

Abstract
Metal-free organic emitters with thermally activated delayed fluorescence (TADF) characteristics are emerging due to the potential applications in optoelectronic devices, time-resolved luminescence imaging, and solid-phase sensing. Herein, we synthesized two (4-bromobenzoyl)pyridine (BPy)-based donor–acceptor (D–A) compounds with varying donor size and strength: the emitter BPy-pTC with tert-butylcarbazole (TC) as the donor and BPy-p3C with bulky tricarbazole (3C) as the donor unit. Both BPy-pTC and BPy-p3C exhibited prominent emission with TADF properties in solution and in the solid phase. The stronger excited-state charge transfer was obtained for BPy-p3C due to the bulkier donor, leading to a more twisted D–A geometry than that of BPy-pTC. Hence, BPy-p3C exhibited aggregation-induced enhanced emission (AIEE) in a THF/water mixture. Interestingly, the singlet–triplet energy gap (ΔEST) was reduced for both compounds in the aggregated state as compared to toluene solution. Consequently, a faster reverse intersystem crossing rate (kRISC) was obtained in the aggregated state, facilitating photon upconversion, leading to enhanced delayed fluorescence. Further, the lone-pair electrons of the pyridinyl nitrogen atom were found to be sensitive to acidic protons. Hence, the exposure to acid and base vapors using trifluoroacetic acid (TFA) and triethylamine (TEA) led to solid-phase fluorescence switching with fatigue resistance. The current study demonstrates the role of the donor strength and size in tuning ΔEST in the aggregated state as well as the relevance for fluorescence-based acid–base sensing.
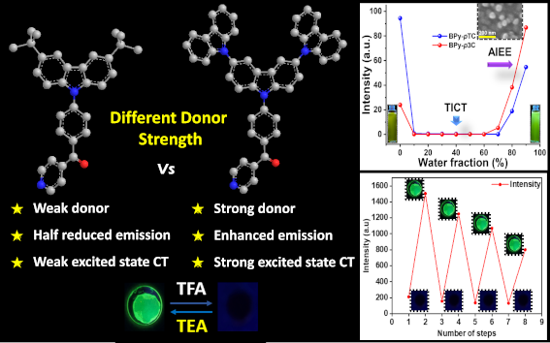
Graphical Abstract
Introduction
Metal-free organic solid-state emitters have gained increasing interest in recent years due to the potential applications in optoelectronic devices [1-4], solid-phase sensing [5], bioimaging [6,7], security [8], and storage devices [9]. The optical and electrochemical properties of the organic emitters can be tuned by external stimuli, such as mechanical force, heat, solvent, acid and base fumes, etc. [10,11]. However, the fluorescence quantum yield of such emitters in the solid state is relatively low due to the aggregation-caused quenching (ACQ) effect [12-14] and limits practical applications. Hence, metal-free emitters with high photoluminescence quantum yield (PLQY) in solution and in the solid state and with multicolor tunability by external stimuli are crucial for task-specific applications.
Recently, donor–acceptor (D–A)-based organic emitters have shown tunable optical and electrochemical properties due to alteration of the intermolecular charge-transfer (ICT) interactions [15]. Several molecular designs were proposed to adjust the ICT interactions through covalent D–A linking. The separation of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of the donor and acceptor molecules leads to a reduction of the singlet–triplet energy gap (ΔEST) [16,17]. The low ΔEST facilitates the exciton upconversion through reverse intersystem crossing (RISC) [2], which is commonly known as thermally activated delayed fluorescence (TADF) [16]. Theoretically, this has up to 100% internal quantum efficiency and could replace heavy metal-based emitters in organic light-emitting diode (OLED) devices [18]. Nevertheless, many TADF emitters suffer from quenching of the emission due to the aggregation-caused quenching effect [19,20]. The strong π–π-stacking interactions in the solid or aggregated state may lead to emission quenching. Therefore, an appropriate molecular design to suppress π–π stacking should help to obtain a strong emission in solution as well as in the solid state.
Solid-state organic emitters with reversible fluorescence switching are emerging for the sensing of pollutant acid vapor [21-23]. However, quickly detecting organic acid vapor at ambient conditions is challenging for solid-state detectors. Therefore, developing a solid-state emitter with reversible switching of the optical properties by external stimuli, such as acid and base vapors, is important. As per the recent literature, such sensors are being developed mainly by focusing on photophysical processes, such as photoinduced electron transfer (PET), excited-state intramolecular proton transfer (ESIPT), fluorescence resonance energy transfer (FRET), etc. [21,22,24,25]. Several literature reports have also demonstrated the switching of fluorescence in designing heteroatom-containing (N, O, S, etc.) chromophores [26]. The lone-pair electrons of heteroatoms can possess a certain degree of basicity, which results in sensitivity to acid. Consequently, the lone pair may be protonated and deprotonated by consecutively adding acid and base. The disruption of the molecular conjugation or ICT interactions upon protonation or deprotonation would lead to switching of the optical properties. While there are many reports on acid–based sensors, TADF emitter-based sensors are rare in the literature.
In this context, we chose the D–A molecular design to demonstrate the solid-phase acid–base sensing process and to determine the RISC rate (kRISC). The selection of different donors with varying donor strength and size should help to understand the charge transfer (CT) interactions and D–A twist. Controlling the D–A twisting by varying the donor unit should be beneficial for suppressing nonradiative deactivation channels, leading to aggregated- or solid-state emission properties. To this end, we chose tert-butylcarbazole (TC) and bulky tricarbazole (3C), respectively, as donor unit in combination with a (4-bromobenzoyl)pyridine (BPy) acceptor core to demonstrate ΔEST tuning in solution and in an aggregated state, including the aggregation-induced enhanced emission (AIEE) characteristics. Further, solid-phase fluorescence switching by alternatingly adding acid and base vapors demonstrated the sensing ability. Therefore, the current study deciphers the role of molecular design and donor size in tuning ΔEST in an aggregated state and in fluorescence switching by acid–base addition.
