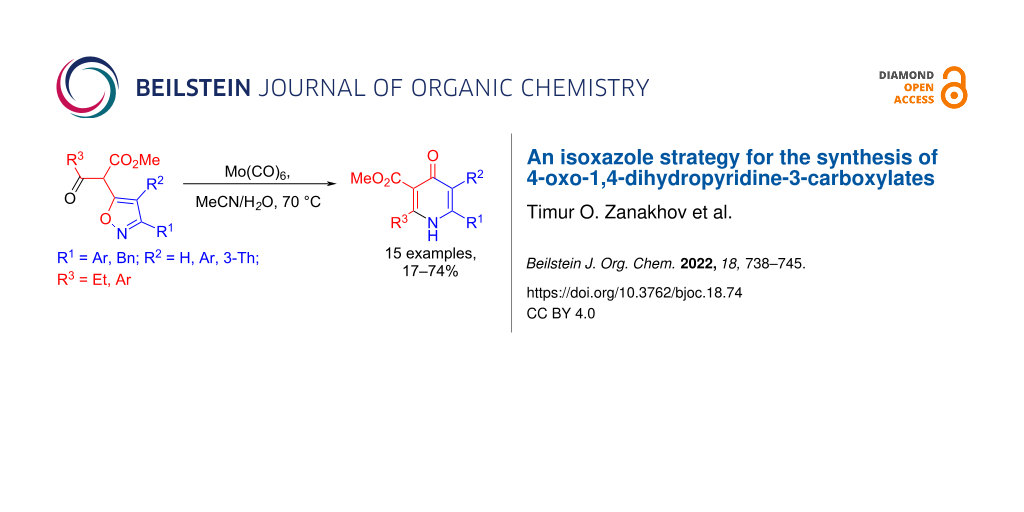
Abstract
A method has been developed for the preparation of 2-alkyl-6-aryl-, 2-aryl-6-aryl and 2,6-diaryl-5-aryl/hetaryl-substituted methyl 4-oxo-1,4-dihydropyridine-3-carboxylates by Mo(CO)6-mediated ring expansion of methyl 2-(isoxazol-5-yl)-3-oxopropanoates. The high reactivity of 4-oxo-1,4-dihydropyridine-3-carboxylates synthesized provide easy access to 2,4,6-triaryl-substituted and 1,2,5,6-tetrasubstituted nicotinates.

Graphical Abstract
Introduction
Pyridine moieties are present in many natural products, drugs, pesticides and industrial materials. Pyridine fragments are used in drugs due to their specific characteristics such as basicity, hydrogen bond forming ability, water solubility, and especially because of pyridine rings are bioisosteres of amines, amides, N-heterocyclic rings and benzene rings [1-5]. A special type of pyridine, the 4-pyridones, is also fairly well represented among drugs [5-12]. Some of bioactive compounds, such as ciprofloxacin, levofloxacin, delafloxacin, and elvitegravir, contain the fragment of 4-oxo-1,4-dihydropyridine-3-carboxylic acid [6,8] and some others, ivacaftor, dolutegravir, bictegravir, aspernigrin B, and 4PYR, contain the fragment of 4-oxo-1,4-dihydropyridine-3-carboxamide [5-7]. Finding new synthetic methods for the preparation of derivatives of 4-oxo-1,4-dihydropyridine-3-carboxylic acid are therefore relevant. Some alkyl 6-aryl-2-methyl-4-oxo-1,4-dihydropyridine-3-carboxylates were prepared by refluxing a xylene solution of methyl/ethyl 3-aminobut-2-enoates and methyl/ethyl 3-aryl-3-oxopropanoates in the presence of molecular sieves, with methyl 3-aryl-3-oxopropanoates giving higher product yields (37–46%) than ethyl derivatives (13–25%) (Scheme 1) [13]. General methods for the preparation of 2,6-diaryl- and 2,5,6-triaryl-substituted derivatives of 4-oxo-1,4-dihydropyridine-3-carboxylic acid, to the best of our knowledge, have not been published to date. We would like to present here a method for the preparation of 2-alkyl-6-aryl-, 2-aryl-6-aryl/hetaryl and 2,6-diaryl-5-aryl-substituted derivatives of 4-oxo-1,4-dihydropyridine-3-carboxylic acid 2 via Mo(CO)6-mediated rearrangement of methyl 2-(isoxazol-5-yl)-3-oxopropanoates 1 (Scheme 1).
