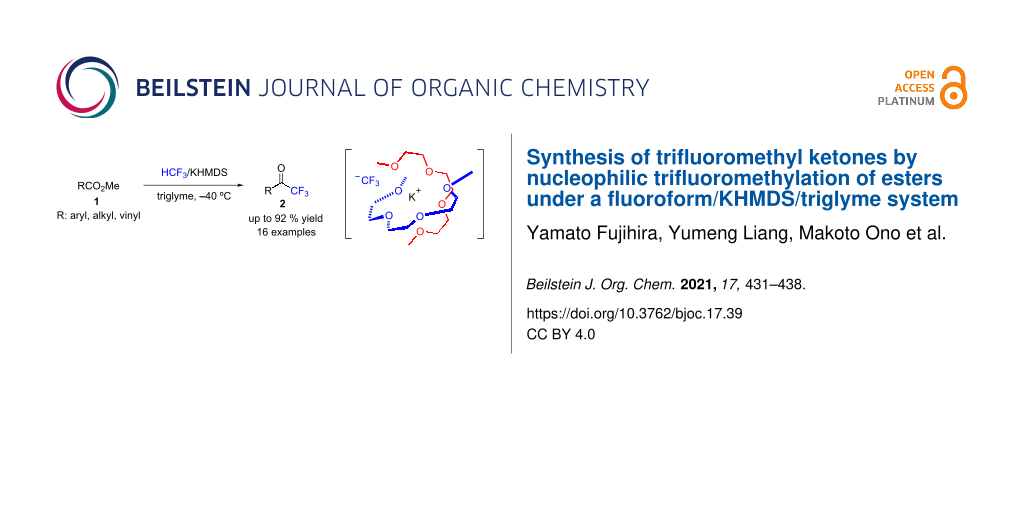
Abstract
A straightforward method that enables the formation of biologically attractive trifluoromethyl ketones from readily available methyl esters using the potent greenhouse gas fluoroform (HCF3, HFC-23) was developed. The combination of fluoroform and KHMDS in triglyme at −40 °C was effective for this transformation, with good yields as high as 92%. Substrate scope of the trifluoromethylation procedure was explored for aromatic, aliphatic, and conjugated methyl esters. This study presents a straightforward trifluoromethylation process of various methyl esters that convert well to the corresponding trifluoromethyl ketones. The tolerance of various pharmacophores under the reaction conditions was also explored.

Graphical Abstract
Introduction
In recent decades, organofluorine molecules have received widespread attention in the field of medicinal chemistry [1-4]. The introduction of fluorine(s) into organic molecules usually leads to significant changes in the chemical and physicochemical properties of the original compounds [5,6]. Hence, the fluorination and related fluoro-functionalization of drug candidates are powerful strategies in drug design to appropriately bias their biological properties, bioavailability, and ADME [7,8]. While tremendous methodologies have been developed for the synthesis of organofluorine compounds [9,10], many of the laboratory methods are not always suitable for industrial production in terms of their synthetic complexity, handling, and cost of target compounds [11-15]. Thus, the development of low-cost and straightforward chemical synthetic technologies, including fluorination and trifluoromethylation, are matters of considerable importance to pharmaceutical and agrochemical industries. Fluoroform (HCF3, HFC-23) is an industrial byproduct of polytetrafluoroethylene synthesis and has become an ideal, economical feedstock for trifluoromethyl (CF3) compounds. Rather than decomposing CF3 compounds, it would be better to maximize the efficiency of their use [16-18]. However, taming HCF3 as a trifluoromethylation agent is a challenge in organic chemistry [19-28], although recent rapid progress in the chemistry of HCF3 by Grushin (for CuCF3) [29-37], Prakash (for KCF3) [38], and others [39-44], including our group [45-50], has dramatically improved the prospects. One of the problems facing the treatment of HCF3 for nucleophilic trifluoromethylation reactions is the low stability of the directly generated CF3 anion (CF3−) for decomposing to difluorocarbene (:CF2) and fluoride (F−) (Scheme 1a). Due to the formation of highly stable fluoride salts (MF), the breakdown of CF3− into difluorocarbene in the presence of alkali (M+) and other metal cations is favored. In earlier studies, the solvent N,N-dimethylformamide (DMF), was essential for nucleophilic trifluoromethylation by HCF3 since DMF acts as a CF3 anion reservoir that is used as a hemiaminaloate adduct [Me2NCH(O)CF3]- (Scheme 1b) [19-28]. Although the taming CF3 anion had been an elusive problem for decades, it has been dramatically progressed in recent years by the substantial works by Grushin [51-53] and Prakash [54]. Our group reported novel DMF-free systems for the nucleophilic trifluoromethylation reaction using HCF3, including the phosphazene base P4-t-Bu (P4-t-Bu), in 2013 (Scheme 1c) [45] and a potassium tert-butoxide (t-BuOK) or potassium hexamethyldisilazide (KHMDS)/glyme combination in 2018 (Scheme 1d) [46]. The success of our DMF-free systems lies in the generation of sterically demanding cationic species, [P4-t-Bu]H+ or glyme capsulized K+, resulting in the stabilization of CF3− from HCF3 by ion separation. The sterically demanding [P4-t-Bu]H+ or encapsulation of K+ by glymes effectively inhibits the contact of CF3− to K+, preventing decomposition into CF2 and KF. The isolated CF3 is rather naked with a highly nucleophilic character, which is suitable for nucleophilic trifluoromethylation reactions. The K+ and glyme combination is particularly useful for the nucleophilic trifluoromethylation of carbonyl compounds to trifluoromethyl carbinols because it does not require any expensive reagents nor very low-temperature conditions. Although the reaction has a broad substrate scope of embracing ketones, chalcones and aldehydes, the transformation of esters to trifluoromethyl ketones by this protocol was never examined [46].
![[1860-5397-17-39-i1]](/bjoc/content/inline/1860-5397-17-39-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Chemistry of the CF3 anion generated from HCF3. a) Decomposition of the trifluoromethyl anion to difluorocarbene and fluoride. b) A hemiaminaloate adduct of CF3 anion to DMF. c) Formation of the [P4-t-Bu]H+ CF3 anion salt. d) Encapsulation of K+ by glymes. Transformation of esters to trifluoromethyl ketones.
Scheme 1: Chemistry of the CF3 anion generated from HCF3. a) Decomposition of the trifluoromethyl anion to di...