Results and Discussion
Design and synthesis
Several BPy derivatives have been reported to lower ΔEST in D–A pairs to promote spin upconversion from the low-lying triplet excited state to the singlet state via RISC [17,27]. Heteroatom lone-pair electrons are sensitive to acid and, once protonated, to base, which would facilitate the tuning of optical properties in such media [21,22]. Herein, (4-(3,6-di-tert-butyl-9H-carbazol-9-yl)phenyl)(pyridin-4-yl)methanone (BPy-pTC) and (4-(9'H-[9,3':6',9''-tercarbazol]-9'-yl)phenyl)(pyridin-4-yl)methanone (BPy-p3C) were synthesized via Ullmann coupling following the reported protocol [28]. These two TADF emitters have already been synthesized and device fabrication had been demonstrated. However, detailed photophysical investigations in the aggregated state have not yet been performed [28]. The acceptor core BPy, coupled with carbazole derivatives, produced BPy-pTC and BPy-p3C, respectively (Scheme 1). The detailed synthetic procedures and characterization data are given in the Experimental section and in Schemes S1 and S2 in Supporting Information File 1. Both compounds were further purified by temperature-gradient vacuum sublimation and characterized by 1H and 13C NMR spectroscopy (Figures S9–S12, Supporting Information File 1) as well as high-resolution mass spectrometry.
![[1860-5397-18-122-i1]](/bjoc/content/inline/1860-5397-18-122-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic schemes of BPy-pTC and BPy-p3C.
Scheme 1: Synthetic schemes of BPy-pTC and BPy-p3C.
Photophysical studies and DFT calculations
The UV–vis absorption and emission spectra of BPy-pTC and BPy-p3C were recorded in solvents with varying polarity (Figure 1 and Table 1). BPy-pTC exhibited prominent absorption bands at λabs = 337, 355, and 383 nm. The peaks at λabs = 337 and 335 nm were due to the π–π* and n–π* transitions [29], whereas the broad peak at λabs = 383 nm was due to the ICT transitions. Similarly, three peaks centered at λabs = 336, 350, and 368 nm were observed for BPy-p3C. The ICT band of BPy-p3C (λabs = 368 nm) was blue-shifted as compared to BPy-pTC (λabs = 383 nm) due to the weaker ground state CT interactions. As a result, a stronger molar extinction coefficient was obtained for the CT band in BPy-pTC (ε373 nm = 17310 M−1⋅cm−1) as compared to BPy-p3C (ε365 nm = 14690 M−1⋅cm−1).
![[1860-5397-18-122-1]](/bjoc/content/figures/1860-5397-18-122-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Normalized absorption spectra of BPy-pTC and BPy-p3C in toluene at room temperature; (b) normalized fluorescence (λex = 375 nm, 10 µM in toluene) spectra at ambient temperature and phosphorescence spectra (λex = 375 nm, 10 µM) of BPy-pTC and BPy-p3C at 77 K; emission spectra (λex = 375 nm) of BPy-pTC (c) and BPy-p3C (d) in various polar solvents. (TL: toluene, DIO: 1,4-dioxane, THF: tetrahydrofuran, DCM: dichloromethane).
Figure 1: (a) Normalized absorption spectra of BPy-pTC and BPy-p3C in toluene at room temperature; (b) normal...
Table 1: Photophysical properties of BPy-pTC and BPy-p3C.a
| compound | λem (nm) | τPF (ns) | τDF (µs) | Φtotal (%) | ΔEST (eV) |
| BPy-pTC | 476 | 6.5 | 0.5 | 19 | 0.121 |
| BPy-p3C | 488 | 5.1 | 0.4 | 26 | 0.047 |
aMeasured in degassed toluene (10 µM).
Computational calculations were performed to understand the ground state electronic communication between the donor and acceptor in BPy-pTC and BPy-p3C using the Gaussian 16 program package (Figure S1 as well as Tables S1 and S2 in Supporting Information File 1). The LUMO of both compounds was predominantly located on the acceptor core, whereas the HOMO was distributed mainly over the donor unit for both compounds and extended to the adjacent phenyl π-spacer ring only for BPy-pTC (Figure S1, Supporting Information File 1). As a consequence, a higher oscillator strength (f = 0.213) was obtained for BPy-pTC as compared to BPy-p3C (f = 0.073, Table S1, Supporting Information File 1). Additionally, a 48° and 51° (59° for the second donor and overall D–A dihedral = 97°) D–A dihedral angle was obtained for BPy-pTC and BPy-p3C, respectively (Figure S1, Supporting Information File 1). Therefore, the coplanarity between the D–A linkage in BPy-pTC facilitated the prominent ground-state communication from the donor to the acceptor core, as compared to the twisted BPy-p3C (Figure S1, Supporting Information File 1) [30].
The steady-state emission of both BPy-pTC and BPy-p3C was recorded in different polar media (Figure 1c and Figure 1d). The BPy-pTC emission lay in the cyan region, and the peak was centered at λem = 476 nm in toluene (Figure 1b). The emission spectra of BPy-pTC were red-shifted in polar media, and the emission maxima changed from λem = 476 nm in toluene to λem = 497 nm in THF and to λem = 545 nm in DCM (Figure 1c). Similarly, blue emission was observed in toluene at λem = 488 nm for BPy-p3C, and it red-shifted to λem = 530 nm in THF and to λem = 582 nm in polar DCM (Figure 1d). As compared to the absorption spectra, the significant solvatochromic shifts in the fluorescence spectra suggested the presence of a highly dipolar excited state with a stronger ICT character, in contrast to the ground state of the molecule. But the Stokes shift of BPy-p3C (![[Graphic 1]](/bjoc/content/inline/1860-5397-18-122-i2.svg?max-width=637&scale=1.18182) = 6640 cm−1 in toluene and
= 6640 cm−1 in toluene and ![[Graphic 2]](/bjoc/content/inline/1860-5397-18-122-i3.svg?max-width=637&scale=1.18182) = 9991 cm−1 in DCM) was always higher than that of BPy-pTC (
= 9991 cm−1 in DCM) was always higher than that of BPy-pTC (![[Graphic 3]](/bjoc/content/inline/1860-5397-18-122-i4.svg?max-width=637&scale=1.18182) = 5145 cm−1 in toluene and
= 5145 cm−1 in toluene and ![[Graphic 4]](/bjoc/content/inline/1860-5397-18-122-i5.svg?max-width=637&scale=1.18182) = 7761 cm−1 in DCM) in all polar media, indicating a highly dipolar excited state (Figure 1c and Figure 1d as well as Tables S3 and S4 in Supporting Information File 1). The large Stokes shift and spectral broadening indicated a highly polarized ICT state in both compounds (Figure 1c and Figure 1d as well as Tables S3 and S4 in Supporting Information File 1) [30]. The excited-state CT characteristics were further probed using the Lippert–Mataga plot for both the compounds (Figure S2 and Tables S3–S5 in Supporting Information File 1) [29]. A larger Stokes shift and transient dipole (µE−µG) than in BPy-pTC were observed in BPy-p3C (Table S5, Supporting Information File 1). This indicated the relatively stronger excited-state CT interactions in BPy-p3C (Figure 1c and Figure 1d as well as Tables S3–S5 in Supporting Information File 1).
