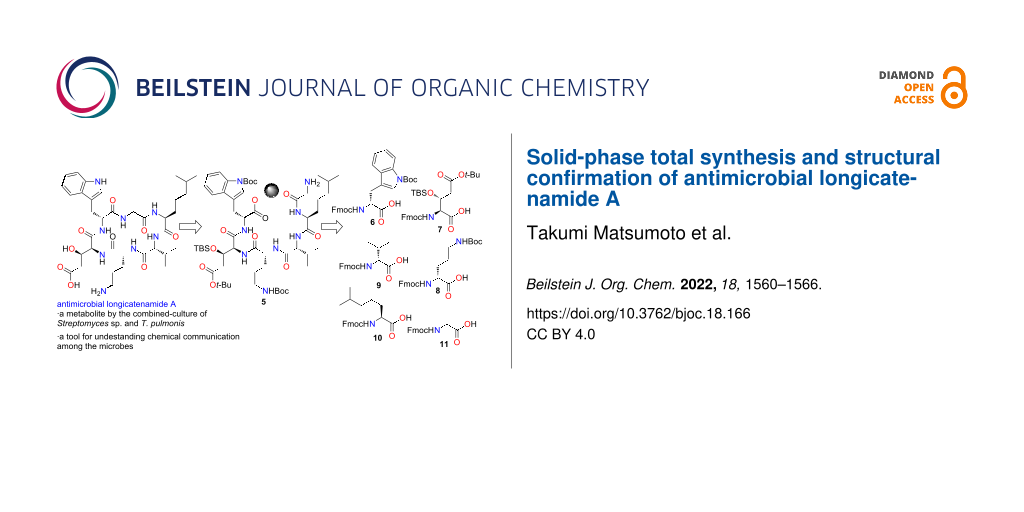
Abstract
Longicatenamides A–D are cyclic hexapeptides isolated from the combined culture of Streptomyces sp. KUSC_F05 and Tsukamurella pulmonis TP-B0596. Because these peptides are not detected in the monoculture broth of the actinomycete, they are key tools for understanding chemical communication in the microbial world. Herein, we report the solid-phase total synthesis and structural confirmation of longicatenamide A. First, commercially unavailable building blocks were chemically synthesized with stereocontrol. Second, the peptide chain was elongated via Fmoc-based solid-phase peptide synthesis. Third, the peptide chain was cyclized in the solution phase, followed by simultaneous cleavage of all protecting groups to afford longicatenamide A. Chromatographic analysis corroborated the chemical structure of longicatenamide A. Furthermore, the antimicrobial activity of synthesized longicatenamide A was confirmed. The developed solid-phase synthesis is expected to facilitate the rapid synthesis of diverse synthetic analogues.

Graphical Abstract
Introduction
Naturally occurring bioactive compounds can serve as both drug leads and research tools for chemical biology [1]. Because the rediscovery rate of these compounds has increased in the last few decades, new approaches to explore natural products are in demand [2]. To this end, the combined-culture strategy has been applied to discover new natural products. For example, the mycolic acid-containing bacterium Tsukamurella pulmonis TP-B0596 can influence the biosynthesis of cryptic natural products [3]. Additionally, we have developed several highly sensitive labeling reagents to detect and identify scarce and cryptic natural products [4]. Integrating the combined-culture strategy and new labeling reagents has led to the detection and structural determination of several unprecedented secondary metabolites [5-7].
Longicatenamides A–D (1–4, Figure 1) are cyclic hexapeptides isolated from the combined-culture of Streptomyces sp. KUSC_F05 and T. pulmonis TP-B0596 [8]. The planar structures were determined by analyzing two-dimensional (2D) nuclear magnetic resonance (NMR) spectra and mass spectrometry (MS) data, and the absolute configurations of their component amino acids were elucidated by using highly sensitive reagents that we recently developed [4]. Among the isolated longicatenamides, compound 1 exhibits weak but preferential antimicrobial activity against Bacillus subtilis. Because peptides 1–4 are not detected in the monoculture broth of Streptomyces sp. KUSC_F05, they are key tools for understanding chemical communication in the microbial world. To elucidate the role of compounds 1–4 in the microbial world, developing a strategy to synthesize compounds 1–4, including future derivatization to produce probe molecules, is required. Herein, we report the total synthesis of peptide 1 by Fmoc-based solid-phase peptide synthesis [9] and the evaluation of its antimicrobial activity. The present study confirmed our proposed structure of 1, which was determined by the use of our original labeling reagents [4].