![[1860-5397-18-74-i1]](/bjoc/content/inline/1860-5397-18-74-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Approaches to the synthesis of alkyl 4-oxo-1,4-dihydropyridine-3-carboxylates.
Scheme 1: Approaches to the synthesis of alkyl 4-oxo-1,4-dihydropyridine-3-carboxylates.
Results and Discussion
Based on our experience of using isoxazoles in the synthesis of heterocyclic compounds [14-18], we hypothesized that isoxazoles 1 can undergo reductive ring opening under the action of Mo(CO)6/H2O/MeCN [14,19-23] to enamines 3, which can be cyclized on acyl R3C(O) group to form pyridones 2 (Scheme 2, route a). The alternative cyclization scenario, which may involve the ester group of enamine 3 and lead to the formation of pyridone 5 (route b), is less likely due to the lower electrophilicity of the ester carbon compared to the acyl carbon. To check this, isoxazole 1g was synthesized and reacted with Mo(CO)6 in wet acetonitrile at 60 °C for 2 d to give pyridone 2g in 45% yield. The reaction at 70 °C was completed in a day to give pyridone 2g in 74% yield. A further increase in temperature, although it led to a reduction in the reaction time, was accompanied by resinification of the reaction mixture and, as a result, led to a decrease in the yields of pyridone 2g (80 °C, 3 h, 63%; 85 °C, 3 h, 42%). Enamine 6, resulted from the deacylation of intermediate 3g, was isolated in the reaction carried out at 70 °C. This side process is probably the reason for the decrease in the yield of the target pyridone 2g. Considering that isoxazoles were previously reduced to enaminones by other reducing systems [24-28], we checked the possibility of using NaBH4/NiSO4·7H2O [25], H2/PtO2/NiRaney [26,27] and H2/NiRaney [28] to obtain pyridone 2g from isoxazole 1g. However, analysis of reaction mixtures, obtained by using the above reductants, showed the formation of a large amount of products, but pyridone 2g was not detected. It is most probable that, in addition to the reductive opening of isoxazole, Mo(CO)6 catalyzes the cyclization of enaminone 3g to pyridine 2g [14]. In the absence of such catalysis, the unstable enaminone intermediate undergoes deacylation and decomposes through other destructive processes.
![[1860-5397-18-74-i2]](/bjoc/content/inline/1860-5397-18-74-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 4-oxo-1,4-dihydropyridine-3-carboxylates.
Scheme 2: Synthesis of 4-oxo-1,4-dihydropyridine-3-carboxylates.
To evaluate the scope of the disclosed reaction a set of isoxazoles 1 was prepared using approaches shown in the Scheme 3 and Scheme 4. Isoxazoles 11a–f were prepared by cycloaddition of nitrile oxides, generated from N-hydroxyimidoyl chlorides 7a–f, to propargyl halides [29], followed by the cyanation of the resulted isoxazoles 8a–f to cyanides 9a–f using Me2C(OH)CN/(Me2N)2C=NH [29], their acid hydrolysis, followed by esterification of the resulting acids 9a–f with diazomethane. 4-Iodoisoxazoles 12a–f, necessary for the preparation of 3,4-disubstituted isoxazoles, were obtained by iodination of 11a,c,e–g with NIS/TFA [30]. The reaction of 11d gave the product of double iodination 12c. 4-Iodoisoxazoles 12a,b,d–f were transformed into 3,4-substituted isoxazoles 13a–g by Suzuki reaction using a published procedure with some modifications [31].
![[1860-5397-18-74-i3]](/bjoc/content/inline/1860-5397-18-74-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Isoxazoles 1, except for isoxazole 1a, were synthesized in 44–98% yield by acylation of isoxazoles 11, 12 and 13 with acyl chlorides in the presence of NaH (Scheme 4). For the preparation of isoxazole 1a propionic anhydride was used.
![[1860-5397-18-74-i4]](/bjoc/content/inline/1860-5397-18-74-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Further, isoxazoles 1 were reacted with Mo(CO)6/H2O/MeCN under the optimal conditions to give pyridones 2 (Scheme 5). Isoxazole 1j with Mo(CO)6/H2O/MeCN gives the corresponding pyridone 2j in trace amounts, while the reaction of isoxazoles 1l,m,r leads to complete resinification. It is likely that one of the reasons for the decrease in the yields of pyridones 2 is the occurrence of a side reaction of deacylation of enamine intermediate 3, as shown for the reaction of isoxazole 1g (Scheme 2).