= 7761 cm−1 in DCM) in all polar media, indicating a highly dipolar excited state (Figure 1c and Figure 1d as well as Tables S3 and S4 in Supporting Information File 1). The large Stokes shift and spectral broadening indicated a highly polarized ICT state in both compounds (Figure 1c and Figure 1d as well as Tables S3 and S4 in Supporting Information File 1) [30]. The excited-state CT characteristics were further probed using the Lippert–Mataga plot for both the compounds (Figure S2 and Tables S3–S5 in Supporting Information File 1) [29]. A larger Stokes shift and transient dipole (µE−µG) than in BPy-pTC were observed in BPy-p3C (Table S5, Supporting Information File 1). This indicated the relatively stronger excited-state CT interactions in BPy-p3C (Figure 1c and Figure 1d as well as Tables S3–S5 in Supporting Information File 1).
The phosphorescence spectra of BPy-pTC and BPy-p3C were recorded in toluene at 77 K (Figure 1b). The structural phosphorescence bands were obtained for both compounds. The nature of the phosphorescence bands indicated the locally excited (3LE) character of the T1 state. The onset of the fluorescence spectrum (energy of S1) in toluene at room temperature and the onset of the phosphorescence spectrum (energy of T1) in toluene at 77 K was chosen to estimate the ΔEST values of the emitters (Figure 1b) [31]. A smaller singlet–triplet splitting was observed for BPy-p3C (ΔEST = 0.047 eV) as compared to BPy-pTC (ΔEST = 0.121 eV) due to the twisted molecular geometry (Figure 1 and Table 1). The small ΔEST values indicated a possible RISC process from the T1 to the S1 state for both the compounds. The photophysical properties of both the emitters in toluene are summarized in Table 1.
Time-resolved spectroscopy
Further, the fluorescence lifetimes on the nanosecond and microsecond timescales of both emitters were recorded in degassed THF to understand the photon upconversion process (Figure 2 and Tables S6 and S7 in Supporting Information File 1). Emission decay on the nanosecond and microsecond timescales was obtained for BPy-pTC, with decay lifetimes of 6.2 ns and 5.8 µs, respectively (Figure 2a as well as Table S6 in Supporting Information File 1). In comparison, a biexponential fluorescence decay with average lifetimes of τavg = 3.5 ns and 6.6 ns was obtained for BPy-p3C (Figure 2b as well as Table S7 in Supporting Information File 1). The short component was due to the prompt fluorescence (PF), and the long component was the delayed fluorescence (DF) [32]. Hence, both emitters displayed prominent DF due to the small ΔEST, facilitating the photon upconversion from the T1 to the S1 state through an RISC process.
![[1860-5397-18-122-2]](/bjoc/content/figures/1860-5397-18-122-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Transient photoluminescence decay (λex = 375 nm) of (a) BPy-pTC and (b) BPy-p3C in degassed THF (10 µM) at room temperature.
Figure 2: Transient photoluminescence decay (λex = 375 nm) of (a) BPy-pTC and (b) BPy-p3C in degassed THF (10...
Aggregation-induced enhanced emission (AIEE)
In order to determine the molecular aggregate formation of BPy-pTC and BPy-p3C and the impact on the optical properties, we carried out aggregation studies in THF/water mixtures (Figure 3) [33]. BPy-pTC emits in the blue-green region at λem = 497 nm in THF with a PLQY of 12.7% (Figure 3a). The emission intensity was reduced to near 0 upon increasing the water fraction from 10 to 70 vol % in the THF solution of BPy-pTC (Figure 3a and Figure 3c). The formation of a dark twisted intramolecular CT state led to fluorescence quenching in the highly polar water/THF mixture (10–70 vol % water, Figure 3a, c, and e) [34-36]. It again started glowing when the water fraction was increased beyond 80 vol % (Figure 3a, c, and e). The formation of molecular aggregates in >80 vol % water/THF mixtures led to the regaining of fluorescence in BPy-pTC (Figure 3a, c, e, and f). A slight blue shift was also observed in 80–90 vol % water/THF mixtures of BPy-pTC due to restriction of D–A rotation in the aggregated state, which led to diminished CT [34]. The nanoaggregates (in 90 vol % water/THF) of BPy-pTC were characterized using scanning electron microscopy (SEM). The SEM image showed the spherical nanoaggregates of BPy-pTC, with 95 nm diameter (Figure 3f). The BPy-p3C compound emitted in the green region at λem = 530 nm in THF, with a PLQY of 3.6% (Figure 3a). Similar to BPy-pTC, for BPy-p3C, a dark state in 10–70 vol % water/THF mixtures and emissive aggregates in 80–90 vol % water/THF mixtures were obtained (Figure 3b, d, and e). Interestingly, AIEE was obtained only for BPy-p3C in a 90 vol % water/THF mixture (Figure 3e).
![[1860-5397-18-122-3]](/bjoc/content/figures/1860-5397-18-122-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: AIEE studies: Emission spectra (λex = 375 nm, 10 µM) of (a) BPy-pTC and (d) BPy-p3C in THF with increasing water fraction (in vol %) at room temperature. Digital photographs of (c) BPy-pTC and (d) BPy-p3C in THF/water solutions under exposure to a UV lamp (λex = 365 nm). (e) Plot of PL intensity vs water fraction of BPy-pTC and BPy-p3C depicting the AIEE phenomenon in BPy-p3C. (f) SEM image of BPy-pTC aggregates formed in a 90 vol % water/THF mixture.
Figure 3: AIEE studies: Emission spectra (λex = 375 nm, 10 µM) of (a) BPy-pTC and (d) BPy-p3C in THF with inc...