![[1860-5397-18-166-1]](/bjoc/content/figures/1860-5397-18-166-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of longicatenamides A–D (1–4).
Figure 1: Structures of longicatenamides A–D (1–4).
Results and Discussion
Although the solution-phase total synthesis of an analogue of longicatenamycin A has been reported [10], a solid-phase strategy can facilitate the production of a wider variety of analogous compounds than solution-phase synthesis [11]. Consequently, in this study, compound 1 was synthesized using a solid-phase strategy for future derivatization to produce probe molecules for deciphering microbial chemical communication.
The retrosynthesis of peptide 1 is displayed in Scheme 1. First, the cyclic peptide 1 was linearized by retrosynthesis, and acid-labile protecting groups were attached onto the reactive side chain. The biomimetic synthesis of cyclic peptides often enables efficient synthesis [12,13] and provides insights into the biosynthesis pathways of these peptides [14]. However, the biosynthetic gene clusters of compounds 1–4 remain unidentified. Therefore, the least sterically hindered amine, namely the amino group of glycine, was selected as a nucleophile of the cyclization reaction in this study. Second, to realize the solid-phase synthesis, the C-terminus of the linear peptide was connected to 2-chlorotrityl resin [15] to give resin-bound peptide 5. Peptide 5 was divided into six building blocks 6–11 by retrosynthesis.
![[1860-5397-18-166-i1]](/bjoc/content/inline/1860-5397-18-166-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthesis of longicatenamycin A (1).
Scheme 1: Retrosynthesis of longicatenamycin A (1).
At the beginning of the total synthesis, commercially unavailable building blocks 7 and 10 were chemically constructed from readily available starting materials. The synthesis of building block 10 commenced with the synthesis of compound 15 through Wittig reaction of Garner’s aldehyde (13) [16], which was readily obtained from tert-butyloxycarbonyl (Boc)-protected ᴅ-serine 12 (Scheme 2). Treatment of the olefin 15 with trifluoroacetic acid (TFA) cleaved the Boc protecting group and the acetonide to deliver unsaturated amino alcohol 16. The amino group in 16 was protected by the fluorenylmethyloxycarbonyl (Fmoc) protecting group for solid-phase peptide synthesis, and then hydrogenation of the double bond in 17 provided intermediate 18. Oxidation of the alcohol 18 to acid 10 was realized with the combination of Dess–Martin oxidation [17,18] and Pinnick oxidation [19].
![[1860-5397-18-166-i2]](/bjoc/content/inline/1860-5397-18-166-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of building block 10.
Scheme 2: Synthesis of building block 10.
Another unusual amino acid 7 was also synthesized from ᴅ-serine (20, Scheme 3). The SnCl2-catalyzed coupling reaction [20] between 21 and 22 afforded β-keto ester 23, which was then reduced to the corresponding β-hydroxy ester 24 by K-Selectride (dr > 20:1), and subsequent acidic removal of the acetonide furnished diol 25. The stereochemistry of the newly generated hydroxy group was determined using the modified Mosher’s method [21]. Protection of diol 25 by tert-butyl(dimethyl)silyl (TBS) group followed by selective deprotection of the primary alcohol led to 27. Finally, acid 7 was obtained from alcohol 27 through the same two-step oxidation used to obtain compound 10.
