; Blanchot, M. Angew. Chem., Int. Ed. 2008, 47, 8492–8495. doi:10.1002/anie.200803068 |
| 16. | Herrero, M. T.; de Sarralde, J. D.; SanMartin, R.; Bravo, L.; Domínguez, E. Adv. Synth. Catal. 2012, 354, 3054–3064. doi:10.1002/adsc.201200430 |
| 30. | Trost, B. M.; Tracy, J. S. ACS Catal. 2019, 9, 1584–1594. doi:10.1021/acscatal.8b04567 |
| 9. | Friedfeld, M. R.; Zhong, H.; Ruck, R. T.; Shevlin, M.; Chirik, P. J. Science 2018, 360, 888–893. doi:10.1126/science.aar6117 |
| 17. | Minko, Y.; Pasco, M.; Lercher, L.; Botoshansky, M.; Marek, I. Nature 2012, 490, 522–526. doi:10.1038/nature11569 |
| 18. | Mallick, R. K.; Prabagar, B.; Sahoo, A. K. J. Org. Chem. 2017, 82, 10583–10594. doi:10.1021/acs.joc.7b02124 |
| 19. | Baldassari, L. L.; de la Torre, A.; Li, J.; Lüdtke, D. S.; Maulide, N. Angew. Chem., Int. Ed. 2017, 56, 15723–15727. doi:10.1002/anie.201709128 |
| 8. | Bai, X.-Y.; Zhao, W.; Sun, X.; Li, B.-J. J. Am. Chem. Soc. 2019, 141, 19870–19878. doi:10.1021/jacs.9b10578 |
| 27. | Chehal, N. K.; Budzelaar, P. H. M.; Hultin, P. G. Org. Biomol. Chem. 2018, 16, 1134–1143. doi:10.1039/c7ob02925j |
| 30. | Trost, B. M.; Tracy, J. S. ACS Catal. 2019, 9, 1584–1594. doi:10.1021/acscatal.8b04567 |
| 7. | Liu, R.-H.; Shen, Z.-Y.; Wang, C.; Loh, T.-P.; Hu, X.-H. Org. Lett. 2020, 22, 944–949. doi:10.1021/acs.orglett.9b04495 |
| 13. | Correa, A.; Bolm, C. Metal-Catalyzed C(sp2)–N Bond Formation. In Amination and Formation of sp2 C-N Bonds; Taillefer, M.; Ma, D., Eds.; Topics in Organometallic Chemistry, Vol. 46; Springer: Berlin, Heidelberg, 2013; pp 55–85. doi:10.1007/3418_2012_57 |
| 25. | Lu, G.-P.; Voigtritter, K. R.; Cai, C.; Lipshutz, B. H. J. Org. Chem. 2012, 77, 3700–3703. doi:10.1021/jo300437t |
| 29. | Li, B. X.; Le, D. N.; Mack, K. A.; McClory, A.; Lim, N.-K.; Cravillion, T.; Savage, S.; Han, C.; Collum, D. B.; Zhang, H.; Gosselin, F. J. Am. Chem. Soc. 2017, 139, 10777–10783. doi:10.1021/jacs.7b05071 |
© 2021 Chvojka et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)