The bulky donor group in BPy-p3C led to facile D–A rotation in solution. As a result, we observed the twisted molecular geometry and a lower PLQY of 3.6% as compared to rigid BPy-pTC (PLQY = 12.7%) in THF solution. In contrast to the THF solution, BPy-p3C showed enhanced PLQY from 3.6% to 9.8% in an aggregated state, while BPy-pTC showed reduced PLQY from 12.7% in THF solution to 7.0% in the aggregated state. The enhanced emission in the aggregated state of BPy-p3C as compared to the THF solution was due to the restricted D–A rotation in the aggregated state [21]. As revealed from the photophysical studies and DFT calculations, BPy-p3C has a twisted molecular geometry with a stronger excited-state ICT than BPy-pTC. As a consequence, it facilitates nonradiative deactivation pathways in solution due to molecular vibrations and facile D–A rotation, as compared to rigid BPy-pTC (strong ground-state CT interactions render the D–A complex rigid). In comparison, due to the ground-state D–A communication on the basis of a +R effect, the weak excited-state CT interactions and rigid molecular geometry in BPy-pTC led to the regain of fluorescence in the aggregated state [30]. However, the fluorescence intensity of aggregates of BPy-pTC did not reach beyond the native THF solution. Another factor that may have been responsible for this were the π–π-stacking interactions in the aggregated or solid phase of BPy-pTC as compared to twisted BPy-p3C [36].
To determine the effect of aggregation on the spin-flipping process (indicated by kRISC) from the low-lying triplet state T1 to the singlet excited state S1, we recorded the fluorescence and phosphorescence spectra of aggregates (90 vol % water/THF) at room temperature and 77 K, respectively (Figure 4). The structural phosphorescence band was obtained from the aggregates in 90 vol % water/THF mixtures (Figure 4). Additionally, the phosphorescence spectra of both compounds were recorded in THF at 77 K (Figure S3 in Supporting Information File 1). The retention of the structural phosphorescence band in THF indicates its origin from the locally excited triplet state 3LE. The ΔEST values calculated based on the onset of the fluorescence and the phosphorescence spectra of BPy-pTC and BPy-p3C in 90 vol % water/THF were 0.06 and −0.047 eV, respectively (Figure 4). The ΔEST values were lower in the aggregated state than in solution for both compounds (ΔEST in toluene for BPy-pTC and BPy-p3C = 0.121 and 0.047 eV, respectively), which indicated aggregation-induced RISC boosting. BPy-p3C showed a negative ΔEST value because the phosphorescence emission came from the locally excited triplet states (3LE-structured emission), not from the 3CT state [37]. This low ΔEST value facilitates fast spin-flipping in the aggregated state. Consequently, a fast kRISC rate (0.74·105 s−1 for BPy-pTC and 2.06·105 s−1 for BPy-p3C) was obtained in the aggregated state (Table 2). Interestingly, a higher reduction of ΔEST as compared to BPy-pTC was obtained for the aggregates of BPy-p3C. This could have been due to the locking of the twisted molecular geometry in the aggregated state and more dipolar excited states in the highly polar water/THF mixtures.
![[1860-5397-18-122-4]](/bjoc/content/figures/1860-5397-18-122-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Normalized fluorescence at room temperature and phosphorescence spectra at 77 K (λex = 375 nm, 10 µM) of (a) BPy-pTC and (b) BPy-p3C aggregates in 90 vol % water/THF mixture.
Figure 4: Normalized fluorescence at room temperature and phosphorescence spectra at 77 K (λex = 375 nm, 10 µ...
Table 2: Photophysical properties of BPy-pTC and BPy-p3C aggregates.
| compound | sample | λema (nm) | ΔESTa (eV) | τPF (ns) | τDF (µs) | Φtotal (%) | ΦPF (%) | ΦDF (%) |
kr
(107 s−1) |
kISC
(107 s−1) |
kRISC
(105 s−1) |
knr, S
(107 s−1) |
| BPy-pTC | 90 vol % water/THF | 496 | 0.06 | 9.0 | 19.1 | 7.0 | 4.8 | 2.2 | 0.5 | 0.36 | 0.74 | 6.9 |
| BPy-p3C | 90 vol % water/THF | 505 | −0.047 | 19.2 | 6.8 | 9.8 | 6.8 | 3.3 | 0.4 | 0.18 | 2.06 | 3.2 |
aMeasured in 90 vol % water/THF mixture.
Further, the PF and DF lifetimes of the aggregates of BPy-pTC and BPy-p3C were measured in degassed solutions (Figure 5 and Table 2). We found an over 1.5- and 3-fold enhancement of the PF and DF lifetimes, respectively, for BPy-pTC in the aggregated state as compared to the THF solutions (Figure 5a and Table 2). However, only the PF lifetime was enhanced in BPy-p3C (3.5-fold), and no significant change was obtained in the DF lifetime of the aggregates (Figure 5b and Table 2). Therefore, it is evident that the aggregation helped to suppress the fluorescence quenching as well as to boost the DF by reducing the ΔEST in BPy-pTC. At the same time, the significant enhancement of the PF lifetime was due to the molecular rigidification and restriction of D–A rotation in BPy-p3C aggregates. Thus, a judicious choice of D–A molecular architecture is necessary to tune the solid- or aggregated-state optical properties for triplet harvesting.
![[1860-5397-18-122-5]](/bjoc/content/figures/1860-5397-18-122-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Transient photoluminescence decay (λex = 375 nm, 20 µM) of (a) BPy-pTC and (b) BPy-p3C aggregates in 90 vol % water/THF mixture.
Figure 5: Transient photoluminescence decay (λex = 375 nm, 20 µM) of (a) BPy-pTC and (b) BPy-p3C aggregates i...
Fluorescence switching
The lone-pair electrons of heteroatoms, such as oxygen and nitrogen, are susceptible to acidic protons [22]. BPy-pTC and BPy-p3C exhibit lone-pair electrons at the pyridinyl nitrogen atom and show prominent emission in the solid and aggregated states. We therefore demonstrated the switching of fluorescence using acid and base vapors in neat film (Figure 6a and Figure 6c). Both emitters exhibited switching of the fluorescence responses upon exposure to acidic and basic fumes on neat film (Figure 6a and Figure 6c). The intense green emission of the BPy-pTC film turned dark upon exposure to trifluoroacetic acid (TFA) vapor for 4 s (Figure 6a). The green fluorescence reappeared after neutralization with triethylamine (TEA) vapor for 6 s (Figure 6a). The reversible color switching was upheld with fatigue resistance for multiple cycles. Similarly, the film of BPy-p3C showed reversible changes of the fluorescence from green to dark upon successive exposure to TFA and TEA vapors (Figure 6c).