Trifluoromethyl ketones (TFMKs) are valuable fluorine-containing synthetic targets of bioactive compounds [55,56] that behave as mimics of the tetrahedral transition-state intermediate of enzymatic hydrolysis of esters and amides by stabilizing their hydrates (Figure 1a) [57]. In fact, the TFMK moiety is a proven effective metal chelator in various enzyme inhibitors (Figure 1b) [58-65].
![[1860-5397-17-39-1]](/bjoc/content/figures/1860-5397-17-39-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Trifluoromethyl ketones. a) Hydrolysis of trifluoromethyl ketones. b) Selected examples of biologically active trifluoromethyl ketones.
Figure 1: Trifluoromethyl ketones. a) Hydrolysis of trifluoromethyl ketones. b) Selected examples of biologic...
Several useful methods exist for preparing trifluoromethyl ketones [66,67], such as the direct trifluoromethylation of esters by the Ruppert–Prakash reagent (Me3SiCF3) [68-71], but the use of HCF3 for this transformation reaction is still limited. In 1998, Russel and Roques examined the transformation of methyl benzoate to trifluoromethyl phenyl ketone with HCF3 in the presence of KHMDS or KH/DMSO in DMF, but the method required DMF and only a single example was indicated (Scheme 2a) [23]. Prakash and co-workers showed the first example of the DMF-free preparation of trifluoromethyl phenyl ketone with HCF3 in the presence of KHMDS in THF, but they did not examined the scope of the reaction (Scheme 2b) [38]. In 2018, Szymczak and co-workers showed a single example of the preparation of phenyl trifluoromethyl ketone using HCF3-derived borazine CF3– in 29% yield (Scheme 2c) [43]. Very recently, Han, Lian, and co-workers reported that a protocol using diisopropylaminosodium (NaDA) was useful for the trifluoromethylation of esters to trifluoromethyl ketones with HCF3 at −60 °C (Scheme 2d) [44]. However, the preparation of NaDA was rather complicated and required pre-mixing of diisopropylamine, tetramethylethylenediamine (TMEDA), isoprene, and even more tedious “dispersion sodium” in n-heptane at 25 °C for 4 h, before the reaction of esters with HCF3 at −60 °C. We herein extend our glyme strategy [50] shown in Scheme 1d, the HCF3/KHMDS/triglyme system, for the synthesis of trifluoromethyl ketones from esters (Scheme 2e). The combination of HCF3 and KHMDS in triglyme at −40 °C was found to be effective for this transformation, with good yields as high as 92%. The substrate scope of the trifluoromethylation procedure was explored for aromatic, aliphatic, and conjugated methyl esters. This study presents a straightforward trifluoromethylation process of various methyl esters that convert well to the corresponding trifluoromethyl ketones. The tolerance of various pharmacophores under the reaction conditions was also explored.
![[1860-5397-17-39-i2]](/bjoc/content/inline/1860-5397-17-39-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Trifluoromethylation of esters by HCF3 by a) Russell and Roques (1998), b) Prakash and co-workers (2012), c) Szymczak and co-workers (2018), d) Han, Lian and co-workers (2019), and e) our group in this work.
Scheme 2: Trifluoromethylation of esters by HCF3 by a) Russell and Roques (1998), b) Prakash and co-workers (...
Results and Discussion
We first examined the trifluoromethylation reaction of methyl 2-naphthoate (1a) as a model substrate for HCF3 to optimize the reaction conditions (Table 1). Following our glymes strategy, we initially used t-BuOK as the base in triglyme, and the desired trifluoromethyl ketone 2a was obtained in 29% yield (Table 1, entry 1). We next carried out the reaction in other solvents, THF (2a, 5%, Table 1, entry 2) and toluene (2a, 0%, Table 1, entry 3), and confirmed the advantage of the triglyme that was used (Table 1, entries 1–3). Increasing the amount of t-BuOK to 4.0 equiv did not improve the yield (25%) of 2a (Table 1, entry 4). When we used KHMDS to replace t-BuOK, the yield of 2a improved significantly to 57% (Table 1, entry 5). As expected, tetraglyme, instead of triglyme, gave a similar good yield of 59% (Table 1, entry 6), while the transformation decreased significantly when diglyme was used (2a, 29%, Table 1, entry 7). Interestingly, when we stopped the reaction after 4 h, the yield increased to 76% (Table 1, entry 8). On this basis, we attempted to reduce the amount of HCF3 to 1.1 equiv and found that the yield was not sacrificed, yielding 75% of 2a (Table 1, entry 9). Other optimized reaction conditions did not improve the yield (see Supporting Information File 1 for an extensive list of reaction conditions, Table S1).
Table 1: Optimized reaction conditions for the conversion of 1a to 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-39-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base (equiv) | Solvent | Time | Yield (%)a |
| 1 | t-BuOK (2.0) | triglyme | overnight | 29 |
| 2 | t-BuOK (2.0) | THF | overnight | 5 |
| 3 | t-BuOK (2.0) | toluene | overnight | 0 |
| 4 | t-BuOK (4.0) | triglyme | overnight | 25 |
| 5 | KHMDS (2.0) | triglyme | overnight | 57 |
| 6 | KHMDS (2.0) | tetraglyme | overnight | 59 |
| 7 | KHMDS (2.0) | diglyme | overnight | 29 |
| 8 | KHMDS (2.0) | triglyme | 4 h | 76 |
| 9b | KHMDS (2.0) | triglyme | 4 h | 75 (71)c |
aDetermined by 19F NMR using a crude mixture with trifluorotoluene as the internal standard. bHCF3 was 1.1 equiv. cIsolated yield.
