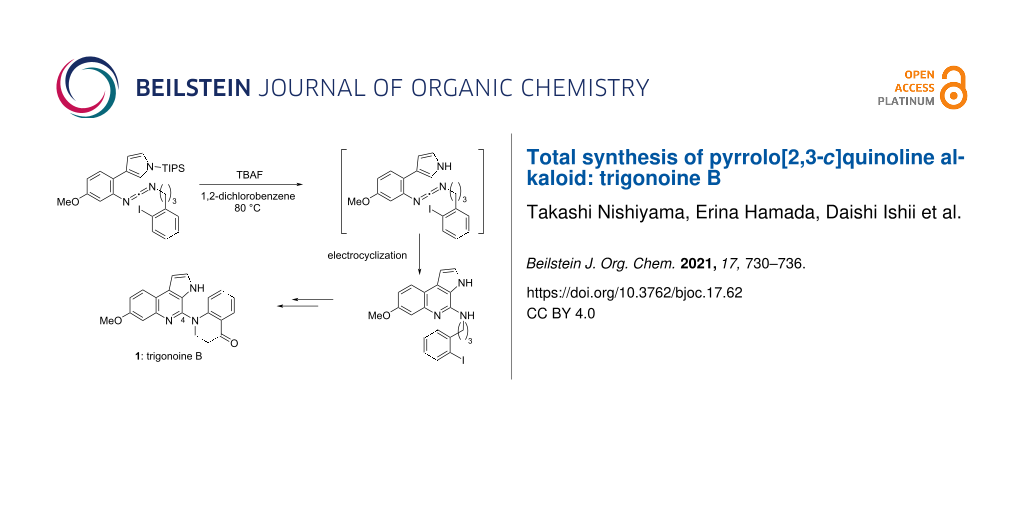
Abstract
The first total synthesis of the pyrrolo[2,3-c]quinoline alkaloid trigonoine B (1) was accomplished via a six-step sequence involving the construction of an N-substituted 4-aminopyrrolo[2,3-c]quinoline framework via electrocyclization of 2-(pyrrol-3-yl)benzene containing a carbodiimide moiety as a 2-azahexatriene system. The employed six-step sequence afforded trigonoine B (1) in 9.2% overall yield. The described route could be employed for the preparation of various N-substituted 4-aminopyrroloquinolines with various biological activities.

Graphical Abstract
Introduction
In 2011, two novel alkaloids, namely trigonoine A and B, were isolated from the leaves of Trigonostemon lii by Hao and co-workers [1]. The structures of the compounds were elucidated through 1D and 2D NMR spectroscopy. It was determined that trigonoine A was a β-carboline alkaloid containing a 2,8-diazabicyclo[3.3.1]nonane ring system, while trigonoine B (1) was formed by a combination of 2,3-dihydroquinolin-4-one and 3H-pyrrolo[2,3-c]quinoline rearranged from a β-carboline skeleton (Figure 1).
![[1860-5397-17-62-1]](/bjoc/content/figures/1860-5397-17-62-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Natural products possessing the pyrrolo[2,3-c]quinoline skeleton.
Figure 1: Natural products possessing the pyrrolo[2,3-c]quinoline skeleton.
Additionally, the antihepatitis B virus (HBV) activity of the alkaloids was evaluated in vitro using an HBV-transfected HepG2 cell line. The antihuman immunodeficiency virus (HIV) properties were also investigated to determine whether trigonoine A and B could prevent the cytopathic effects of HIV-1IIIB in C8166 cells. Notably, it was found that both natural products exhibited anti-HIV activity.
In addition to trigonoine B, aplidiopsamine A (2) [2] and marinoquinolines 3 [3] are natural products that possess the pyrrolo[2,3-c]quinoline skeleton (Figure 1). These pyrrolo[2,3-c]quinolines have been demonstrated to show antimalarial and antibacterial biological activities [2-4]. Thus, the development of a convenient and efficient synthetic route to the pyrrolo[2,3-c]quinoline skeleton has attracted considerable attention from organic and medicinal chemists. The total syntheses of aplidiopsamine A (2) [5-9] and marinoquinolines 3 [10-13] have been achieved by various synthetic strategies. Nevertheless, the total synthesis of trigonoine B (1) has not yet been reported.