![[1860-5397-18-166-i3]](/bjoc/content/inline/1860-5397-18-166-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Having synthesized the necessary building blocks, we turned our attention to construct resin-bound peptide 5 (Scheme 4). The assembly of this hexapeptide started with the loading of Fmoc-ᴅ-Trp(Boc)-OH (6) onto 2-chlorotrityl resin with iPr2NEt, which was followed by piperidine treatment to liberate resin-bound amine 29. Then, five rounds of DIC/Oxyma-mediated amidation [22] and Nα-deprotection with piperidine led to resin-bound peptide 5. Treatment of 5 with TFA/CH2Cl2 1:99 released 30 into the solution without unmasking the acid-labile protecting groups of the side chains. Subsequently, peptide 30 was cyclized by the action of PyBOP/HOAt [23,24] followed by treatment with TFA/iPr3SiH/H2O 95:2.5:2.5 to provide crude 1. After reversed-phase high-performance liquid chromatography (HPLC) purification, longicatenamide A (1) was obtained with 36% yield over 15 steps starting from 6.
![[1860-5397-18-166-i4]](/bjoc/content/inline/1860-5397-18-166-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Total synthesis of longicatenamycin A (1).
Scheme 4: Total synthesis of longicatenamycin A (1).
The NMR spectra of synthesized compound 1 agreed with those of natural 1. At this stage the confirmation of the identity of natural and synthesized compounds by structural determination using NMR spectroscopy is often difficult because the NMR spectra of peptidic products vary depending on the conditions in the NMR tube [25-29], such as concentration, pH, and purity. Thus, analyses of the NMR spectra of peptides sometimes lead to incorrect structural determination even though total synthesis of the proposed structures is successful [30]. In this study, synthesized and natural 1 were compared by collecting LC–MS data. As displayed in Figure 2, the retention time of synthesized 1 was identical to that of natural 1. These results confirmed total synthesis of 1 and supported our proposed structure of isolated natural 1.
![[1860-5397-18-166-2]](/bjoc/content/figures/1860-5397-18-166-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: LC–MS extracted ion chromatograms (EICs) of synthesized and natural 1. Column: Imtakt Cadenza CD-C18 3 × 150 mm; eluent: MeCN/H2O/TFA 30:70:0.05, isocratic, 0.2 mL/min; 40 °C.
Figure 2: LC–MS extracted ion chromatograms (EICs) of synthesized and natural 1. Column: Imtakt Cadenza CD-C1...
Total synthesis sometimes invalidates the reported biological activity of the isolated natural product, which could be due to the presence of uncharacterized impurities. To verify the bioactivity of 1, the antimicrobial activity of synthesized 1 was preliminary tested in this study following a previously reported method [8], revealing that chemically synthesized 1 (minimum inhibitory concentration (MIC) = 50 μM) exhibited moderate but selective antimicrobial activity against Bacillus subtilis similar to natural 1 (MIC = 100 μM).
Conclusion
We accomplished solid-phase total synthesis of longicatenamide A (1). Initially, commercially unavailable building blocks 7 and 10 were chemically synthesized with stereocontrol. Then, the peptide chain was elongated by Fmoc-based solid-phase peptide synthesis. Finally, the cyclization of the peptide chain followed by simultaneous cleavage of all protecting groups in the solution phase afforded target compound 1. The comparison of the chromatograms of synthesized and natural 1 corroborated the chemical structure of 1. Further studies of the structure–activity relationship, identification of biosynthetic gene clusters, and detailed investigations of the combined-culture production of longicatenamides are currently underway in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedures and compound characterization data. | ||
| Format: PDF | Size: 2.9 MB | Download |
Funding
This work was financially supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan [17H06401 (H.K.), 18K14396 (T.K.), 19H02840 (H.K.), 22K05112 (T.K.)], in addition to the Platform Project for Supporting Drug Discovery and Life Science Research from the Japan Agency for Medical Research and Development (AMED) (H.K.). Contributions from the Takeda Science Foundation, The Tokyo Biochemical Research Foundation, and the foundation of Tokyo Chemical Industry to T.K. are greatly appreciated.