We explored the substrate scope of this trifluoromethylation reaction with the optimized conditions in hand (entry 9, Table 1). Various carboxylic esters were investigated in the presence of 1.1 equiv of HCF3 and two equiv of KHMDS (Scheme 3). Methyl 2-naphthoate (1a) gave 2a in 75% yield, but sterically demanding methyl 1-naphthoate (1b) gave the desired trifluoromethyl ketone 2b in only lower yield (37%). Functionalities on the benzene ring at the para-position were well-tolerated in the KHMDS/glyme system. Halogen groups, such as chloro (1c), bromo (1d), and reactive iodo (1e) substitutions were also tolerated, resulting in the corresponding trifluoromethyl aryl ketones in moderate yields (56–63%) under basic conditions. The alkyl groups of tert-butyl (1f)- and cyclohexyl (1g)-substituted methyl benzoate derivatives, biphenyl benzoate (1h), and electron-donating 4-methoxybenzoate, were nicely transformed into aryl trifluoromethyl ketones in moderate to high yields (45–92%). Aryl substrates with a halogen attached at the meta- and ortho-positions were also accepted to furnish the desired products (2j–m) in good yields (66–82%). Moreover, di-substituted benzoate (1n), sterically demanding methyl adamantly carboxylate (1o), and conjugated methyl ester (1p) transformed effectively into trifluoromethyl ketones (2o–p) in moderate yields (50–62%). A gram-scale reaction was also carried out for 1h, 1n, and 1p to furnish 2h, 2p, and 2n in similar isolated yields, 43%, 40%, and 36%, respectively. The double CF3 addition product 3 was not observed due to the preferential formation of stable tetrahedral species I instead of the CF3 ketones 2 in the reaction mixture. However, all the yields were moderate to good. This fact could be explained by the appearance of hydrate products 4 in the 19F NMR spectrum of the crude reaction mixture [72], while the hydrates 4 disappeared completely after purification by silica gel column chromatography [73].
![[1860-5397-17-39-i3]](/bjoc/content/inline/1860-5397-17-39-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope of esters 1 for trifluoromethylation by HCF3 under the optimized conditions. aDetermined by 19F NMR of the crude 2 with trifluorotoluene as an internal standard. bIsolated yield. cIsolated yield of gram-scale reaction by using 1 g of substrate.
Scheme 3: Substrate scope of esters 1 for trifluoromethylation by HCF3 under the optimized conditions. aDeter...
Given the relevance of this trifluoromethylation reaction system for drug discovery, we conducted a robustness screening experiment to gain further information on its tolerance to various pharmacophores (Table 2). A range of common nitrogen-containing compounds such as pyridine, pyrazine, 1H-pyrazole, 1H-indole, 1-methyl-1H-indole, piperidine, and piperazine were subjected to screening. Pyridine and piperidine slightly hamper the reaction of 1g (Table 2, entries 2 and 7, 80–82%). Other nitrogen-containing compounds have more effect on the yield of the reaction of 1g (Table 2, entries 3–6, 58–72%). Next, a range of common oxygen and sulfur-containing compounds such as furan, tetrahydrofuran, 1,4-dioxane, thiophene, benzo[b]thiophene, dibenzo[b,d]thiophene, and diphenylsulfane were also screened. These substances also have some effect on the reaction (Table 2, entries 9–15, 63–87%). Besides, silicon-containing compound, trimethyl(phenyl)silane that is more sensitive to fluorine was screened, 79% yield were obtained in this test. To consider the frequency of these motifs in modern pharmaceutical drugs, these tests are necessary, and the resistance of the reaction was also verified from various pharmacophores to be acceptable.
Table 2: Tolerance of various pharmacophores under the trifluoromethylation conditions.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-39-i5.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | Additive | Yielda (%) | Entry | Additive | Yielda (%) | Entry | Additive | Yielda (%) |
| 1 | – | 92 | 7 | piperidine | 80 | 13 | benzo[b]thiophene | 78 |
| 2 | pyridine | 82 | 8 | piperazine | 64 | 14 | dibenzo[b,d]thiophene | 64 |
| 3 | pyrazine | 72 | 9 | furan | 87 | 15 | PhSPh | 75 |
| 4 | pyrazole | 66 | 10 | THF | 64 | 16 | Ph-SiMe3 | 79 |
| 5 | indole | 58 | 11 | 1,4-dioxane | 63 | |||
| 6 | 1-methyl-1H-indole | 67 | 12 | thiophene | 75 | |||
aDetermined by 19F NMR using the crude 2g with trifluorotoluene as an internal standard.
Conclusion
In conclusion, the trifluoromethylation of methyl carboxylates to trifluoromethyl ketones is accomplished under basic conditions with fluoroform in triglyme at −40 °C. An equivalent amount of fluoroform was sufficient for this transformation. A wide variety of medicinally attractive aryl and alkyl trifluoromethyl ketones are obtained in good yields by a relatively simple procedure, although the protocol is not applicable to enolizable esters. Fluoroform is an economical feedstock, and methyl esters are readily available inexpensive precursors. Besides, glymes are versatile solvents for chemical processes in industry [74] would the protocol be useful for the industrial extension, although there are still many points to be overcome such as requirements of low temperature, two equivalents of KHMDS. Further application of this “batch protocol” for a “continuous-flow microreactor” reaction is now ongoing in our laboratory towards industrial collaboration.
Experimental
A test tube containing 1 (0.4 mmol) in triglyme (0.7 mL) was charged with HCF3 (9.9 mL, 1.1 equiv, measured by a syringe, see the picture in Supporting Information File 1, Figure S1) by cooling in liquid nitrogen under vacuum. KHMDS (160 mg, 2.0 equiv) in triglyme (commercial grade, without drying, 0.3 mL) was added at −40 °C under nitrogen atmosphere, and the reaction mixture was stirred at the same temperature for 4 h. Thereafter, 1 M HCl aq (1.0 mL) was added, and the aqueous layer was extracted with CH2Cl2 (1.0 mL × 3). The combined organic layer was washed with brine, dried over Na2SO4, concentrated under reduced pressure, and purified by column chromatography on silica gel to give products 2.
Supporting Information
| Supporting Information File 1: Optimization of reaction conditions, general procedure and product characterization data. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830
Return to citation in text: [1] -
Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467
Return to citation in text: [1] -
Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258
Return to citation in text: [1] -
Park, B. K.; Kitteringham, N. R.; O'Neill, P. M. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. doi:10.1146/annurev.pharmtox.41.1.443
Return to citation in text: [1] -
Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Kirsch, P. Modern Fluoroorganic Chemistry Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393x
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Mazloomi, Z.; Bansode, A.; Benavente, P.; Lishchynskyi, A.; Urakawa, A.; Grushin, V. V. Org. Process Res. Dev. 2014, 18, 1020–1026. doi:10.1021/op500109v
Return to citation in text: [1] -
Musio, B.; Gala, E.; Ley, S. V. ACS Sustainable Chem. Eng. 2018, 6, 1489–1495. doi:10.1021/acssuschemeng.7b04012
Return to citation in text: [1] -
Harsanyi, A.; Sandford, G. Org. Process Res. Dev. 2014, 18, 981–992. doi:10.1021/op500141c
Return to citation in text: [1] -
Caron, S. Org. Process Res. Dev. 2020, 24, 470–480. doi:10.1021/acs.oprd.0c00030
Return to citation in text: [1] -
Glenadel, Q.; Alazet, S.; Baert, F.; Billard, T. Org. Process Res. Dev. 2016, 20, 960–964. doi:10.1021/acs.oprd.6b00062
Return to citation in text: [1] -
McCulloch, A.; Lindley, A. A. Atmos. Environ. 2007, 41, 1560–1566. doi:10.1016/j.atmosenv.2006.02.021
Return to citation in text: [1] -
Han, W.; Li, Y.; Tang, H.; Liu, H. J. Fluorine Chem. 2012, 140, 7–16. doi:10.1016/j.jfluchem.2012.04.012
Return to citation in text: [1] -
Grushin, V. V. Chim. Oggi 2014, 32 (3), 81–90.