We have been interested in the synthesis of heterocyclic compounds by constructing fused pyridine ring systems based on a thermal electrocyclization of an azahexatriene moiety [14,15]. It has been hoped that the development of compounds with enhanced biological activity would be possible using these natural products and their derivatives [16-18]. We have previously reported the total syntheses of indolo[3,2-c]quinoline (isocryptolepine) [19], azaanthracenones (kalasinamide, marcanine A, and geovanine) [20], imidazo[4',5':4,5]pyrido[2,3-b]indole (grossularine-1 and -2) [21,22], imidazo[4,5-b]pyridine (2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 2-amino-1,6-dimethylimidazo[4,5-b]pyridine) [23], and imidazo[4,5-c]quinoline (imiquimod) [24] based on the electrocyclization of 2-azahexatriene involving an isocyanate moiety as the key intermediate.
In addition, we recently reported the total syntheses of marinoquinolines A (3a), B (3b), and E (3e) comprising the pyrrolo[2,3-c]quinoline skeleton [25]. As demonstrated in Scheme 1, Curtius rearrangement of carboxylic acid 4 resulted in the formation of isocyanate 5, which, upon electrocyclization furnished pyrrolo[2,3-c]quinoline 6. Intermediate 6 was subsequently transformed into triflate 7, and the total syntheses of marinoquinolines 3a, 3b, and 3e were accomplished by introducing different substituents at the C4 position. However, despite our efforts to introduce dihydroquinoline derivatives into triflate 7 using various conditions, the synthesis of 8 could not be achieved. Consequently, we decided to develop a new synthetic strategy for the preparation of trigonoine B (1).
![[1860-5397-17-62-i1]](/bjoc/content/inline/1860-5397-17-62-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Total synthesis of marinoquinolines and the failure of the introduction of a tetrahydroquinoline moiety by direct amination to triflate 7.
Scheme 1: Total synthesis of marinoquinolines and the failure of the introduction of a tetrahydroquinoline mo...
Carbodiimides are valuable synthetic intermediates that can be obtained by an aza-Wittig reaction of isocyanates with iminophosphoranes or by dehydration of urea [26]. Molina et al. previously reported the synthesis of 2-aminopyridine derivatives through an electrocyclization of conjugated carbodiimides derived from an aza-Wittig reaction of iminophosphoranes and isocyanates [27]. Hibino and co-workers also achieved the preparation of mutagenic amino-α-carbolines (2-amino-9H-pyrido[2,3-b]indole, AαC) and MeAαC by the electrocyclization of 3-alkenyl-2-carbodiimidoindole derivatives obtained by an aza-Wittig reaction of indol-2-yl-iminophosphoranes and isocyanates [28].
In this work, we report the first total synthesis of trigonoine B (1) involving the construction of a pyrrolo[2,3-c]quinoline framework by electrocyclization of 2-(pyrrol-3-yl)benzene containing a carbodiimide moiety as a 2-azahexatriene system.
Results and Discussion
Scheme 2 illustrates the retrosynthetic strategy designed to synthesize triogonoine B (1). It was speculated that the dihydroquinoline moiety of trigonoine B (1) could be constructed through a cycloamination reaction between positions C1a and N1. The synthesis of the precursor pyrroloquinoline 9 possessing a substituted amino group at the 4-position could be achieved by the electrocyclization of pyrrol-3-ylbenzene 10 containing a carbodiimide moiety as a 2-azahexatriene system. Lastly, it was proposed that the carbodiimide 10 could be derived from urea 11.
![[1860-5397-17-62-i2]](/bjoc/content/inline/1860-5397-17-62-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Retrosynthetic analysis of the pyrrolo[2,3-c]quinoline ring construction.
Scheme 2: Retrosynthetic analysis of the pyrrolo[2,3-c]quinoline ring construction.