![[1860-5397-18-74-i5]](/bjoc/content/inline/1860-5397-18-74-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Taking into account that compounds 2 can potentially exist in pyridone and pyridole tautomeric forms, we performed calculations at the DFT B3LYP-D3/6-311+G(d,p) level of theory with SMD solvent model to evaluate this tautomeric equilibrium. According to the calculation results compounds 2 exist in the pyridone form in acetonitrile or chloroform solution (Table 1).
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-74-i7.svg?max-width=637&scale=1.0)
All new compounds were characterized by 1H, 13C NMR and HRMS methods. Moreover, the structure of 2k was also confirmed by single-crystal X-ray diffraction analysis (Figure S1, Supporting Information File 1).
Compounds 2 have several reactive centers that make them attractive starting materials for the synthesis of new libraries of 4-pyridone and pyridine derivatives (Scheme 6). The methoxycarbonyl group of compound 2c was selectively reduced to give 3-(hydroxymethyl)pyridin-4(1H)-one 14 in 89% yield. The reaction of pyridone 2e with PBr3 afforded methyl 4-bromo-6-(4-fluorophenyl)-2-phenylnicotinate (15) in 80% yield. The latter was employed in the Suzuki reaction in the high-yield synthesis of 2,4,6-triaryl-substituted nicotinate 16. The nitrogen of 2,3,4,6-tetrasubstituted pyridone 2n can be quantitatively benzylated by benzyl chloride to give derivative 17.
![[1860-5397-18-74-i6]](/bjoc/content/inline/1860-5397-18-74-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Transformations of pyridones 2.
Scheme 6: Transformations of pyridones 2.
Conclusion
A method has been developed for the preparation of 2-alkyl-6-aryl-, 2-aryl-6-aryl and 2,6-diaryl-5-aryl/hetaryl-substituted derivatives of 4-oxo-1,4-dihydropyridine-3-carboxylic acid 2 via Mo(CO)6-mediated rearrangement of methyl 2-(isoxazol-5-yl)-3-oxopropanoates 1. High yield transformations of compound 2 provide easy access to 2,4,6-triaryl-substituted and 1,2,5,6-tetrasubstituted nicotinates.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, X-ray diffraction experiment, and copies of NMR spectra of new compounds. | ||
| Format: PDF | Size: 25.7 MB | Download |
| Supporting Information File 2: Crystallographic information file for compound 2k. | ||
| Format: CIF | Size: 275.9 KB | Download |
Funding
We gratefully acknowledge the financial support of the Russian Science Foundation project 21-73-00019. This research was carried out using resources of the Centre for Magnetic Resonance, the Research Centre for X-ray Diffraction Studies, the Centre for Chemical Analysis and Materials, and the Computer Centre of the Science Park of Saint Petersburg State University.
References
-
Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2013, 9, 2265–2319. doi:10.3762/bjoc.9.265
Return to citation in text: [1] -
Heravi, M. M.; Zadsirjan, V. RSC Adv. 2020, 10, 44247–44311. doi:10.1039/d0ra09198g
Return to citation in text: [1] -
Sahu, R.; Mishra, R.; Kumar, R.; Majee, C.; Salahuddin; Mazumder, A.; Kumar, A. Indian J. Pharm. Sci. 2021, 83, 162–185. doi:10.36468/pharmaceutical-sciences.763
Return to citation in text: [1] -