Return to citation in text: [1] -
Shono, T.; Ishifune, M.; Okada, T.; Kashimura, S. J. Org. Chem. 1991, 56, 2–4. doi:10.1021/jo00001a002
Return to citation in text: [1] [2] -
Symons, E. A.; Clermont, M. J. J. Am. Chem. Soc. 1981, 103, 3127–3130. doi:10.1021/ja00401a034
Return to citation in text: [1] [2] -
Barhdadi, R.; Troupel, M.; Périchon, J. Chem. Commun. 1998, 1251–1252. doi:10.1039/a801406j
Return to citation in text: [1] [2] -
Folléas, B.; Marek, I.; Normant, J.-F.; Jalmes, L. S. Tetrahedron Lett. 1998, 39, 2973–2976. doi:10.1016/s0040-4039(98)00391-8
Return to citation in text: [1] [2] -
Russell, J.; Roques, N. Tetrahedron 1998, 54, 13771–13782. doi:10.1016/s0040-4020(98)00846-1
Return to citation in text: [1] [2] [3] -
Mispelaere, C.; Roques, N. Tetrahedron Lett. 1999, 40, 6411–6414. doi:10.1016/s0040-4039(99)01369-6
Return to citation in text: [1] [2] -
Folléas, B.; Marek, I.; Normant, J.-F.; Saint-Jalmes, L. Tetrahedron 2000, 56, 275–283. doi:10.1016/s0040-4020(99)00951-5
Return to citation in text: [1] [2] -
Large, S.; Roques, N.; Langlois, B. R. J. Org. Chem. 2000, 65, 8848–8856. doi:10.1021/jo000150s
Return to citation in text: [1] [2] -
Billard, T.; Bruns, S.; Langlois, B. R. Org. Lett. 2000, 2, 2101–2103. doi:10.1021/ol005987o
Return to citation in text: [1] [2] -
van der Born, D.; Herscheid, J. D. M.; Orru, R. V. A.; Vugts, D. J. Chem. Commun. 2013, 49, 4018–4020. doi:10.1039/c3cc37833k
Return to citation in text: [1] [2] -
Tomashenko, O. A.; Escudero-Adán, E. C.; Martínez Belmonte, M.; Grushin, V. V. Angew. Chem., Int. Ed. 2011, 50, 7655–7659. doi:10.1002/anie.201101577
Return to citation in text: [1] -
Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026
Return to citation in text: [1] -
Lishchynskyi, A.; Novikov, M. A.; Martin, E.; Escudero-Adán, E. C.; Novák, P.; Grushin, V. V. J. Org. Chem. 2013, 78, 11126–11146. doi:10.1021/jo401423h
Return to citation in text: [1] -
Lishchynskyi, A.; Berthon, G.; Grushin, V. V. Chem. Commun. 2014, 50, 10237–10240. doi:10.1039/c4cc04930f
Return to citation in text: [1] -
Novák, P.; Lishchynskyi, A.; Grushin, V. V. J. Am. Chem. Soc. 2012, 134, 16167–16170. doi:10.1021/ja307783w
Return to citation in text: [1] -
Novák, P.; Lishchynskyi, A.; Grushin, V. V. Angew. Chem., Int. Ed. 2012, 51, 7767–7770. doi:10.1002/anie.201201613
Return to citation in text: [1] -
Konovalov, A. I.; Benet-Buchholz, J.; Martin, E.; Grushin, V. V. Angew. Chem., Int. Ed. 2013, 52, 11637–11641. doi:10.1002/anie.201306272
Return to citation in text: [1] -
Konovalov, A. I.; Lishchynskyi, A.; Grushin, V. V. J. Am. Chem. Soc. 2014, 136, 13410–13425. doi:10.1021/ja507564p
Return to citation in text: [1] -
Romine, A. M.; Nebra, N.; Konovalov, A. I.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. Angew. Chem., Int. Ed. 2015, 54, 2745–2749. doi:10.1002/anie.201411348
Return to citation in text: [1] -
Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859
Return to citation in text: [1] [2] -
He, L.; Tsui, G. C. Org. Lett. 2016, 18, 2800–2803. doi:10.1021/acs.orglett.6b00999
Return to citation in text: [1] -
Xiang, J.-X.; Ouyang, Y.; Xu, X.-H.; Qing, F.-L. Angew. Chem., Int. Ed. 2019, 58, 10320–10324. doi:10.1002/anie.201905782
Return to citation in text: [1] -
Geri, J. B.; Szymczak, N. K. J. Am. Chem. Soc. 2017, 139, 9811–9814. doi:10.1021/jacs.7b05408
Return to citation in text: [1] -
Biggadike, K.; Boudjelal, M.; Clackers, M.; Coe, D. M.; Demaine, D. A.; Hardy, G. W.; Humphreys, D.; Inglis, G. G. A.; Johnston, M. J.; Jones, H. T.; House, D.; Loiseau, R.; Needham, D.; Skone, P. A.; Uings, I.; Veitch, G.; Weingarten, G. G.; McLay, I. M.; Macdonald, S. J. F. J. Med. Chem. 2007, 50, 6519–6534. doi:10.1021/jm070778w
Return to citation in text: [1] -
Geri, J. B.; Wade Wolfe, M. M.; Szymczak, N. K. Angew. Chem., Int. Ed. 2018, 57, 1381–1385. doi:10.1002/anie.201711316
Return to citation in text: [1] [2] -
Han, Z.; Chen, S.; Tu, Y.; Lian, X.; Li, G. Eur. J. Org. Chem. 2019, 4658–4661. doi:10.1002/ejoc.201900250
Return to citation in text: [1] [2] -
Kawai, H.; Yuan, Z.; Tokunaga, E.; Shibata, N. Org. Biomol. Chem. 2013, 11, 1446–1450. doi:10.1039/c3ob27368g
Return to citation in text: [1] [2] -