![[1860-5397-18-122-6]](/bjoc/content/figures/1860-5397-18-122-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Fluorescence switching by acid and base fumes exposure: Emission spectra (λex = 375 nm) of (a) BPy-pTC and (c) BPy-p3C in neat film upon exposure to TFA and TEA vapor at room temperature. Fluorescence switching by addition of TFA and KOH to (b) BPy-pTC and (d) BPy-p3C in toluene solution.
Figure 6: Fluorescence switching by acid and base fumes exposure: Emission spectra (λex = 375 nm) of (a) BPy-p...
To understand the effect of an acidic solution on the optical properties, we carried out the same experiment in toluene solution (Figure 6b and Figure 6d). The protonation of the pyridinyl nitrogen atom upon addition of TFA quenched the fluorescence of both compounds (Figure 6b and Figure 6d). In turn, neutralization of the toluene solution by TEA or KOH addition led to the regain of fluorescence (Figure 6b and Figure 6d). Further, the Stern–Volmer plots were analyzed for both compounds to obtain the quenching and recovery constants (Figures S4 and S5 in Supporting Information File 1). The linear fitted Stern–Volmer plots for quenching and recovery were obtained for both compounds upon addition of acid and base. Thereby, the protonation–deprotonation events were confirmed using absorption spectroscopy (Figure S6, Supporting Information File 1). The new peak at λabs = 425 nm in the absorption spectrum of BPy-pTC upon adding TFA, indicated the protonation of the pyridinyl nitrogen atom (Figure S6a, Supporting Information File 1). This enhanced the electron-deficient character of the BPy acceptor core and the ground-state communication between the donor and acceptor units. As a result, the red-shifted CT absorption band in the acidic medium was obtained (Figure S6a in Supporting Information File 1). The absorption spectra upon addition of TEA again matched with the absorption spectra in toluene (Figure S6a, Supporting Information File 1).
Further, TDDFT calculations for a protonated pyridinyl nitrogen atom (N–H) and carbonyl oxygen atom (CO–H) were performed in comparison to neutral BPy-pTC to determine the protonation site (Figure S7 in Supporting Information File 1). The computed absorption band of BPy-pTC and protonated pyridinyl resembled the change observed experimentally (Figures S6 and S7 in Supporting Information File 1). Additionally, the 1H NMR study upon the addition of TFA in BPy-pTC suggested protonation of the pyridinyl nitrogen atom (Figure S8, Supporting Information File 1). Thus, the facile protonation of the pyridinyl nitrogen atom affected the optical properties of both compounds. The reversible change of absorption and emission by successive addition of acid and base in solution as well as in the solid state indicated that both compounds are suitable for acid sensing in solution and in the solid phase. Reversible fluorescence for thin emitter films was observed in the presence of TFA and TEA. Essentially, quenching occurs in the presence of acid and regaining of the fluorescence intensity happens under basic conditions (Figure 7).
![[1860-5397-18-122-7]](/bjoc/content/figures/1860-5397-18-122-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Fluorescence intensity vs number of exposures for (a) BPy-p3C and (b) BPy-pTC thin films upon exposure to TFA and TEA vapors.
Figure 7: Fluorescence intensity vs number of exposures for (a) BPy-p3C and (b) BPy-pTC thin films upon expos...
Conclusion
In summary, we designed and synthesized two D–A-based TADF emitters, BPy-pTC and BPy-p3C. Both emitters showed excited-state ICT characteristics, leading to positive solvatochromism in solution. The Lippert–Mataga plot and DFT calculations indicated that BPy-p3C had more excited-state CT properties than BPy-pTC due to the bulkier donor group. Both compounds displayed strong emission in the aggregated state in a highly polar medium (80–90 vol % water/THF mixtures). As compared to the native solution, BPy-p3C showed AIEE. Moreover, as compared to the solution, ΔEST was reduced in the aggregated form. Consequently, a fast RISC rate was obtained for the aggregates of both compounds. Further, these two TADF emitters were used to demonstrate solid-state fluorescence switching upon exposure to TFA and TEA vapors. The current study helps to understand the enhancement of DF in the aggregated state, which is important for the fabrication of efficient OLED devices and reversible switching of fluorescence by acid–base exposure.
Supporting Information
| Supporting Information File 1: General information, synthesis and characterization data including NMR spectra, computational details, UV–vis data, Lippert–Mataga plot, and acid–base switching. | ||
| Format: PDF | Size: 1.4 MB | Download |
Funding
A. K. M. is grateful to the Prime Minister's Research Fellowship (PMRF), Ministry of Education, Government of India (Application No: PMRF-2122-3210) for fellowship support, and G. P. N. thanks the Council of Scientific & Industrial Research (CSIR) India for the Junior Research Fellowship (JRF). Bahadur thanks the Indian Institute of Science (IISc) Bangalore, India, for C. V. Raman Fellowship. U. D. and N. Y. thank IISc for doctoral fellowship. P. R. thanks IISc for generous financial support and the Science & Engineering Research Board (SERB), India, for the SERB-Power Grant (SPG, Grant No: SPG/2020/000107). P. R. also acknowledges Rekha Rao Young Investigator award (2022-24) for the financial support.