Therefore, we investigated the electrocyclization of a pyrrol-3-ylbenzene containing a carbodiimide moiety. First, 2-(pyrrol-3-yl)aniline 14 was synthesized by a Suzuki–Miyaura coupling reaction of 2-iodoaniline (12) and 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-[tris(1-methylethyl)silyl]-1H-pyrrole (13) according to Pratt’s conditions (Scheme 3) [29]. Subsequently, to remove the triisopropylsilyl (TIPS) protecting group, compound 14 was treated with tetra-n-butylammonium fluoride (TBAF) in THF, affording aniline 15 in 65% yield. Treatment of 15 with phenyl isocyanate in CH2Cl2 gave urea 16a in 54% yield. To obtain carbodiimide 17a, 16a was treated with carbon tetrabromide (CBr4), PPh3, and Et3N in CH2Cl2. The reaction was monitored by TLC, which confirmed the complete consumption of the starting material. However, after the workup, extraction, and removal of solvent (in vacuo), the appearance of the TLC plate changed due to the formation of various byproducts. Then, the reaction mixture was purified, but the desired carbodiimide 17a was not obtained, but aminopyrroloquinoline 18 formed through electrocyclization of 17a in 25% yield. Notably, it was found that the electrocyclization of 17a proceeded easily at low temperature (≈60 °C).
![[1860-5397-17-62-i3]](/bjoc/content/inline/1860-5397-17-62-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of N-substituted 4-aminopyrrolo[3,2-c]quinoline 18.
Scheme 3: Synthesis of N-substituted 4-aminopyrrolo[3,2-c]quinoline 18.
Although we tried to optimize the reaction conditions to improve the yield of 18, the yield could not be improved owing to the generation of numerous byproducts in the reaction of 16a with CBr4, PPh3, and Et3N. Using the same procedure, carbodiimide 17b was then synthesized in 58% yield starting from aniline 14 via urea 16b. Then, the electrocyclization of 17b was attempted, but the cyclization did not proceed at all. As possible reason we considered a steric hindrance of the TIPS group. Based on the above results, it was speculated that the electrocyclization of 17b could proceed continuously following the removal of the TIPS group. Indeed, when 17b was heated with TBAF in 1,2-dichlorobenzene (1,2-DCB) at 80 °C, the expected reaction proceeded and the desired pyrroloquinoline 18 was obtained in 58% yield.
We subsequently examined the versatility of the electrocyclization of carbodiimides 20a–d as 2-azahexatriene systems (Table 1). First, urea derivatives 19a–d were synthesized by reacting 2-(pyrrol-3-yl)aniline 14 with isocyanates, which were commercially available or prepared from an appropriate carboxylic acid through a Curtius rearrangement reaction (50–98% yield). Treatment of urea derivatives 19a–d with CBr4, PPh3, and Et3N afforded carbodiimides 20a–d in 64–75% yield. Compounds 21a–d were obtained in situ following the removal of the TIPS protecting group in 20a–d by TBAF in 1,2-DCB at 80 °C. The electrocyclization then proceeded immediately, affording the desired pyrroloquinolines 22a–c in 49–90% yield (Table 1, entries 1–3). However, the cyclization of 20d only gave a mixture of unidentified products (Table 1, entry 4).
Table 1: Synthesis of N-substituted 4-aminopyrrolo[3,2-c]quinolines 22 by electrocyclization of carbodiimides 20.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-62-i6.svg?max-width=637&scale=1.0)
|
||||||||
| entry | R | 19 | 20 | 22 | ||||
|
time
(h) |
yield
(%) |
time
(h) |
yield
(%) |
time
(h) |
yield
(%) |
|||
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-62-i7.svg?max-width=637&scale=1.0)
|
a | 12 | 50 | 4 | 64 | 2 | 90 |
| 2 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-62-i8.svg?max-width=637&scale=1.0)
|
b | 12 | 98 | 1 | –a | 2 | 49b |
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-62-i9.svg?max-width=637&scale=1.0)
|
c | 20 | 64 | 2 | 75 | 1 | 61 |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-62-i10.svg?max-width=637&scale=1.0)
|
d | 18 | 98 | 0.5 | 68 | 1 | –c |
aSince carbodiimide 20b was unstable, the next reaction was carried out without purification. bYield from 19b. cUnknown compounds.
Hence, we decided to evaluate the synthesis of the 2,3-dihydroquinolin-4-one moiety of trigonoine B (1) by cycloamination of 22c (Scheme 4). The Buchwald–Hartwig amination of 22c was conducted in the presence of t-BuONa, BINAP, and Pd2(dba)3·CHCl3; however, the desired tetrahydroquinoline 23 was not obtained and only 22c was recovered. We then examined the conditions reported by Orito and co-workers [30]. Gratifyingly, the treatment of 22c with Pd(OAc)2, Cu(OAc)2, and K2CO3 afforded the cyclized product 23 in 34% yield.