References
-
Kakeya, H. Nat. Prod. Rep. 2016, 33, 648–654. doi:10.1039/c5np00120j
Return to citation in text: [1] -
Pye, C. R.; Bertin, M. J.; Lokey, R. S.; Gerwick, W. H.; Linington, R. G. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 5601–5606. doi:10.1073/pnas.1614680114
Return to citation in text: [1] -
Onaka, H.; Mori, Y.; Igarashi, Y.; Furumai, T. Appl. Environ. Microbiol. 2011, 77, 400–406. doi:10.1128/aem.01337-10
Return to citation in text: [1] -
Kuranaga, T.; Minote, M.; Morimoto, R.; Pan, C.; Ogawa, H.; Kakeya, H. ACS Chem. Biol. 2020, 15, 2499–2506. doi:10.1021/acschembio.0c00517
Return to citation in text: [1] [2] [3] -
Pan, C.; Kuranaga, T.; Liu, C.; Lu, S.; Shinzato, N.; Kakeya, H. Org. Lett. 2020, 22, 3014–3017. doi:10.1021/acs.orglett.0c00776
Return to citation in text: [1] -
Pan, C.; Kuranaga, T.; Cao, X.; Suzuki, T.; Dohmae, N.; Shinzato, N.; Onaka, H.; Kakeya, H. J. Org. Chem. 2021, 86, 1843–1849. doi:10.1021/acs.joc.0c02660
Return to citation in text: [1] -
Kuranaga, T.; Tamura, M.; Ikeda, H.; Terada, S.; Nakagawa, Y.; Kakeya, H. Org. Lett. 2021, 23, 7106–7111. doi:10.1021/acs.orglett.1c02506
Return to citation in text: [1] -
Jiang, Y.; Matsumoto, T.; Kuranaga, T.; Lu, S.; Wang, W.; Onaka, H.; Kakeya, H. J. Antibiot. 2021, 74, 307–316. doi:10.1038/s41429-020-00400-3
Return to citation in text: [1] [2] -
Sheppard, R. J. Pept. Sci. 2003, 9, 545–552. doi:10.1002/psc.479
Return to citation in text: [1] -
von Nussbaum, F.; Anlauf, S.; Freiberg, C.; Benet-Buchholz, J.; Schamberger, J.; Henkel, T.; Schiffer, G.; Häbich, D. ChemMedChem 2008, 3, 619–626. doi:10.1002/cmdc.200700297
Return to citation in text: [1] -
Behrendt, R.; White, P.; Offer, J. J. Pept. Sci. 2016, 22, 4–27. doi:10.1002/psc.2836
Return to citation in text: [1] -
Kuranaga, T.; Enomoto, A.; Tan, H.; Fujita, K.; Wakimoto, T. Org. Lett. 2017, 19, 1366–1369. doi:10.1021/acs.orglett.7b00249
Return to citation in text: [1] -
Kuranaga, T.; Matsuda, K.; Sano, A.; Kobayashi, M.; Ninomiya, A.; Takada, K.; Matsunaga, S.; Wakimoto, T. Angew. Chem., Int. Ed. 2018, 57, 9447–9451. doi:10.1002/anie.201805541
Return to citation in text: [1] -
Matsuda, K.; Kobayashi, M.; Kuranaga, T.; Takada, K.; Ikeda, H.; Matsunaga, S.; Wakimoto, T. Org. Biomol. Chem. 2019, 17, 1058–1061. doi:10.1039/c8ob02867b
Return to citation in text: [1] -
Chatzi, K. B. O.; Gatos, D.; Stavropoulos, G. Int. J. Pept. Protein Res. 1991, 37, 513–520. doi:10.1111/j.1399-3011.1991.tb00769.x
Return to citation in text: [1] -
Garner, P. Tetrahedron Lett. 1984, 25, 5855–5858. doi:10.1016/s0040-4039(01)81703-2
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070
Return to citation in text: [1] -
Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277–7287. doi:10.1021/ja00019a027
Return to citation in text: [1] -
Bal, B. S.; Childers, W. E., Jr.; Pinnick, H. W. Tetrahedron 1981, 37, 2091–2096. doi:10.1016/s0040-4020(01)97963-3