References
-
Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M. R. Nat. Rev. Mater. 2018, 3, 18020. doi:10.1038/natrevmats.2018.20
Return to citation in text: [1] -
Data, P.; Takeda, Y. Chem. – Asian J. 2019, 14, 1613–1636. doi:10.1002/asia.201801791
Return to citation in text: [1] [2] -
Rajamalli, P.; Senthilkumar, N.; Huang, P.-Y.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. J. Am. Chem. Soc. 2017, 139, 10948–10951. doi:10.1021/jacs.7b03848
Return to citation in text: [1] -
Wong, M. Y.; Zysman-Colman, E. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1605444. doi:10.1002/adma.201605444
Return to citation in text: [1] -
Zhou, Y.; Qin, W.; Du, C.; Gao, H.; Zhu, F.; Liang, G. Angew. Chem., Int. Ed. 2019, 58, 12102–12106. doi:10.1002/anie.201906312
Return to citation in text: [1] -
Xiong, X.; Song, F.; Wang, J.; Zhang, Y.; Xue, Y.; Sun, L.; Jiang, N.; Gao, P.; Tian, L.; Peng, X. J. Am. Chem. Soc. 2014, 136, 9590–9597. doi:10.1021/ja502292p
Return to citation in text: [1] -
Zhang, K. Y.; Yu, Q.; Wei, H.; Liu, S.; Zhao, Q.; Huang, W. Chem. Rev. 2018, 118, 1770–1839. doi:10.1021/acs.chemrev.7b00425
Return to citation in text: [1] -
Sk, B.; Sharma, S.; James, A.; Kundu, S.; Patra, A. J. Mater. Chem. C 2020, 8, 12943–12950. doi:10.1039/d0tc02520h
Return to citation in text: [1] -
Cafferty, B. J.; Ten, A. S.; Fink, M. J.; Morey, S.; Preston, D. J.; Mrksich, M.; Whitesides, G. M. ACS Cent. Sci. 2019, 5, 911–916. doi:10.1021/acscentsci.9b00210
Return to citation in text: [1] -
Paisley, N. R.; Tonge, C. M.; Hudson, Z. M. Front. Chem. (Lausanne, Switz.) 2020, 8, 229. doi:10.3389/fchem.2020.00229
Return to citation in text: [1] -
Huang, L.; Qian, C.; Ma, Z. Chem. – Eur. J. 2020, 26, 11914–11930. doi:10.1002/chem.202000526
Return to citation in text: [1] -
Förster, T.; Kasper, M. Z. Phys. Chem. 1954, 1, 275–277. doi:10.1524/zpch.1954.1.5_6.275
Return to citation in text: [1] -
Ma, X.; Sun, R.; Cheng, J.; Liu, J.; Gou, F.; Xiang, H.; Zhou, X. J. Chem. Educ. 2016, 93, 345–350. doi:10.1021/acs.jchemed.5b00483
Return to citation in text: [1] -
Li, Q.; Li, Z. Adv. Sci. 2017, 4, 1600484. doi:10.1002/advs.201600484
Return to citation in text: [1] -
Sasaki, S.; Drummen, G. P. C.; Konishi, G.-i. J. Mater. Chem. C 2016, 4, 2731–2743. doi:10.1039/c5tc03933a
Return to citation in text: [1] -
Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Nature 2012, 492, 234–238. doi:10.1038/nature11687
Return to citation in text: [1] [2] -
Rajamalli, P.; Thangaraji, V.; Senthilkumar, N.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. J. Mater. Chem. C 2017, 5, 2919–2926. doi:10.1039/c7tc00457e
Return to citation in text: [1] [2] -
Tao, Y.; Yuan, K.; Chen, T.; Xu, P.; Li, H.; Chen, R.; Zheng, C.; Zhang, L.; Huang, W. Adv. Mater. (Weinheim, Ger.) 2014, 26, 7931–7958. doi:10.1002/adma.201402532
Return to citation in text: [1] -
Tsujimoto, H.; Ha, D.-G.; Markopoulos, G.; Chae, H. S.; Baldo, M. A.; Swager, T. M. J. Am. Chem. Soc. 2017, 139, 4894–4900. doi:10.1021/jacs.7b00873
Return to citation in text: [1] -
Chen, H.; Liu, H.; Xiong, Y.; He, J.; Zhao, Z.; Tang, B. Z. Mater. Chem. Front. 2022, 6, 924–932. doi:10.1039/d1qm01625c
Return to citation in text: [1] -
Yang, Z.; Qin, W.; Lam, J. W. Y.; Chen, S.; Sung, H. H. Y.; Williams, I. D.; Tang, B. Z. Chem. Sci. 2013, 4, 3725–3730. doi:10.1039/c3sc50648g
Return to citation in text: [1] [2] [3] [4] -
Steinegger, A.; Wolfbeis, O. S.; Borisov, S. M. Chem. Rev. 2020, 120, 12357–12489. doi:10.1021/acs.chemrev.0c00451
Return to citation in text: [1] [2] [3] [4] -
Fan, S.; Dennison, G. H.; FitzGerald, N.; Burn, P. L.; Gentle, I. R.; Shaw, P. E. Commun. Chem. 2021, 4, 45. doi:10.1038/s42004-021-00482-6
Return to citation in text: [1] -
Chen, L.; Fu, P.-Y.; Wang, H.-P.; Pan, M. Adv. Opt. Mater. 2021, 9, 2001952. doi:10.1002/adom.202001952
Return to citation in text: [1] -
Zhang, Q.; Yang, L.; Han, Y.; Wang, Z.; Li, H.; Sun, S.; Xu, Y. Chem. Eng. J. 2022, 428, 130986. doi:10.1016/j.cej.2021.130986
Return to citation in text: [1] -
Putra, A. U.; Çakmaz, D.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Beilstein J. Org. Chem. 2020, 16, 2282–2296. doi:10.3762/bjoc.16.189
Return to citation in text: [1] -
Rajamalli, P.; Senthilkumar, N.; Gandeepan, P.; Ren-Wu, C.-Z.; Lin, H.-W.; Cheng, C.-H. J. Mater. Chem. C 2016, 4, 900–904. doi:10.1039/c5tc03943f
Return to citation in text: [1] -