Ling, Y.; Hao, Z.-Y.; Liang, D.; Zhang, C.-L.; Liu, Y.-F.; Wang, Y. Drug Des., Dev. Ther. 2021, 15, 4289–4338. doi:10.2147/dddt.s329547
Return to citation in text: [1] [2] [3] -
Zhang, Y.; Pike, A. Bioorg. Med. Chem. Lett. 2021, 38, 127849. doi:10.1016/j.bmcl.2021.127849
Return to citation in text: [1] [2] [3] -
Mierzejewska, P.; Kunc, M.; Zabielska-Kaczorowska, M. A.; Kutryb-Zajac, B.; Pelikant-Malecka, I.; Braczko, A.; Jablonska, P.; Romaszko, P.; Koszalka, P.; Szade, J.; Smolenski, R. T.; Slominska, E. M. Exp. Mol. Med. 2021, 53, 1402–1412. doi:10.1038/s12276-021-00669-w
Return to citation in text: [1] [2] -
Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639
Return to citation in text: [1] [2] -
Bueno, J. M.; Calderon, F.; Chicharro, J.; De la Rosa, J. C.; Díaz, B.; Fernández, J.; Fiandor, J. M.; Fraile, M. T.; García, M.; Herreros, E.; García-Pérez, A.; Lorenzo, M.; Mallo, A.; Puente, M.; Saadeddin, A.; Ferrer, S.; Angulo-Barturen, I.; Burrows, J. N.; León, M. L. J. Med. Chem. 2018, 61, 3422–3435. doi:10.1021/acs.jmedchem.7b01256
Return to citation in text: [1] -
Gordon, J. A.; Fishwick, C. W. G.; McPhillie, M. J. Curr. Top. Med. Chem. 2017, 17, 79–90. doi:10.2174/1568026616666160719164542
Return to citation in text: [1] -
Miyagawa, M.; Akiyama, T.; Mikamiyama-Iwata, M.; Hattori, K.; Kurihara, N.; Taoda, Y.; Takahashi-Kageyama, C.; Kurose, N.; Mikamiyama, H.; Suzuki, N.; Takaya, K.; Tomita, K.; Matsuo, K.; Morimoto, K.; Yoshida, R.; Shishido, T.; Yoshinaga, T.; Sato, A.; Kawai, M. Bioorg. Med. Chem. Lett. 2016, 26, 4739–4742. doi:10.1016/j.bmcl.2016.08.038
Return to citation in text: [1] -
Bueno, J. M.; Herreros, E.; Angulo-Barturen, I.; Ferrer, S.; Fiandor, J. M.; Gamo, F. J.; Gargallo-Viola, D.; Derimanov, G. Future Med. Chem. 2012, 4, 2311–2323. doi:10.4155/fmc.12.177
Return to citation in text: [1] -
Makabe, O.; Murai, Y.; Fukatsu, S. Heterocycles 1979, 13, 239–246. doi:10.3987/s-1979-01-0239
Return to citation in text: [1] -
Galenko, E. E.; Linnik, S. A.; Khoroshilova, O. V.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2019, 84, 11275–11285. doi:10.1021/acs.joc.9b01634
Return to citation in text: [1] [2] [3] -
Galenko, E. E.; Novikov, M. S.; Shakirova, F. M.; Shakirova, J. R.; Kornyakov, I. V.; Bodunov, V. A.; Khlebnikov, A. F. J. Org. Chem. 2019, 84, 3524–3536. doi:10.1021/acs.joc.9b00115
Return to citation in text: [1] -
Mikhailov, K. I.; Galenko, E. E.; Galenko, A. V.; Novikov, M. S.; Ivanov, A. Y.; Starova, G. L.; Khlebnikov, A. F. J. Org. Chem. 2018, 83, 3177–3187. doi:10.1021/acs.joc.8b00069
Return to citation in text: [1] -
Galenko, E. E.; Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Shakirova, J. R. J. Org. Chem. 2016, 81, 8495–8507. doi:10.1021/acs.joc.6b01662
Return to citation in text: [1] -
Galenko, E. E.; Tomashenko, O. A.; Khlebnikov, A. F.; Novikov, M. S. Org. Biomol. Chem. 2015, 13, 9825–9833. doi:10.1039/c5ob01537e
Return to citation in text: [1] -
Nitta, M.; Kobayashi, T. J. Chem. Soc., Chem. Commun. 1982, 877–878. doi:10.1039/c39820000877
Return to citation in text: [1] -
Nitta, M.; Kobayashi, T. J. Chem. Soc., Perkin Trans. 1 1985, 1401–1406. doi:10.1039/p19850001401
Return to citation in text: [1] -