Return to citation in text: [1] -
Holmquist, C. R.; Roskamp, E. J. J. Org. Chem. 1989, 54, 3258–3260. doi:10.1021/jo00275a006
Return to citation in text: [1] -
Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006
Return to citation in text: [1] -
Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Chem. – Eur. J. 2009, 15, 9394–9403. doi:10.1002/chem.200900614
Return to citation in text: [1] -
Coste, J.; Le-Nguyen, D.; Castro, B. Tetrahedron Lett. 1990, 31, 205–208. doi:10.1016/s0040-4039(00)94371-5
Return to citation in text: [1] -
Carpino, L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. doi:10.1021/ja00063a082
Return to citation in text: [1] -
Zou, B.; Long, K.; Ma, D. Org. Lett. 2005, 7, 4237–4240. doi:10.1021/ol051685g
Return to citation in text: [1] -
Sugiyama, H.; Watanabe, A.; Teruya, T.; Suenaga, K. Tetrahedron Lett. 2009, 50, 7343–7345. doi:10.1016/j.tetlet.2009.10.059
Return to citation in text: [1] -
Ma, B.; Litvinov, D. N.; He, L.; Banerjee, B.; Castle, S. L. Angew. Chem., Int. Ed. 2009, 48, 6104–6107. doi:10.1002/anie.200902425
Return to citation in text: [1] -
Ma, B.; Banerjee, B.; Litvinov, D. N.; He, L.; Castle, S. L. J. Am. Chem. Soc. 2010, 132, 1159–1171. doi:10.1021/ja909870g
Return to citation in text: [1] -
Kuranaga, T.; Sesoko, Y.; Sakata, K.; Maeda, N.; Hayata, A.; Inoue, M. J. Am. Chem. Soc. 2013, 135, 5467–5474. doi:10.1021/ja401457h
Return to citation in text: [1] -
Kuranaga, T.; Mutoh, H.; Sesoko, Y.; Goto, T.; Matsunaga, S.; Inoue, M. J. Am. Chem. Soc. 2015, 137, 9443–9451. doi:10.1021/jacs.5b05550
Return to citation in text: [1]
| 21. | Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem. Soc. 1991, 113, 4092–4096. doi:10.1021/ja00011a006 |
| 19. | Bal, B. S.; Childers, W. E., Jr.; Pinnick, H. W. Tetrahedron 1981, 37, 2091–2096. doi:10.1016/s0040-4020(01)97963-3 |
| 20. | Holmquist, C. R.; Roskamp, E. J. J. Org. Chem. 1989, 54, 3258–3260. doi:10.1021/jo00275a006 |
| 5. | Pan, C.; Kuranaga, T.; Liu, C.; Lu, S.; Shinzato, N.; Kakeya, H. Org. Lett. 2020, 22, 3014–3017. doi:10.1021/acs.orglett.0c00776 |
| 6. | Pan, C.; Kuranaga, T.; Cao, X.; Suzuki, T.; Dohmae, N.; Shinzato, N.; Onaka, H.; Kakeya, H. J. Org. Chem. 2021, 86, 1843–1849. doi:10.1021/acs.joc.0c02660 |
| 7. | Kuranaga, T.; Tamura, M.; Ikeda, H.; Terada, S.; Nakagawa, Y.; Kakeya, H. Org. Lett. 2021, 23, 7106–7111. doi:10.1021/acs.orglett.1c02506 |
| 16. | Garner, P. Tetrahedron Lett. 1984, 25, 5855–5858. doi:10.1016/s0040-4039(01)81703-2 |
| 4. | Kuranaga, T.; Minote, M.; Morimoto, R.; Pan, C.; Ogawa, H.; Kakeya, H. ACS Chem. Biol. 2020, 15, 2499–2506. doi:10.1021/acschembio.0c00517 |
| 17. | Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156. doi:10.1021/jo00170a070 |
| 18. | Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277–7287. doi:10.1021/ja00019a027 |
| 3. | Onaka, H.; Mori, Y.; Igarashi, Y.; Furumai, T. Appl. Environ. Microbiol. 2011, 77, 400–406. doi:10.1128/aem.01337-10 |