Rajamalli, P.; Senthilkumar, N.; Gandeepan, P.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. ACS Appl. Mater. Interfaces 2016, 8, 27026–27034. doi:10.1021/acsami.6b10678
Return to citation in text: [1] [2] -
Lakowicz, J. R., Ed. Principles of fluorescence spectroscopy, 3rd ed.; Springer Science and Business Media: New York, NY, USA, 2006. doi:10.1007/978-0-387-46312-4
Return to citation in text: [1] [2] -
Sk, B.; Khodia, S.; Patra, A. Chem. Commun. 2018, 54, 1786–1789. doi:10.1039/c7cc09261j
Return to citation in text: [1] [2] [3] -
Ward, J. S.; Kukhta, N. A.; dos Santos, P. L.; Congrave, D. G.; Batsanov, A. S.; Monkman, A. P.; Bryce, M. R. Chem. Mater. 2019, 31, 6684–6695. doi:10.1021/acs.chemmater.9b01184
Return to citation in text: [1] -
Sk, B.; Ravindran, E.; Deori, U.; Yadav, N.; Nanda, G. P.; Rajamalli, P. J. Mater. Chem. C 2022, 10, 4886–4893. doi:10.1039/d1tc05027c
Return to citation in text: [1] -
Thulaseedharan Nair Sailaja, S.; Maisuls, I.; Kösters, J.; Hepp, A.; Faust, A.; Voskuhl, J.; Strassert, C. A. Beilstein J. Org. Chem. 2020, 16, 2960–2970. doi:10.3762/bjoc.16.246
Return to citation in text: [1] -
Wang, J.; Zhang, J.; Jiang, C.; Yao, C.; Xi, X. ACS Appl. Mater. Interfaces 2021, 13, 57713–57724. doi:10.1021/acsami.1c17449
Return to citation in text: [1] [2] -
Wang, Y.-F.; Liu, X.; Zhu, Y.; Li, M.; Chen, C.-F. J. Mater. Chem. C 2022, 10, 4805–4812. doi:10.1039/d1tc04893g
Return to citation in text: [1] -
Wang, J.; Yang, Y.; Jiang, C.; He, M.; Yao, C.; Zhang, J. J. Mater. Chem. C 2022, 10, 3163–3171. doi:10.1039/d1tc05497j
Return to citation in text: [1] [2] -
Noda, H.; Nakanotani, H.; Adachi, C. Sci. Adv. 2018, 4, eaao6910. doi:10.1126/sciadv.aao6910
Return to citation in text: [1]
| 34. | Wang, J.; Zhang, J.; Jiang, C.; Yao, C.; Xi, X. ACS Appl. Mater. Interfaces 2021, 13, 57713–57724. doi:10.1021/acsami.1c17449 |
| 35. | Wang, Y.-F.; Liu, X.; Zhu, Y.; Li, M.; Chen, C.-F. J. Mater. Chem. C 2022, 10, 4805–4812. doi:10.1039/d1tc04893g |
| 36. | Wang, J.; Yang, Y.; Jiang, C.; He, M.; Yao, C.; Zhang, J. J. Mater. Chem. C 2022, 10, 3163–3171. doi:10.1039/d1tc05497j |
| 34. | Wang, J.; Zhang, J.; Jiang, C.; Yao, C.; Xi, X. ACS Appl. Mater. Interfaces 2021, 13, 57713–57724. doi:10.1021/acsami.1c17449 |
| 21. | Yang, Z.; Qin, W.; Lam, J. W. Y.; Chen, S.; Sung, H. H. Y.; Williams, I. D.; Tang, B. Z. Chem. Sci. 2013, 4, 3725–3730. doi:10.1039/c3sc50648g |
| 1. | Liu, Y.; Li, C.; Ren, Z.; Yan, S.; Bryce, M. R. Nat. Rev. Mater. 2018, 3, 18020. doi:10.1038/natrevmats.2018.20 |
| 2. | Data, P.; Takeda, Y. Chem. – Asian J. 2019, 14, 1613–1636. doi:10.1002/asia.201801791 |
| 3. | Rajamalli, P.; Senthilkumar, N.; Huang, P.-Y.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. J. Am. Chem. Soc. 2017, 139, 10948–10951. doi:10.1021/jacs.7b03848 |
| 4. | Wong, M. Y.; Zysman-Colman, E. Adv. Mater. (Weinheim, Ger.) 2017, 29, 1605444. doi:10.1002/adma.201605444 |
| 9. | Cafferty, B. J.; Ten, A. S.; Fink, M. J.; Morey, S.; Preston, D. J.; Mrksich, M.; Whitesides, G. M. ACS Cent. Sci. 2019, 5, 911–916. doi:10.1021/acscentsci.9b00210 |
| 21. | Yang, Z.; Qin, W.; Lam, J. W. Y.; Chen, S.; Sung, H. H. Y.; Williams, I. D.; Tang, B. Z. Chem. Sci. 2013, 4, 3725–3730. doi:10.1039/c3sc50648g |
| 22. | Steinegger, A.; Wolfbeis, O. S.; Borisov, S. M. Chem. Rev. 2020, 120, 12357–12489. doi:10.1021/acs.chemrev.0c00451 |
| 24. | Chen, L.; Fu, P.-Y.; Wang, H.-P.; Pan, M. Adv. Opt. Mater. 2021, 9, 2001952. doi:10.1002/adom.202001952 |
| 25. | Zhang, Q.; Yang, L.; Han, Y.; Wang, Z.; Li, H.; Sun, S.; Xu, Y. Chem. Eng. J. 2022, 428, 130986. doi:10.1016/j.cej.2021.130986 |
| 8. | Sk, B.; Sharma, S.; James, A.; Kundu, S.; Patra, A. J. Mater. Chem. C 2020, 8, 12943–12950. doi:10.1039/d0tc02520h |
| 26. | Putra, A. U.; Çakmaz, D.; Seferoğlu, N.; Barsella, A.; Seferoğlu, Z. Beilstein J. Org. Chem. 2020, 16, 2282–2296. doi:10.3762/bjoc.16.189 |
| 6. | Xiong, X.; Song, F.; Wang, J.; Zhang, Y.; Xue, Y.; Sun, L.; Jiang, N.; Gao, P.; Tian, L.; Peng, X. J. Am. Chem. Soc. 2014, 136, 9590–9597. doi:10.1021/ja502292p |
| 7. | Zhang, K. Y.; Yu, Q.; Wei, H.; Liu, S.; Zhao, Q.; Huang, W. Chem. Rev. 2018, 118, 1770–1839. doi:10.1021/acs.chemrev.7b00425 |
| 19. | Tsujimoto, H.; Ha, D.-G.; Markopoulos, G.; Chae, H. S.; Baldo, M. A.; Swager, T. M. J. Am. Chem. Soc. 2017, 139, 4894–4900. doi:10.1021/jacs.7b00873 |
| 20. | Chen, H.; Liu, H.; Xiong, Y.; He, J.; Zhao, Z.; Tang, B. Z. Mater. Chem. Front. 2022, 6, 924–932. doi:10.1039/d1qm01625c |