![[1860-5397-17-62-i4]](/bjoc/content/inline/1860-5397-17-62-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the tetrahydroquinoline moiety through cycloamination.
Scheme 4: Synthesis of the tetrahydroquinoline moiety through cycloamination.
The focus subsequently shifted to the total synthesis of trigonoine B (1) (Scheme 5). The key starting material, 2-iodo-5-methoxyaniline (24), was synthesized according to the procedure previously reported by Wetzel and co-workers [31]. The Suzuki–Miyaura coupling of 2-iodoaniline derivative 24 and pyrrole-3-boronic acid pinacol ester 13 was carried out in the presence of Pd(OAc)2 and SPhos, followed by the treatment of the resulting 2-(pyrrol-3-yl)aniline 25 with 3-(2-bromophenyl)propyl isocyanate (26a), which afforded urea 27a in 60% yield. The treatment of urea 27a with CBr4 and PPh3 in the presence of Et3N then gave carbodiimide 28a in a good yield of 74%. Following the reaction of 28a with TBAF in 1,2-DCB and desilylation, the electrocyclization of 29a proceeded smoothly to afford the desired 4-aminopyrroloquinoline 30a in 68% yield. Subsequently, the cycloamination of 30a in the presence of Pd(OAc)2, Cu(OAc)2, and K2CO3 gave tetrahydroquinoline 31 in 25% yield. However, although attempts were made to optimize the cycloamination reaction conditions, the yield could not be improved. We hypothesized that the low yield could be attributed to a too low reactivity of the bromo group. Thus, the same reaction was performed using a compound bearing a more reactive iodo group.
![[1860-5397-17-62-i5]](/bjoc/content/inline/1860-5397-17-62-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Accordingly, 4-aminopyrroloquinoline 30b was synthesized from 2-(pyrrol-3-yl)aniline 25 and 3-(2-iodophenyl)propyl isocyanate (26b) in 3 steps using the same procedure as described for 30a. The subsequent cycloamination of 30b gave tetrahydroquinoline 31 in 73% yield, thus resulting in a significantly improved yield.
Finally, the oxidation of tetrahydroquinoline 31 was examined. Following the evaluation of various reaction conditions [32-34], the best outcome was obtained upon treatment of 31 with KMnO4 and MgSO4 in an acetone–H2O solvent system [35]. Employing these conditions, trigonoine B (1) was obtained in 43% yield. The acquired physical and spectroscopic data of the herein synthesized compound 1 were consistent with those of the natural trigonoine B [1]. Thus, in this study, we successfully achieved the first total synthesis of trigonoine B (1).
Conclusion
The total synthesis of a pyrrolo[2,3-c]quinoline alkaloid, trigonoine B (1), was achieved for the first time through the electrocyclization of 2-(pyrrol-3-yl)benzene bearing a carbodiimide moiety as a 2-azahexatriene system. The employed six-step sequence afforded the desired product 1 in 9.2% overall yield. Notably, the developed synthetic route could be used for the synthesis of various N-substituted 4-aminopyrroloquinolines. The investigation of the biological activity of trigonoine B and its derivatives is under evaluation.