Donati, D.; Ferrini, S.; Fusi, S.; Ponticelli, F. J. Heterocycl. Chem. 2004, 41, 761–766. doi:10.1002/jhet.5570410518
Return to citation in text: [1] -
Pérez, J. M.; Ramón, D. J. ACS Sustainable Chem. Eng. 2015, 3, 2343–2349. doi:10.1021/acssuschemeng.5b00689
Return to citation in text: [1] -
Llopis, Q.; Guillamot, G.; Phansavath, P.; Ratovelomanana-Vidal, V. Org. Lett. 2017, 19, 6428–6431. doi:10.1021/acs.orglett.7b03332
Return to citation in text: [1] -
Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/rcr4503
Return to citation in text: [1] -
Koroleva, E. V.; Lakhvich, F. A.; Yankova, T. V. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1987, 23, 1267. doi:10.1007/bf00479385
Return to citation in text: [1] [2] -
Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron Lett. 1984, 25, 4313–4316. doi:10.1016/s0040-4039(01)81426-x
Return to citation in text: [1] [2] -
Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron 1987, 43, 235–242. doi:10.1016/s0040-4020(01)89949-x
Return to citation in text: [1] [2] -
Calle, M.; Calvo, L. A.; González-Ortega, A.; González-Nogal, A. M. Tetrahedron 2006, 62, 611–618. doi:10.1016/j.tet.2005.10.001
Return to citation in text: [1] [2] -
Serebryannikova, A. V.; Galenko, E. E.; Novikov, M. S.; Khlebnikov, A. F. Tetrahedron 2021, 88, 132153. doi:10.1016/j.tet.2021.132153
Return to citation in text: [1] [2] -
Chalyk, B. A.; Hrebeniuk, K. V.; Fil, Y. V.; Gavrilenko, K. S.; Rozhenko, A. B.; Vashchenko, B. V.; Borysov, O. V.; Biitseva, A. V.; Lebed, P. S.; Bakanovych, I.; Moroz, Y. S.; Grygorenko, O. O. J. Org. Chem. 2019, 84, 15877–15899. doi:10.1021/acs.joc.9b02264
Return to citation in text: [1] -
Brough, P. A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J. E.; Cheung, K.-M. J.; Collins, I.; Davies, N. G. M.; Drysdale, M. J.; Dymock, B.; Eccles, S. A.; Finch, H.; Fink, A.; Hayes, A.; Howes, R.; Hubbard, R. E.; James, K.; Jordan, A. M.; Lockie, A.; Martins, V.; Massey, A.; Matthews, T. P.; McDonald, E.; Northfield, C. J.; Pearl, L. H.; Prodromou, C.; Ray, S.; Raynaud, F. I.; Roughley, S. D.; Sharp, S. Y.; Surgenor, A.; Walmsley, D. L.; Webb, P.; Wood, M.; Workman, P.; Wrightt, L. J. Med. Chem. 2008, 51, 196–218. doi:10.1021/jm701018h
Return to citation in text: [1]
| 1. | Taylor, R. D.; MacCoss, M.; Lawson, A. D. G. J. Med. Chem. 2014, 57, 5845–5859. doi:10.1021/jm4017625 |
| 2. | Baumann, M.; Baxendale, I. R. Beilstein J. Org. Chem. 2013, 9, 2265–2319. doi:10.3762/bjoc.9.265 |
| 3. | Heravi, M. M.; Zadsirjan, V. RSC Adv. 2020, 10, 44247–44311. doi:10.1039/d0ra09198g |
| 4. | Sahu, R.; Mishra, R.; Kumar, R.; Majee, C.; Salahuddin; Mazumder, A.; Kumar, A. Indian J. Pharm. Sci. 2021, 83, 162–185. doi:10.36468/pharmaceutical-sciences.763 |
| 5. | Ling, Y.; Hao, Z.-Y.; Liang, D.; Zhang, C.-L.; Liu, Y.-F.; Wang, Y. Drug Des., Dev. Ther. 2021, 15, 4289–4338. doi:10.2147/dddt.s329547 |
| 13. | Makabe, O.; Murai, Y.; Fukatsu, S. Heterocycles 1979, 13, 239–246. doi:10.3987/s-1979-01-0239 |
| 30. | Chalyk, B. A.; Hrebeniuk, K. V.; Fil, Y. V.; Gavrilenko, K. S.; Rozhenko, A. B.; Vashchenko, B. V.; Borysov, O. V.; Biitseva, A. V.; Lebed, P. S.; Bakanovych, I.; Moroz, Y. S.; Grygorenko, O. O. J. Org. Chem. 2019, 84, 15877–15899. doi:10.1021/acs.joc.9b02264 |