Saito, T.; Wang, J.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Sci. Rep. 2018, 8, 11501. doi:10.1038/s41598-018-29748-1
Return to citation in text: [1] [2] [3] -
Okusu, S.; Tokunaga, E.; Shibata, N. Org. Lett. 2015, 17, 3802–3805. doi:10.1021/acs.orglett.5b01778
Return to citation in text: [1] -
Okusu, S.; Hirano, K.; Tokunaga, E.; Shibata, N. ChemistryOpen 2015, 4, 581–585. doi:10.1002/open.201500160
Return to citation in text: [1] -
Punna, N.; Saito, T.; Kosobokov, M.; Tokunaga, E.; Sumii, Y.; Shibata, N. Chem. Commun. 2018, 54, 4294–4297. doi:10.1039/c8cc01526k
Return to citation in text: [1] -
Hirano, K.; Saito, T.; Fujihira, Y.; Sedgwick, D. M.; Fustero, S.; Shibata, N. J. Org. Chem. 2020, 85, 7976–7985. doi:10.1021/acs.joc.0c00796
Return to citation in text: [1] [2] -
Lishchynskyi, A.; Miloserdov, F. M.; Martin, E.; Benet-Buchholz, J.; Escudero-Adán, E. C.; Konovalov, A. I.; Grushin, V. V. Angew. Chem., Int. Ed. 2015, 54, 15289–15293. doi:10.1002/anie.201507356
Return to citation in text: [1] -
Miloserdov, F. M.; Konovalov, A. I.; Martin, E.; Benet-Buchholz, J.; Escudero-Adán, E. C.; Lishchynskyi, A.; Grushin, V. V. Helv. Chim. Acta 2017, 100, e1700032. doi:10.1002/hlca.201700032
Return to citation in text: [1] -
Harlow, R. L.; Benet-Buchholz, J.; Miloserdov, F. M.; Konovalov, A. I.; Marshall, W. J.; Escudero-Adán, E. C.; Martin, E.; Lishchynskyi, A.; Grushin, V. V. Helv. Chim. Acta 2018, 101, e1800015. doi:10.1002/hlca.201800015
Return to citation in text: [1] -
Prakash, G. K. S.; Wang, F.; Zhang, Z.; Haiges, R.; Rahm, M.; Christe, K. O.; Mathew, T.; Olah, G. A. Angew. Chem., Int. Ed. 2014, 53, 11575–11578. doi:10.1002/anie.201406505
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. Tetrahedron 1991, 47, 3207–3258. doi:10.1016/s0040-4020(01)86391-2
Return to citation in text: [1] -
Kawase, M. J. Synth. Org. Chem., Jpn. 2001, 59, 755–765. doi:10.5059/yukigoseikyokaishi.59.755
Return to citation in text: [1] -
Gelb, M. H.; Svaren, J. P.; Abeles, R. H. Biochemistry 1985, 24, 1813–1817. doi:10.1021/bi00329a001
Return to citation in text: [1] -
Frey, R. R.; Wada, C. K.; Garland, R. B.; Curtin, M. L.; Michaelides, M. R.; Li, J.; Pease, L. J.; Glaser, K. B.; Marcotte, P. A.; Bouska, J. J.; Murphy, S. S.; Davidsen, S. K. Bioorg. Med. Chem. Lett. 2002, 12, 3443–3447. doi:10.1016/s0960-894x(02)00754-0
Return to citation in text: [1] -
Patricelli, M. P.; Patterson, J. E.; Boger, D. L.; Cravatt, B. F. Bioorg. Med. Chem. Lett. 1998, 8, 613–618. doi:10.1016/s0960-894x(98)00073-0
Return to citation in text: [1] -
Lehn, M.; Griessbach, K. Pharm. Pharmacol. Commun. 1999, 5, 389–393. doi:10.1111/j.1469-0691.1999.tb00846.x
Return to citation in text: [1] -
Hornsperger, J.-M.; Collard, J.-N.; Heydt, J.-G.; Giacobini, E.; Funes, S.; Dow, J.; Schirlin, D. Biochem. Soc. Trans. 1994, 22, 758–763. doi:10.1042/bst0220758
Return to citation in text: [1] -
Parrilla, A.; Villuendas, I.; Guerrero, A. Bioorg. Med. Chem. 1994, 2, 243–252. doi:10.1016/s0968-0896(00)82167-7
Return to citation in text: [1] -
Neises, B.; Broersma, R. J.; Tarnus, C.; Piriou, F.; Remy, J. M.; Lintz, C.; Heminger, E. F.; Kutcher, L. W. Bioorg. Med. Chem. 1995, 3, 1049–1061. doi:10.1016/0968-0896(95)00097-z
Return to citation in text: [1] -
Patel, D. V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C. A.; Smith, S. A.; Petrillo, E. W., Jr. J. Med. Chem. 1993, 36, 2431–2447. doi:10.1021/jm00069a001
Return to citation in text: [1] -
Kawase, M.; Harada, H.; Saito, S.; Cui, J.; Tani, S. Bioorg. Med. Chem. Lett. 1999, 9, 193–194. doi:10.1016/s0960-894x(98)00726-4
Return to citation in text: [1] -
Wu, W.; Weng, Z. Synthesis 2018, 50, 1958–1964. doi:10.1055/s-0036-1591971
Return to citation in text: [1] -
Kelly, C. B.; Mercadante, M. A.; Leadbeater, N. E. Chem. Commun. 2013, 49, 11133–11148. doi:10.1039/c3cc46266h
Return to citation in text: [1] -
Wiedemann, J.; Heiner, T.; Mloston, G.; Prakash, G. K. S.; Olah, G. A. Angew. Chem., Int. Ed. 1998, 37, 820–821. doi:10.1002/(sici)1521-3773(19980403)37:6<820::aid-anie820>3.0.co;2-m
Return to citation in text: [1] -
Singh, R. P.; Cao, G.; Kirchmeier, R. L.; Shreeve, J. M. J. Org. Chem. 1999, 64, 2873–2876. doi:10.1021/jo982494c
Return to citation in text: [1] -