Supporting Information
| Supporting Information File 1: 1H NMR and 13C NMR spectra of all new compounds. | ||
| Format: PDF | Size: 3.9 MB | Download |
References
-
Li, S.-F.; Di, Y.-T.; He, H.-P.; Zhang, Y.; Wang, Y.-H.; Yin, J.-L.; Tan, C.-J.; Li, S.-L.; Hao, X.-J. Tetrahedron Lett. 2011, 52, 3186–3188. doi:10.1016/j.tetlet.2011.03.015
Return to citation in text: [1] [2] -
Carroll, A. R.; Duffy, S.; Avery, V. M. J. Org. Chem. 2010, 75, 8291–8294. doi:10.1021/jo101695v
Return to citation in text: [1] [2] -
Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K. Mar. Drugs 2008, 6, 578–586. doi:10.3390/md6040578
Return to citation in text: [1] [2] -
Okanya, P. W.; Mohr, K. I.; Gerth, K.; Jansen, R.; Müller, R. J. Nat. Prod. 2011, 74, 603–608. doi:10.1021/np100625a
Return to citation in text: [1] -
Ni, L.; Li, Z.; Wu, F.; Xu, J.; Wu, X.; Kong, L.; Yao, H. Tetrahedron Lett. 2012, 53, 1271–1274. doi:10.1016/j.tetlet.2011.12.124
Return to citation in text: [1] -
Schwalm, C. S.; Correia, C. R. D. Tetrahedron Lett. 2012, 53, 4836–4840. doi:10.1016/j.tetlet.2012.06.115
Return to citation in text: [1] -
Ma, X.; Vo, Y.; Banwell, M. G.; Willis, A. C. Asian J. Org. Chem. 2012, 1, 160–165. doi:10.1002/ajoc.201200037
Return to citation in text: [1] -
Lindsay, A. C.; Sperry, J. Synlett 2013, 24, 461–464. doi:10.1055/s-0032-1318137
Return to citation in text: [1] -
Aguiar, A. C. C.; Panciera, M.; Simão dos Santos, E. F.; Singh, M. K.; Garcia, M. L.; de Souza, G. E.; Nakabashi, M.; Costa, J. L.; Garcia, C. R. S.; Oliva, G.; Correia, C. R. D.; Guido, R. V. C. J. Med. Chem. 2018, 61, 5547–5568. doi:10.1021/acs.jmedchem.8b00143
Return to citation in text: [1] -
Mahajan, J. P.; Suryawanshi, Y. R.; Mhaske, S. B. Org. Lett. 2012, 14, 5804–5807. doi:10.1021/ol302676v
Return to citation in text: [1] -
Yamaoka, Y.; Yoshida, T.; Shinozaki, M.; Yamada, K.-i.; Takasu, K. J. Org. Chem. 2015, 80, 957–964. doi:10.1021/jo502467m
Return to citation in text: [1] -
Bolte, B.; Bryan, C. S.; Sharp, P. P.; Sayyahi, S.; Rihouey, C.; Kendrick, A.; Lan, P.; Banwell, M. G.; Jackson, C. J.; Fraser, N. J.; Willis, A. C.; Ward, J. S. J. Org. Chem. 2020, 85, 650–663. doi:10.1021/acs.joc.9b02725
Return to citation in text: [1] -
Panarese, J. D.; Lindsley, C. W. Org. Lett. 2012, 14, 5808–5810. doi:10.1021/ol3024665
Return to citation in text: [1] -
Choshi, T.; Hibino, S. Heterocycles 2011, 83, 1205–1239. doi:10.3987/rev-10-691
Return to citation in text: [1] -
Hibino, S. Yakugaku Zasshi 2016, 136, 607–648. doi:10.1248/yakushi.15-00273
and related references cited.
Return to citation in text: [1] -
Furukawa, Y.; Sawamoto, A.; Yamaoka, M.; Nakaya, M.; Hieda, Y.; Choshi, T.; Hatae, N.; Okuyama, S.; Nakajima, M.; Hibino, S. Molecules 2019, 24, 1366. doi:10.3390/molecules24071366
Return to citation in text: [1] -