| 14. | Matsuda, K.; Kobayashi, M.; Kuranaga, T.; Takada, K.; Ikeda, H.; Matsunaga, S.; Wakimoto, T. Org. Biomol. Chem. 2019, 17, 1058–1061. doi:10.1039/c8ob02867b |
| 8. | Jiang, Y.; Matsumoto, T.; Kuranaga, T.; Lu, S.; Wang, W.; Onaka, H.; Kakeya, H. J. Antibiot. 2021, 74, 307–316. doi:10.1038/s41429-020-00400-3 |
| 2. | Pye, C. R.; Bertin, M. J.; Lokey, R. S.; Gerwick, W. H.; Linington, R. G. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 5601–5606. doi:10.1073/pnas.1614680114 |
| 15. | Chatzi, K. B. O.; Gatos, D.; Stavropoulos, G. Int. J. Pept. Protein Res. 1991, 37, 513–520. doi:10.1111/j.1399-3011.1991.tb00769.x |
| 4. | Kuranaga, T.; Minote, M.; Morimoto, R.; Pan, C.; Ogawa, H.; Kakeya, H. ACS Chem. Biol. 2020, 15, 2499–2506. doi:10.1021/acschembio.0c00517 |
| 11. | Behrendt, R.; White, P.; Offer, J. J. Pept. Sci. 2016, 22, 4–27. doi:10.1002/psc.2836 |
| 25. | Zou, B.; Long, K.; Ma, D. Org. Lett. 2005, 7, 4237–4240. doi:10.1021/ol051685g |
| 26. | Sugiyama, H.; Watanabe, A.; Teruya, T.; Suenaga, K. Tetrahedron Lett. 2009, 50, 7343–7345. doi:10.1016/j.tetlet.2009.10.059 |
| 27. | Ma, B.; Litvinov, D. N.; He, L.; Banerjee, B.; Castle, S. L. Angew. Chem., Int. Ed. 2009, 48, 6104–6107. doi:10.1002/anie.200902425 |
| 28. | Ma, B.; Banerjee, B.; Litvinov, D. N.; He, L.; Castle, S. L. J. Am. Chem. Soc. 2010, 132, 1159–1171. doi:10.1021/ja909870g |
| 29. | Kuranaga, T.; Sesoko, Y.; Sakata, K.; Maeda, N.; Hayata, A.; Inoue, M. J. Am. Chem. Soc. 2013, 135, 5467–5474. doi:10.1021/ja401457h |
| 12. | Kuranaga, T.; Enomoto, A.; Tan, H.; Fujita, K.; Wakimoto, T. Org. Lett. 2017, 19, 1366–1369. doi:10.1021/acs.orglett.7b00249 |
| 13. | Kuranaga, T.; Matsuda, K.; Sano, A.; Kobayashi, M.; Ninomiya, A.; Takada, K.; Matsunaga, S.; Wakimoto, T. Angew. Chem., Int. Ed. 2018, 57, 9447–9451. doi:10.1002/anie.201805541 |
| 30. | Kuranaga, T.; Mutoh, H.; Sesoko, Y.; Goto, T.; Matsunaga, S.; Inoue, M. J. Am. Chem. Soc. 2015, 137, 9443–9451. doi:10.1021/jacs.5b05550 |
| 4. | Kuranaga, T.; Minote, M.; Morimoto, R.; Pan, C.; Ogawa, H.; Kakeya, H. ACS Chem. Biol. 2020, 15, 2499–2506. doi:10.1021/acschembio.0c00517 |
| 22. | Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Chem. – Eur. J. 2009, 15, 9394–9403. doi:10.1002/chem.200900614 |
| 8. | Jiang, Y.; Matsumoto, T.; Kuranaga, T.; Lu, S.; Wang, W.; Onaka, H.; Kakeya, H. J. Antibiot. 2021, 74, 307–316. doi:10.1038/s41429-020-00400-3 |
| 10. | von Nussbaum, F.; Anlauf, S.; Freiberg, C.; Benet-Buchholz, J.; Schamberger, J.; Henkel, T.; Schiffer, G.; Häbich, D. ChemMedChem 2008, 3, 619–626. doi:10.1002/cmdc.200700297 |
| 23. | Coste, J.; Le-Nguyen, D.; Castro, B. Tetrahedron Lett. 1990, 31, 205–208. doi:10.1016/s0040-4039(00)94371-5 |
| 24. | Carpino, L. A. J. Am. Chem. Soc. 1993, 115, 4397–4398. doi:10.1021/ja00063a082 |
© 2022 Matsumoto et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.