Kawano, Y.; Kaneko, N.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 1133–1145. doi:10.1246/bcsj.79.1133
Return to citation in text: [1] -
Cui, B.; Sun, H.; Xu, Y.; Duan, L.; Li, Y.-M. Tetrahedron 2017, 73, 6754–6762. doi:10.1016/j.tet.2017.10.021
Return to citation in text: [1] -
Kadoh, Y.; Tashiro, M.; Oisaki, K.; Kanai, M. Adv. Synth. Catal. 2015, 357, 2193–2198. doi:10.1002/adsc.201500131
Return to citation in text: [1] -
Jiang, X.-D.; Matsukawa, S.; Kakuda, K.-i.; Fukuzaki, Y.; Zhao, W.-L.; Li, L.-S.; Shen, H.-B.; Kojima, S.; Yamamoto, Y. Dalton Trans. 2010, 39, 9823–9829. doi:10.1039/c0dt00539h
Return to citation in text: [1] -
Tang, S.; Zhao, H. RSC Adv. 2014, 4, 11251–11287. doi:10.1039/c3ra47191h
Return to citation in text: [1]
| 72. | Kadoh, Y.; Tashiro, M.; Oisaki, K.; Kanai, M. Adv. Synth. Catal. 2015, 357, 2193–2198. doi:10.1002/adsc.201500131 |
| 73. | Jiang, X.-D.; Matsukawa, S.; Kakuda, K.-i.; Fukuzaki, Y.; Zhao, W.-L.; Li, L.-S.; Shen, H.-B.; Kojima, S.; Yamamoto, Y. Dalton Trans. 2010, 39, 9823–9829. doi:10.1039/c0dt00539h |
| 1. | Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778 |
| 2. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 3. | Inoue, M.; Sumii, Y.; Shibata, N. ACS Omega 2020, 5, 10633–10640. doi:10.1021/acsomega.0c00830 |
| 4. | Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. iScience 2020, 23, 101467. doi:10.1016/j.isci.2020.101467 |
| 11. | Mazloomi, Z.; Bansode, A.; Benavente, P.; Lishchynskyi, A.; Urakawa, A.; Grushin, V. V. Org. Process Res. Dev. 2014, 18, 1020–1026. doi:10.1021/op500109v |
| 12. | Musio, B.; Gala, E.; Ley, S. V. ACS Sustainable Chem. Eng. 2018, 6, 1489–1495. doi:10.1021/acssuschemeng.7b04012 |
| 13. | Harsanyi, A.; Sandford, G. Org. Process Res. Dev. 2014, 18, 981–992. doi:10.1021/op500141c |
| 14. | Caron, S. Org. Process Res. Dev. 2020, 24, 470–480. doi:10.1021/acs.oprd.0c00030 |
| 15. | Glenadel, Q.; Alazet, S.; Baert, F.; Billard, T. Org. Process Res. Dev. 2016, 20, 960–964. doi:10.1021/acs.oprd.6b00062 |
| 45. | Kawai, H.; Yuan, Z.; Tokunaga, E.; Shibata, N. Org. Biomol. Chem. 2013, 11, 1446–1450. doi:10.1039/c3ob27368g |
| 9. | Kirsch, P. Modern Fluoroorganic Chemistry Synthesis, Reactivity, Applications; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/352760393x |
| 10. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 46. | Saito, T.; Wang, J.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Sci. Rep. 2018, 8, 11501. doi:10.1038/s41598-018-29748-1 |
| 7. | Isanbor, C.; O’Hagan, D. J. Fluorine Chem. 2006, 127, 303–319. doi:10.1016/j.jfluchem.2006.01.011 |
| 8. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 51. | Lishchynskyi, A.; Miloserdov, F. M.; Martin, E.; Benet-Buchholz, J.; Escudero-Adán, E. C.; Konovalov, A. I.; Grushin, V. V. Angew. Chem., Int. Ed. 2015, 54, 15289–15293. doi:10.1002/anie.201507356 |
| 52. | Miloserdov, F. M.; Konovalov, A. I.; Martin, E.; Benet-Buchholz, J.; Escudero-Adán, E. C.; Lishchynskyi, A.; Grushin, V. V. Helv. Chim. Acta 2017, 100, e1700032. doi:10.1002/hlca.201700032 |
| 53. | Harlow, R. L.; Benet-Buchholz, J.; Miloserdov, F. M.; Konovalov, A. I.; Marshall, W. J.; Escudero-Adán, E. C.; Martin, E.; Lishchynskyi, A.; Grushin, V. V. Helv. Chim. Acta 2018, 101, e1800015. doi:10.1002/hlca.201800015 |
| 5. | Gillis, E. P.; Eastman, K. J.; Hill, M. D.; Donnelly, D. J.; Meanwell, N. A. J. Med. Chem. 2015, 58, 8315–8359. doi:10.1021/acs.jmedchem.5b00258 |
| 6. | Park, B. K.; Kitteringham, N. R.; O'Neill, P. M. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. doi:10.1146/annurev.pharmtox.41.1.443 |
| 54. | Prakash, G. K. S.; Wang, F.; Zhang, Z.; Haiges, R.; Rahm, M.; Christe, K. O.; Mathew, T.; Olah, G. A. Angew. Chem., Int. Ed. 2014, 53, 11575–11578. doi:10.1002/anie.201406505 |
| 38. | Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859 |
| 45. | Kawai, H.; Yuan, Z.; Tokunaga, E.; Shibata, N. Org. Biomol. Chem. 2013, 11, 1446–1450. doi:10.1039/c3ob27368g |
| 46. | Saito, T.; Wang, J.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Sci. Rep. 2018, 8, 11501. doi:10.1038/s41598-018-29748-1 |
| 47. | Okusu, S.; Tokunaga, E.; Shibata, N. Org. Lett. 2015, 17, 3802–3805. doi:10.1021/acs.orglett.5b01778 |