Itoh, T.; Hatae, N.; Nishiyama, T.; Choshi, T.; Hibino, S.; Yoshimura, T.; Ishikura, M. Med. Chem. Res. 2018, 27, 412–419. doi:10.1007/s00044-017-2068-6
Return to citation in text: [1] -
Nishiyama, T.; Hatae, N.; Mizutani, M.; Yoshimura, T.; Kitamura, T.; Miyano, M.; Fujii, M.; Satsuki, N.; Ishikura, M.; Hibino, S.; Choshi, T. Eur. J. Med. Chem. 2017, 136, 1–13. doi:10.1016/j.ejmech.2017.04.071
Return to citation in text: [1] -
Hayashi, K.; Choshi, T.; Chikaraishi, K.; Oda, A.; Yoshinaga, R.; Hatae, N.; Ishikura, M.; Hibino, S. Tetrahedron 2012, 68, 4274–4279. doi:10.1016/j.tet.2012.03.055
Return to citation in text: [1] -
Nishiyama, T.; Hatae, N.; Chikaraishi, K.; Uchida, K.; Yokoyama, C.; Hibino, S.; Choshi, T. Heterocycles 2019, 99, 415–424. doi:10.3987/com-18-s(f)36
Return to citation in text: [1] -
Choshi, T.; Yamada, S.; Sugino, E.; Kuwada, T.; Hibino, S. J. Org. Chem. 1995, 60, 5899–5904. doi:10.1021/jo00123a028
Return to citation in text: [1] -
Choshi, T.; Yamada, S.; Sugino, E.; Kuwada, T.; Hibino, S. Synlett 1995, 147–148. doi:10.1055/s-1995-4910
Return to citation in text: [1] -
Choshi, T.; Tonari, A.; Yoshioka, H.; Harada, K.; Sugino, E.; Hibino, S. J. Org. Chem. 1993, 58, 7952–7954. doi:10.1021/jo00079a055
Return to citation in text: [1] -
Yoshioka, H.; Matsuya, Y.; Choshi, T.; Sugino, E.; Hibino, S. Chem. Pharm. Bull. 1996, 44, 709–714. doi:10.1248/cpb.44.709
Return to citation in text: [1] -
Nishiyama, T.; Murakami, M.; Taninaka, K.; Hamada, E.; Endo, M.; Kinou, D.; Hatae, N.; Choshi, T. Heterocycles 2021, in press. doi:10.3987/com-20-s(k)14
Return to citation in text: [1] -
Tsukano, C.; Nakajima, M.; Hande, S. M.; Takemoto, Y. Org. Biomol. Chem. 2019, 17, 1731–1735. doi:10.1039/c8ob02224k
Return to citation in text: [1] -
Molina, P.; Fresneda, P. M.; Alarcón, P. Tetrahedron Lett. 1988, 29, 379–380. doi:10.1016/s0040-4039(00)80101-x
Return to citation in text: [1] -
Hibino, S.; Sugino, E.; Kuwada, T.; Ogura, N.; Shintani, Y.; Satoh, K. Chem. Pharm. Bull. 1991, 39, 79–80. doi:10.1248/cpb.39.79
Return to citation in text: [1] -
Morrison, M. D.; Hanthorn, J. J.; Pratt, D. A. Org. Lett. 2009, 11, 1051–1054. doi:10.1021/ol8026957
Return to citation in text: [1] -
Harada, R.; Nishida, N.; Uchiito, S.; Onozaki, Y.; Kurono, N.; Senboku, H.; Masao, T.; Ohkuma, T.; Orito, K. Eur. J. Org. Chem. 2012, 366–379. doi:10.1002/ejoc.201101214
Return to citation in text: [1] -
Wetzel, A.; Gagosz, F. Angew. Chem., Int. Ed. 2011, 50, 7354–7358. doi:10.1002/anie.201102707
Return to citation in text: [1] -
Nakanishi, M.; Bolm, C. Adv. Synth. Catal. 2007, 349, 861–864. doi:10.1002/adsc.200600553
Return to citation in text: [1] -
Bonvin, Y.; Callens, E.; Larrosa, I.; Henderson, D. A.; Oldham, J.; Burton, A. J.; Barrett, A. G. M. Org. Lett. 2005, 7, 4549–4552. doi:10.1021/ol051765k
Return to citation in text: [1] -
Wu, S.; Liu, C.; Luo, G.; Jin, Z.; Zheng, P.; Chi, Y. R. Angew. Chem., Int. Ed. 2019, 58, 18410–18413. doi:10.1002/anie.201909479