| 5. | Ling, Y.; Hao, Z.-Y.; Liang, D.; Zhang, C.-L.; Liu, Y.-F.; Wang, Y. Drug Des., Dev. Ther. 2021, 15, 4289–4338. doi:10.2147/dddt.s329547 |
| 6. | Zhang, Y.; Pike, A. Bioorg. Med. Chem. Lett. 2021, 38, 127849. doi:10.1016/j.bmcl.2021.127849 |
| 7. | Mierzejewska, P.; Kunc, M.; Zabielska-Kaczorowska, M. A.; Kutryb-Zajac, B.; Pelikant-Malecka, I.; Braczko, A.; Jablonska, P.; Romaszko, P.; Koszalka, P.; Szade, J.; Smolenski, R. T.; Slominska, E. M. Exp. Mol. Med. 2021, 53, 1402–1412. doi:10.1038/s12276-021-00669-w |
| 31. | Brough, P. A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J. E.; Cheung, K.-M. J.; Collins, I.; Davies, N. G. M.; Drysdale, M. J.; Dymock, B.; Eccles, S. A.; Finch, H.; Fink, A.; Hayes, A.; Howes, R.; Hubbard, R. E.; James, K.; Jordan, A. M.; Lockie, A.; Martins, V.; Massey, A.; Matthews, T. P.; McDonald, E.; Northfield, C. J.; Pearl, L. H.; Prodromou, C.; Ray, S.; Raynaud, F. I.; Roughley, S. D.; Sharp, S. Y.; Surgenor, A.; Walmsley, D. L.; Webb, P.; Wood, M.; Workman, P.; Wrightt, L. J. Med. Chem. 2008, 51, 196–218. doi:10.1021/jm701018h |
| 6. | Zhang, Y.; Pike, A. Bioorg. Med. Chem. Lett. 2021, 38, 127849. doi:10.1016/j.bmcl.2021.127849 |
| 8. | Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639 |
| 29. | Serebryannikova, A. V.; Galenko, E. E.; Novikov, M. S.; Khlebnikov, A. F. Tetrahedron 2021, 88, 132153. doi:10.1016/j.tet.2021.132153 |
| 5. | Ling, Y.; Hao, Z.-Y.; Liang, D.; Zhang, C.-L.; Liu, Y.-F.; Wang, Y. Drug Des., Dev. Ther. 2021, 15, 4289–4338. doi:10.2147/dddt.s329547 |
| 6. | Zhang, Y.; Pike, A. Bioorg. Med. Chem. Lett. 2021, 38, 127849. doi:10.1016/j.bmcl.2021.127849 |
| 7. | Mierzejewska, P.; Kunc, M.; Zabielska-Kaczorowska, M. A.; Kutryb-Zajac, B.; Pelikant-Malecka, I.; Braczko, A.; Jablonska, P.; Romaszko, P.; Koszalka, P.; Szade, J.; Smolenski, R. T.; Slominska, E. M. Exp. Mol. Med. 2021, 53, 1402–1412. doi:10.1038/s12276-021-00669-w |
| 8. | Han, J.; Remete, A. M.; Dobson, L. S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V. A.; O’Hagan, D. J. Fluorine Chem. 2020, 239, 109639. doi:10.1016/j.jfluchem.2020.109639 |
| 9. | Bueno, J. M.; Calderon, F.; Chicharro, J.; De la Rosa, J. C.; Díaz, B.; Fernández, J.; Fiandor, J. M.; Fraile, M. T.; García, M.; Herreros, E.; García-Pérez, A.; Lorenzo, M.; Mallo, A.; Puente, M.; Saadeddin, A.; Ferrer, S.; Angulo-Barturen, I.; Burrows, J. N.; León, M. L. J. Med. Chem. 2018, 61, 3422–3435. doi:10.1021/acs.jmedchem.7b01256 |
| 10. | Gordon, J. A.; Fishwick, C. W. G.; McPhillie, M. J. Curr. Top. Med. Chem. 2017, 17, 79–90. doi:10.2174/1568026616666160719164542 |
| 11. | Miyagawa, M.; Akiyama, T.; Mikamiyama-Iwata, M.; Hattori, K.; Kurihara, N.; Taoda, Y.; Takahashi-Kageyama, C.; Kurose, N.; Mikamiyama, H.; Suzuki, N.; Takaya, K.; Tomita, K.; Matsuo, K.; Morimoto, K.; Yoshida, R.; Shishido, T.; Yoshinaga, T.; Sato, A.; Kawai, M. Bioorg. Med. Chem. Lett. 2016, 26, 4739–4742. doi:10.1016/j.bmcl.2016.08.038 |
| 12. | Bueno, J. M.; Herreros, E.; Angulo-Barturen, I.; Ferrer, S.; Fiandor, J. M.; Gamo, F. J.; Gargallo-Viola, D.; Derimanov, G. Future Med. Chem. 2012, 4, 2311–2323. doi:10.4155/fmc.12.177 |