| 5. | Zhou, Y.; Qin, W.; Du, C.; Gao, H.; Zhu, F.; Liang, G. Angew. Chem., Int. Ed. 2019, 58, 12102–12106. doi:10.1002/anie.201906312 |
| 21. | Yang, Z.; Qin, W.; Lam, J. W. Y.; Chen, S.; Sung, H. H. Y.; Williams, I. D.; Tang, B. Z. Chem. Sci. 2013, 4, 3725–3730. doi:10.1039/c3sc50648g |
| 22. | Steinegger, A.; Wolfbeis, O. S.; Borisov, S. M. Chem. Rev. 2020, 120, 12357–12489. doi:10.1021/acs.chemrev.0c00451 |
| 23. | Fan, S.; Dennison, G. H.; FitzGerald, N.; Burn, P. L.; Gentle, I. R.; Shaw, P. E. Commun. Chem. 2021, 4, 45. doi:10.1038/s42004-021-00482-6 |
| 16. | Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Nature 2012, 492, 234–238. doi:10.1038/nature11687 |
| 17. | Rajamalli, P.; Thangaraji, V.; Senthilkumar, N.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. J. Mater. Chem. C 2017, 5, 2919–2926. doi:10.1039/c7tc00457e |
| 16. | Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Nature 2012, 492, 234–238. doi:10.1038/nature11687 |
| 37. | Noda, H.; Nakanotani, H.; Adachi, C. Sci. Adv. 2018, 4, eaao6910. doi:10.1126/sciadv.aao6910 |
| 15. | Sasaki, S.; Drummen, G. P. C.; Konishi, G.-i. J. Mater. Chem. C 2016, 4, 2731–2743. doi:10.1039/c5tc03933a |
| 18. | Tao, Y.; Yuan, K.; Chen, T.; Xu, P.; Li, H.; Chen, R.; Zheng, C.; Zhang, L.; Huang, W. Adv. Mater. (Weinheim, Ger.) 2014, 26, 7931–7958. doi:10.1002/adma.201402532 |
| 22. | Steinegger, A.; Wolfbeis, O. S.; Borisov, S. M. Chem. Rev. 2020, 120, 12357–12489. doi:10.1021/acs.chemrev.0c00451 |
| 12. | Förster, T.; Kasper, M. Z. Phys. Chem. 1954, 1, 275–277. doi:10.1524/zpch.1954.1.5_6.275 |
| 13. | Ma, X.; Sun, R.; Cheng, J.; Liu, J.; Gou, F.; Xiang, H.; Zhou, X. J. Chem. Educ. 2016, 93, 345–350. doi:10.1021/acs.jchemed.5b00483 |
| 14. | Li, Q.; Li, Z. Adv. Sci. 2017, 4, 1600484. doi:10.1002/advs.201600484 |
| 30. | Sk, B.; Khodia, S.; Patra, A. Chem. Commun. 2018, 54, 1786–1789. doi:10.1039/c7cc09261j |
| 10. | Paisley, N. R.; Tonge, C. M.; Hudson, Z. M. Front. Chem. (Lausanne, Switz.) 2020, 8, 229. doi:10.3389/fchem.2020.00229 |
| 11. | Huang, L.; Qian, C.; Ma, Z. Chem. – Eur. J. 2020, 26, 11914–11930. doi:10.1002/chem.202000526 |
| 2. | Data, P.; Takeda, Y. Chem. – Asian J. 2019, 14, 1613–1636. doi:10.1002/asia.201801791 |
| 36. | Wang, J.; Yang, Y.; Jiang, C.; He, M.; Yao, C.; Zhang, J. J. Mater. Chem. C 2022, 10, 3163–3171. doi:10.1039/d1tc05497j |
| 28. | Rajamalli, P.; Senthilkumar, N.; Gandeepan, P.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. ACS Appl. Mater. Interfaces 2016, 8, 27026–27034. doi:10.1021/acsami.6b10678 |
| 17. | Rajamalli, P.; Thangaraji, V.; Senthilkumar, N.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. J. Mater. Chem. C 2017, 5, 2919–2926. doi:10.1039/c7tc00457e |
| 27. | Rajamalli, P.; Senthilkumar, N.; Gandeepan, P.; Ren-Wu, C.-Z.; Lin, H.-W.; Cheng, C.-H. J. Mater. Chem. C 2016, 4, 900–904. doi:10.1039/c5tc03943f |
| 21. | Yang, Z.; Qin, W.; Lam, J. W. Y.; Chen, S.; Sung, H. H. Y.; Williams, I. D.; Tang, B. Z. Chem. Sci. 2013, 4, 3725–3730. doi:10.1039/c3sc50648g |
| 22. | Steinegger, A.; Wolfbeis, O. S.; Borisov, S. M. Chem. Rev. 2020, 120, 12357–12489. doi:10.1021/acs.chemrev.0c00451 |
| 32. | Sk, B.; Ravindran, E.; Deori, U.; Yadav, N.; Nanda, G. P.; Rajamalli, P. J. Mater. Chem. C 2022, 10, 4886–4893. doi:10.1039/d1tc05027c |
| 33. | Thulaseedharan Nair Sailaja, S.; Maisuls, I.; Kösters, J.; Hepp, A.; Faust, A.; Voskuhl, J.; Strassert, C. A. Beilstein J. Org. Chem. 2020, 16, 2960–2970. doi:10.3762/bjoc.16.246 |
| 29. | Lakowicz, J. R., Ed. Principles of fluorescence spectroscopy, 3rd ed.; Springer Science and Business Media: New York, NY, USA, 2006. doi:10.1007/978-0-387-46312-4 |
| 31. | Ward, J. S.; Kukhta, N. A.; dos Santos, P. L.; Congrave, D. G.; Batsanov, A. S.; Monkman, A. P.; Bryce, M. R. Chem. Mater. 2019, 31, 6684–6695. doi:10.1021/acs.chemmater.9b01184 |
| 30. | Sk, B.; Khodia, S.; Patra, A. Chem. Commun. 2018, 54, 1786–1789. doi:10.1039/c7cc09261j |
| 30. | Sk, B.; Khodia, S.; Patra, A. Chem. Commun. 2018, 54, 1786–1789. doi:10.1039/c7cc09261j |
| 28. | Rajamalli, P.; Senthilkumar, N.; Gandeepan, P.; Ren-Wu, C.-C.; Lin, H.-W.; Cheng, C.-H. ACS Appl. Mater. Interfaces 2016, 8, 27026–27034. doi:10.1021/acsami.6b10678 |
| 29. | Lakowicz, J. R., Ed. Principles of fluorescence spectroscopy, 3rd ed.; Springer Science and Business Media: New York, NY, USA, 2006. doi:10.1007/978-0-387-46312-4 |
© 2022 Mazumdar et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.