Return to citation in text: [1] -
Silva, S. B. L.; Della Torre, A.; de Carvalho, J. E.; Ruiz, A. L. T. G.; Silva, L. F., Jr. Molecules 2015, 20, 1475–1494. doi:10.3390/molecules20011475
Return to citation in text: [1]
| 30. | Harada, R.; Nishida, N.; Uchiito, S.; Onozaki, Y.; Kurono, N.; Senboku, H.; Masao, T.; Ohkuma, T.; Orito, K. Eur. J. Org. Chem. 2012, 366–379. doi:10.1002/ejoc.201101214 |
| 28. | Hibino, S.; Sugino, E.; Kuwada, T.; Ogura, N.; Shintani, Y.; Satoh, K. Chem. Pharm. Bull. 1991, 39, 79–80. doi:10.1248/cpb.39.79 |
| 29. | Morrison, M. D.; Hanthorn, J. J.; Pratt, D. A. Org. Lett. 2009, 11, 1051–1054. doi:10.1021/ol8026957 |
| 1. | Li, S.-F.; Di, Y.-T.; He, H.-P.; Zhang, Y.; Wang, Y.-H.; Yin, J.-L.; Tan, C.-J.; Li, S.-L.; Hao, X.-J. Tetrahedron Lett. 2011, 52, 3186–3188. doi:10.1016/j.tetlet.2011.03.015 |
| 5. | Ni, L.; Li, Z.; Wu, F.; Xu, J.; Wu, X.; Kong, L.; Yao, H. Tetrahedron Lett. 2012, 53, 1271–1274. doi:10.1016/j.tetlet.2011.12.124 |
| 6. | Schwalm, C. S.; Correia, C. R. D. Tetrahedron Lett. 2012, 53, 4836–4840. doi:10.1016/j.tetlet.2012.06.115 |
| 7. | Ma, X.; Vo, Y.; Banwell, M. G.; Willis, A. C. Asian J. Org. Chem. 2012, 1, 160–165. doi:10.1002/ajoc.201200037 |
| 8. | Lindsay, A. C.; Sperry, J. Synlett 2013, 24, 461–464. doi:10.1055/s-0032-1318137 |
| 9. | Aguiar, A. C. C.; Panciera, M.; Simão dos Santos, E. F.; Singh, M. K.; Garcia, M. L.; de Souza, G. E.; Nakabashi, M.; Costa, J. L.; Garcia, C. R. S.; Oliva, G.; Correia, C. R. D.; Guido, R. V. C. J. Med. Chem. 2018, 61, 5547–5568. doi:10.1021/acs.jmedchem.8b00143 |
| 26. | Tsukano, C.; Nakajima, M.; Hande, S. M.; Takemoto, Y. Org. Biomol. Chem. 2019, 17, 1731–1735. doi:10.1039/c8ob02224k |
| 2. | Carroll, A. R.; Duffy, S.; Avery, V. M. J. Org. Chem. 2010, 75, 8291–8294. doi:10.1021/jo101695v |
| 3. | Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K. Mar. Drugs 2008, 6, 578–586. doi:10.3390/md6040578 |
| 4. | Okanya, P. W.; Mohr, K. I.; Gerth, K.; Jansen, R.; Müller, R. J. Nat. Prod. 2011, 74, 603–608. doi:10.1021/np100625a |
| 27. | Molina, P.; Fresneda, P. M.; Alarcón, P. Tetrahedron Lett. 1988, 29, 379–380. doi:10.1016/s0040-4039(00)80101-x |
| 3. | Sangnoi, Y.; Sakulkeo, O.; Yuenyongsawad, S.; Kanjana-opas, A.; Ingkaninan, K.; Plubrukarn, A.; Suwanborirux, K. Mar. Drugs 2008, 6, 578–586. doi:10.3390/md6040578 |
| 24. | Yoshioka, H.; Matsuya, Y.; Choshi, T.; Sugino, E.; Hibino, S. Chem. Pharm. Bull. 1996, 44, 709–714. doi:10.1248/cpb.44.709 |
| 2. | Carroll, A. R.; Duffy, S.; Avery, V. M. J. Org. Chem. 2010, 75, 8291–8294. doi:10.1021/jo101695v |
| 25. | Nishiyama, T.; Murakami, M.; Taninaka, K.; Hamada, E.; Endo, M.; Kinou, D.; Hatae, N.; Choshi, T. Heterocycles 2021, in press. doi:10.3987/com-20-s(k)14 |
| 19. | Hayashi, K.; Choshi, T.; Chikaraishi, K.; Oda, A.; Yoshinaga, R.; Hatae, N.; Ishikura, M.; Hibino, S. Tetrahedron 2012, 68, 4274–4279. doi:10.1016/j.tet.2012.03.055 |