| 48. | Okusu, S.; Hirano, K.; Tokunaga, E.; Shibata, N. ChemistryOpen 2015, 4, 581–585. doi:10.1002/open.201500160 |
| 49. | Punna, N.; Saito, T.; Kosobokov, M.; Tokunaga, E.; Sumii, Y.; Shibata, N. Chem. Commun. 2018, 54, 4294–4297. doi:10.1039/c8cc01526k |
| 50. | Hirano, K.; Saito, T.; Fujihira, Y.; Sedgwick, D. M.; Fustero, S.; Shibata, N. J. Org. Chem. 2020, 85, 7976–7985. doi:10.1021/acs.joc.0c00796 |
| 29. | Tomashenko, O. A.; Escudero-Adán, E. C.; Martínez Belmonte, M.; Grushin, V. V. Angew. Chem., Int. Ed. 2011, 50, 7655–7659. doi:10.1002/anie.201101577 |
| 30. | Zanardi, A.; Novikov, M. A.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. J. Am. Chem. Soc. 2011, 133, 20901–20913. doi:10.1021/ja2081026 |
| 31. | Lishchynskyi, A.; Novikov, M. A.; Martin, E.; Escudero-Adán, E. C.; Novák, P.; Grushin, V. V. J. Org. Chem. 2013, 78, 11126–11146. doi:10.1021/jo401423h |
| 32. | Lishchynskyi, A.; Berthon, G.; Grushin, V. V. Chem. Commun. 2014, 50, 10237–10240. doi:10.1039/c4cc04930f |
| 33. | Novák, P.; Lishchynskyi, A.; Grushin, V. V. J. Am. Chem. Soc. 2012, 134, 16167–16170. doi:10.1021/ja307783w |
| 34. | Novák, P.; Lishchynskyi, A.; Grushin, V. V. Angew. Chem., Int. Ed. 2012, 51, 7767–7770. doi:10.1002/anie.201201613 |
| 35. | Konovalov, A. I.; Benet-Buchholz, J.; Martin, E.; Grushin, V. V. Angew. Chem., Int. Ed. 2013, 52, 11637–11641. doi:10.1002/anie.201306272 |
| 36. | Konovalov, A. I.; Lishchynskyi, A.; Grushin, V. V. J. Am. Chem. Soc. 2014, 136, 13410–13425. doi:10.1021/ja507564p |
| 37. | Romine, A. M.; Nebra, N.; Konovalov, A. I.; Martin, E.; Benet-Buchholz, J.; Grushin, V. V. Angew. Chem., Int. Ed. 2015, 54, 2745–2749. doi:10.1002/anie.201411348 |
| 19. | Shono, T.; Ishifune, M.; Okada, T.; Kashimura, S. J. Org. Chem. 1991, 56, 2–4. doi:10.1021/jo00001a002 |
| 20. | Symons, E. A.; Clermont, M. J. J. Am. Chem. Soc. 1981, 103, 3127–3130. doi:10.1021/ja00401a034 |
| 21. | Barhdadi, R.; Troupel, M.; Périchon, J. Chem. Commun. 1998, 1251–1252. doi:10.1039/a801406j |
| 22. | Folléas, B.; Marek, I.; Normant, J.-F.; Jalmes, L. S. Tetrahedron Lett. 1998, 39, 2973–2976. doi:10.1016/s0040-4039(98)00391-8 |
| 23. | Russell, J.; Roques, N. Tetrahedron 1998, 54, 13771–13782. doi:10.1016/s0040-4020(98)00846-1 |
| 24. | Mispelaere, C.; Roques, N. Tetrahedron Lett. 1999, 40, 6411–6414. doi:10.1016/s0040-4039(99)01369-6 |
| 25. | Folléas, B.; Marek, I.; Normant, J.-F.; Saint-Jalmes, L. Tetrahedron 2000, 56, 275–283. doi:10.1016/s0040-4020(99)00951-5 |
| 26. | Large, S.; Roques, N.; Langlois, B. R. J. Org. Chem. 2000, 65, 8848–8856. doi:10.1021/jo000150s |
| 27. | Billard, T.; Bruns, S.; Langlois, B. R. Org. Lett. 2000, 2, 2101–2103. doi:10.1021/ol005987o |
| 28. | van der Born, D.; Herscheid, J. D. M.; Orru, R. V. A.; Vugts, D. J. Chem. Commun. 2013, 49, 4018–4020. doi:10.1039/c3cc37833k |
| 19. | Shono, T.; Ishifune, M.; Okada, T.; Kashimura, S. J. Org. Chem. 1991, 56, 2–4. doi:10.1021/jo00001a002 |
| 20. | Symons, E. A.; Clermont, M. J. J. Am. Chem. Soc. 1981, 103, 3127–3130. doi:10.1021/ja00401a034 |
| 21. | Barhdadi, R.; Troupel, M.; Périchon, J. Chem. Commun. 1998, 1251–1252. doi:10.1039/a801406j |
| 22. | Folléas, B.; Marek, I.; Normant, J.-F.; Jalmes, L. S. Tetrahedron Lett. 1998, 39, 2973–2976. doi:10.1016/s0040-4039(98)00391-8 |
| 23. | Russell, J.; Roques, N. Tetrahedron 1998, 54, 13771–13782. doi:10.1016/s0040-4020(98)00846-1 |
| 24. | Mispelaere, C.; Roques, N. Tetrahedron Lett. 1999, 40, 6411–6414. doi:10.1016/s0040-4039(99)01369-6 |
| 25. | Folléas, B.; Marek, I.; Normant, J.-F.; Saint-Jalmes, L. Tetrahedron 2000, 56, 275–283. doi:10.1016/s0040-4020(99)00951-5 |
| 26. | Large, S.; Roques, N.; Langlois, B. R. J. Org. Chem. 2000, 65, 8848–8856. doi:10.1021/jo000150s |
| 27. | Billard, T.; Bruns, S.; Langlois, B. R. Org. Lett. 2000, 2, 2101–2103. doi:10.1021/ol005987o |
| 28. | van der Born, D.; Herscheid, J. D. M.; Orru, R. V. A.; Vugts, D. J. Chem. Commun. 2013, 49, 4018–4020. doi:10.1039/c3cc37833k |
| 16. | McCulloch, A.; Lindley, A. A. Atmos. Environ. 2007, 41, 1560–1566. doi:10.1016/j.atmosenv.2006.02.021 |
| 17. | Han, W.; Li, Y.; Tang, H.; Liu, H. J. Fluorine Chem. 2012, 140, 7–16. doi:10.1016/j.jfluchem.2012.04.012 |