| 29. | Serebryannikova, A. V.; Galenko, E. E.; Novikov, M. S.; Khlebnikov, A. F. Tetrahedron 2021, 88, 132153. doi:10.1016/j.tet.2021.132153 |
| 25. | Koroleva, E. V.; Lakhvich, F. A.; Yankova, T. V. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1987, 23, 1267. doi:10.1007/bf00479385 |
| 28. | Calle, M.; Calvo, L. A.; González-Ortega, A.; González-Nogal, A. M. Tetrahedron 2006, 62, 611–618. doi:10.1016/j.tet.2005.10.001 |
| 24. | Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/rcr4503 |
| 25. | Koroleva, E. V.; Lakhvich, F. A.; Yankova, T. V. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1987, 23, 1267. doi:10.1007/bf00479385 |
| 26. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron Lett. 1984, 25, 4313–4316. doi:10.1016/s0040-4039(01)81426-x |
| 27. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron 1987, 43, 235–242. doi:10.1016/s0040-4020(01)89949-x |
| 28. | Calle, M.; Calvo, L. A.; González-Ortega, A.; González-Nogal, A. M. Tetrahedron 2006, 62, 611–618. doi:10.1016/j.tet.2005.10.001 |
| 14. | Galenko, E. E.; Linnik, S. A.; Khoroshilova, O. V.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2019, 84, 11275–11285. doi:10.1021/acs.joc.9b01634 |
| 14. | Galenko, E. E.; Linnik, S. A.; Khoroshilova, O. V.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2019, 84, 11275–11285. doi:10.1021/acs.joc.9b01634 |
| 19. | Nitta, M.; Kobayashi, T. J. Chem. Soc., Chem. Commun. 1982, 877–878. doi:10.1039/c39820000877 |
| 20. | Nitta, M.; Kobayashi, T. J. Chem. Soc., Perkin Trans. 1 1985, 1401–1406. doi:10.1039/p19850001401 |
| 21. | Donati, D.; Ferrini, S.; Fusi, S.; Ponticelli, F. J. Heterocycl. Chem. 2004, 41, 761–766. doi:10.1002/jhet.5570410518 |
| 22. | Pérez, J. M.; Ramón, D. J. ACS Sustainable Chem. Eng. 2015, 3, 2343–2349. doi:10.1021/acssuschemeng.5b00689 |
| 23. | Llopis, Q.; Guillamot, G.; Phansavath, P.; Ratovelomanana-Vidal, V. Org. Lett. 2017, 19, 6428–6431. doi:10.1021/acs.orglett.7b03332 |
| 14. | Galenko, E. E.; Linnik, S. A.; Khoroshilova, O. V.; Novikov, M. S.; Khlebnikov, A. F. J. Org. Chem. 2019, 84, 11275–11285. doi:10.1021/acs.joc.9b01634 |
| 15. | Galenko, E. E.; Novikov, M. S.; Shakirova, F. M.; Shakirova, J. R.; Kornyakov, I. V.; Bodunov, V. A.; Khlebnikov, A. F. J. Org. Chem. 2019, 84, 3524–3536. doi:10.1021/acs.joc.9b00115 |
| 16. | Mikhailov, K. I.; Galenko, E. E.; Galenko, A. V.; Novikov, M. S.; Ivanov, A. Y.; Starova, G. L.; Khlebnikov, A. F. J. Org. Chem. 2018, 83, 3177–3187. doi:10.1021/acs.joc.8b00069 |
| 17. | Galenko, E. E.; Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Shakirova, J. R. J. Org. Chem. 2016, 81, 8495–8507. doi:10.1021/acs.joc.6b01662 |
| 18. | Galenko, E. E.; Tomashenko, O. A.; Khlebnikov, A. F.; Novikov, M. S. Org. Biomol. Chem. 2015, 13, 9825–9833. doi:10.1039/c5ob01537e |
| 26. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron Lett. 1984, 25, 4313–4316. doi:10.1016/s0040-4039(01)81426-x |
| 27. | Baraldi, P. G.; Barco, A.; Benetti, S.; Manfredini, S.; Pollini, G. P.; Simoni, D. Tetrahedron 1987, 43, 235–242. doi:10.1016/s0040-4020(01)89949-x |
© 2022 Zanakhov et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.