| 21. | Choshi, T.; Yamada, S.; Sugino, E.; Kuwada, T.; Hibino, S. J. Org. Chem. 1995, 60, 5899–5904. doi:10.1021/jo00123a028 |
| 22. | Choshi, T.; Yamada, S.; Sugino, E.; Kuwada, T.; Hibino, S. Synlett 1995, 147–148. doi:10.1055/s-1995-4910 |
| 35. | Silva, S. B. L.; Della Torre, A.; de Carvalho, J. E.; Ruiz, A. L. T. G.; Silva, L. F., Jr. Molecules 2015, 20, 1475–1494. doi:10.3390/molecules20011475 |
| 16. | Furukawa, Y.; Sawamoto, A.; Yamaoka, M.; Nakaya, M.; Hieda, Y.; Choshi, T.; Hatae, N.; Okuyama, S.; Nakajima, M.; Hibino, S. Molecules 2019, 24, 1366. doi:10.3390/molecules24071366 |
| 17. | Itoh, T.; Hatae, N.; Nishiyama, T.; Choshi, T.; Hibino, S.; Yoshimura, T.; Ishikura, M. Med. Chem. Res. 2018, 27, 412–419. doi:10.1007/s00044-017-2068-6 |
| 18. | Nishiyama, T.; Hatae, N.; Mizutani, M.; Yoshimura, T.; Kitamura, T.; Miyano, M.; Fujii, M.; Satsuki, N.; Ishikura, M.; Hibino, S.; Choshi, T. Eur. J. Med. Chem. 2017, 136, 1–13. doi:10.1016/j.ejmech.2017.04.071 |
| 23. | Choshi, T.; Tonari, A.; Yoshioka, H.; Harada, K.; Sugino, E.; Hibino, S. J. Org. Chem. 1993, 58, 7952–7954. doi:10.1021/jo00079a055 |
| 1. | Li, S.-F.; Di, Y.-T.; He, H.-P.; Zhang, Y.; Wang, Y.-H.; Yin, J.-L.; Tan, C.-J.; Li, S.-L.; Hao, X.-J. Tetrahedron Lett. 2011, 52, 3186–3188. doi:10.1016/j.tetlet.2011.03.015 |
| 14. | Choshi, T.; Hibino, S. Heterocycles 2011, 83, 1205–1239. doi:10.3987/rev-10-691 |
| 15. |
Hibino, S. Yakugaku Zasshi 2016, 136, 607–648. doi:10.1248/yakushi.15-00273
and related references cited. |
| 31. | Wetzel, A.; Gagosz, F. Angew. Chem., Int. Ed. 2011, 50, 7354–7358. doi:10.1002/anie.201102707 |
| 10. | Mahajan, J. P.; Suryawanshi, Y. R.; Mhaske, S. B. Org. Lett. 2012, 14, 5804–5807. doi:10.1021/ol302676v |
| 11. | Yamaoka, Y.; Yoshida, T.; Shinozaki, M.; Yamada, K.-i.; Takasu, K. J. Org. Chem. 2015, 80, 957–964. doi:10.1021/jo502467m |
| 12. | Bolte, B.; Bryan, C. S.; Sharp, P. P.; Sayyahi, S.; Rihouey, C.; Kendrick, A.; Lan, P.; Banwell, M. G.; Jackson, C. J.; Fraser, N. J.; Willis, A. C.; Ward, J. S. J. Org. Chem. 2020, 85, 650–663. doi:10.1021/acs.joc.9b02725 |
| 13. | Panarese, J. D.; Lindsley, C. W. Org. Lett. 2012, 14, 5808–5810. doi:10.1021/ol3024665 |
| 20. | Nishiyama, T.; Hatae, N.; Chikaraishi, K.; Uchida, K.; Yokoyama, C.; Hibino, S.; Choshi, T. Heterocycles 2019, 99, 415–424. doi:10.3987/com-18-s(f)36 |
| 32. | Nakanishi, M.; Bolm, C. Adv. Synth. Catal. 2007, 349, 861–864. doi:10.1002/adsc.200600553 |
| 33. | Bonvin, Y.; Callens, E.; Larrosa, I.; Henderson, D. A.; Oldham, J.; Burton, A. J.; Barrett, A. G. M. Org. Lett. 2005, 7, 4549–4552. doi:10.1021/ol051765k |
| 34. | Wu, S.; Liu, C.; Luo, G.; Jin, Z.; Zheng, P.; Chi, Y. R. Angew. Chem., Int. Ed. 2019, 58, 18410–18413. doi:10.1002/anie.201909479 |
© 2021 Nishiyama et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)