| 18. | Grushin, V. V. Chim. Oggi 2014, 32 (3), 81–90. |
| 39. | He, L.; Tsui, G. C. Org. Lett. 2016, 18, 2800–2803. doi:10.1021/acs.orglett.6b00999 |
| 40. | Xiang, J.-X.; Ouyang, Y.; Xu, X.-H.; Qing, F.-L. Angew. Chem., Int. Ed. 2019, 58, 10320–10324. doi:10.1002/anie.201905782 |
| 41. | Geri, J. B.; Szymczak, N. K. J. Am. Chem. Soc. 2017, 139, 9811–9814. doi:10.1021/jacs.7b05408 |
| 42. | Biggadike, K.; Boudjelal, M.; Clackers, M.; Coe, D. M.; Demaine, D. A.; Hardy, G. W.; Humphreys, D.; Inglis, G. G. A.; Johnston, M. J.; Jones, H. T.; House, D.; Loiseau, R.; Needham, D.; Skone, P. A.; Uings, I.; Veitch, G.; Weingarten, G. G.; McLay, I. M.; Macdonald, S. J. F. J. Med. Chem. 2007, 50, 6519–6534. doi:10.1021/jm070778w |
| 43. | Geri, J. B.; Wade Wolfe, M. M.; Szymczak, N. K. Angew. Chem., Int. Ed. 2018, 57, 1381–1385. doi:10.1002/anie.201711316 |
| 44. | Han, Z.; Chen, S.; Tu, Y.; Lian, X.; Li, G. Eur. J. Org. Chem. 2019, 4658–4661. doi:10.1002/ejoc.201900250 |
| 57. | Gelb, M. H.; Svaren, J. P.; Abeles, R. H. Biochemistry 1985, 24, 1813–1817. doi:10.1021/bi00329a001 |
| 46. | Saito, T.; Wang, J.; Tokunaga, E.; Tsuzuki, S.; Shibata, N. Sci. Rep. 2018, 8, 11501. doi:10.1038/s41598-018-29748-1 |
| 55. | Bégué, J.-P.; Bonnet-Delpon, D. Tetrahedron 1991, 47, 3207–3258. doi:10.1016/s0040-4020(01)86391-2 |
| 56. | Kawase, M. J. Synth. Org. Chem., Jpn. 2001, 59, 755–765. doi:10.5059/yukigoseikyokaishi.59.755 |
| 44. | Han, Z.; Chen, S.; Tu, Y.; Lian, X.; Li, G. Eur. J. Org. Chem. 2019, 4658–4661. doi:10.1002/ejoc.201900250 |
| 50. | Hirano, K.; Saito, T.; Fujihira, Y.; Sedgwick, D. M.; Fustero, S.; Shibata, N. J. Org. Chem. 2020, 85, 7976–7985. doi:10.1021/acs.joc.0c00796 |
| 38. | Prakash, G. K. S.; Jog, P. V.; Batamack, P. T. D.; Olah, G. A. Science 2012, 338, 1324–1327. doi:10.1126/science.1227859 |
| 43. | Geri, J. B.; Wade Wolfe, M. M.; Szymczak, N. K. Angew. Chem., Int. Ed. 2018, 57, 1381–1385. doi:10.1002/anie.201711316 |
| 68. | Wiedemann, J.; Heiner, T.; Mloston, G.; Prakash, G. K. S.; Olah, G. A. Angew. Chem., Int. Ed. 1998, 37, 820–821. doi:10.1002/(sici)1521-3773(19980403)37:6<820::aid-anie820>3.0.co;2-m |
| 69. | Singh, R. P.; Cao, G.; Kirchmeier, R. L.; Shreeve, J. M. J. Org. Chem. 1999, 64, 2873–2876. doi:10.1021/jo982494c |
| 70. | Kawano, Y.; Kaneko, N.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2006, 79, 1133–1145. doi:10.1246/bcsj.79.1133 |
| 71. | Cui, B.; Sun, H.; Xu, Y.; Duan, L.; Li, Y.-M. Tetrahedron 2017, 73, 6754–6762. doi:10.1016/j.tet.2017.10.021 |
| 23. | Russell, J.; Roques, N. Tetrahedron 1998, 54, 13771–13782. doi:10.1016/s0040-4020(98)00846-1 |
| 58. | Frey, R. R.; Wada, C. K.; Garland, R. B.; Curtin, M. L.; Michaelides, M. R.; Li, J.; Pease, L. J.; Glaser, K. B.; Marcotte, P. A.; Bouska, J. J.; Murphy, S. S.; Davidsen, S. K. Bioorg. Med. Chem. Lett. 2002, 12, 3443–3447. doi:10.1016/s0960-894x(02)00754-0 |
| 59. | Patricelli, M. P.; Patterson, J. E.; Boger, D. L.; Cravatt, B. F. Bioorg. Med. Chem. Lett. 1998, 8, 613–618. doi:10.1016/s0960-894x(98)00073-0 |
| 60. | Lehn, M.; Griessbach, K. Pharm. Pharmacol. Commun. 1999, 5, 389–393. doi:10.1111/j.1469-0691.1999.tb00846.x |
| 61. | Hornsperger, J.-M.; Collard, J.-N.; Heydt, J.-G.; Giacobini, E.; Funes, S.; Dow, J.; Schirlin, D. Biochem. Soc. Trans. 1994, 22, 758–763. doi:10.1042/bst0220758 |
| 62. | Parrilla, A.; Villuendas, I.; Guerrero, A. Bioorg. Med. Chem. 1994, 2, 243–252. doi:10.1016/s0968-0896(00)82167-7 |
| 63. | Neises, B.; Broersma, R. J.; Tarnus, C.; Piriou, F.; Remy, J. M.; Lintz, C.; Heminger, E. F.; Kutcher, L. W. Bioorg. Med. Chem. 1995, 3, 1049–1061. doi:10.1016/0968-0896(95)00097-z |
| 64. | Patel, D. V.; Rielly-Gauvin, K.; Ryono, D. E.; Free, C. A.; Smith, S. A.; Petrillo, E. W., Jr. J. Med. Chem. 1993, 36, 2431–2447. doi:10.1021/jm00069a001 |
| 65. | Kawase, M.; Harada, H.; Saito, S.; Cui, J.; Tani, S. Bioorg. Med. Chem. Lett. 1999, 9, 193–194. doi:10.1016/s0960-894x(98)00726-4 |
| 66. | Wu, W.; Weng, Z. Synthesis 2018, 50, 1958–1964. doi:10.1055/s-0036-1591971 |
| 67. | Kelly, C. B.; Mercadante, M. A.; Leadbeater, N. E. Chem. Commun. 2013, 49, 11133–11148. doi:10.1039/c3cc46266h |
© 2021 Fujihira et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)