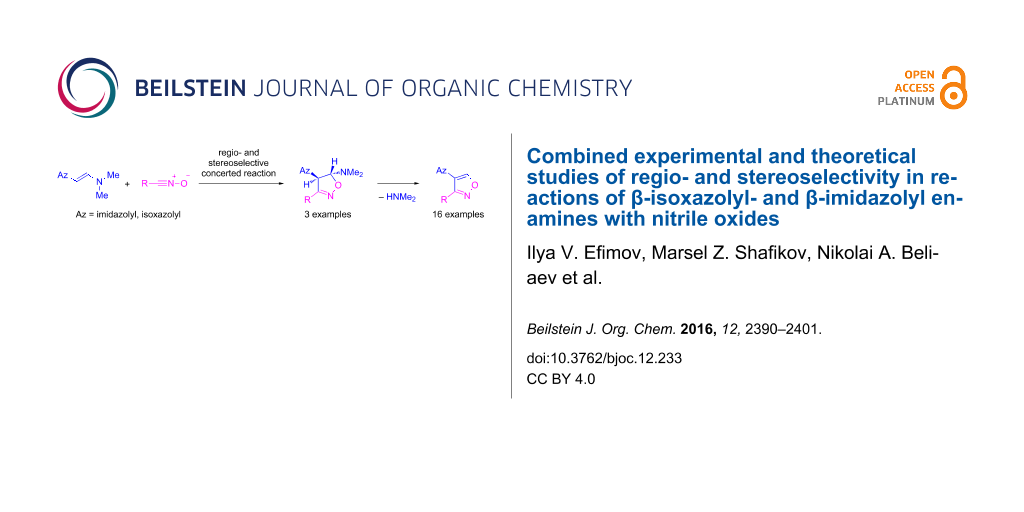
Abstract
Reactions of β-azolyl enamines and nitrile oxides were studied by both experimental and theoretical methods. (E)-β-(4-Nitroimidazol-5-yl), (5-nitroimidazol-4-yl) and isoxazol-5-yl enamines smoothly react regioselectively at room temperature in dioxane solution with aryl, pyridyl, and cyclohexylhydroxamoyl chlorides without a catalyst or a base to form 4-azolylisoxazoles as the only products in good yields. The intermediate 4,5-dihydroisoxazolines were isolated as trans isomers during the reaction of (E)-β-imidazol-4-yl enamines with aryl and cyclohexylhydroxamoyl chlorides. Stepwise and concerted pathways for the reaction of β-azolyl enamines with hydroxamoyl chlorides were considered and studied at the B3LYP/Def2-TZVP level of theory combined with D3BJ dispersion correction. The reactions of benzonitrile oxide with both E- and Z-imidazolyl enamines have been shown to proceed stereoselectively to form trans- and cis-isoxazolines, respectively. The preference of E-isomers over Z-isomers, driven by the higher stability of the former, apparently controls the stereoselectivity of the investigated cycloaddition reaction with benzonitrilе oxide. Based on the reactivity of azolyl enamines towards hydroxamoyl chlorides, a novel, effective catalyst-free method was elaborated to prepare 4-azolyl-5-substituted isoxazoles that are otherwise difficult to obtain.

Graphical Abstract
Introduction
The biological activity and technically useful properties of isoxazoles have made them the focus of both medicinal and materials chemistry over the years [1]. Isoxazoles have been found in natural products [1], and they exhibit anticancer [2], antiviral [1], anti-inflammatory [3], antidiabetic [4], anti-Alzheimer [5] and many other types of biological activity [6]. Isoxazolines and isoxazoles have been applied as chemosensors, liquid crystalline compounds, ligands for asymmetric synthesis, and they are also convenient reagents in organic synthesis [1]. Although bicyclic assemblies of azoles exhibit interesting chemical properties and biological activities [1,7-11] isoxazoles conjugated to other azole rings are poorly presented in the literature in comparison with monocyclic and fused derivatives [1,6].
Recently isoxazoles conjugated to pyrazole A [12], imidazole B [13] and tetrazole C [4] rings were found as promising candidates for anticancer and antidiabetic drugs and for the treatment of cognitive disorder (Figure 1). It makes prospective the synthesis of new derivatives of isoxazoles conjugated with other azole rings.
![[1860-5397-12-233-1]](/bjoc/content/figures/1860-5397-12-233-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active isoxazoles conjugated to other azole rings.
Figure 1: Biologically active isoxazoles conjugated to other azole rings.
The few synthetic methods for 4-(azol-5-yl)isoxazoles published in the literature involve the formation of either azole or isoxazole rings in the final step [1,13,14]. Cyclization reactions of 2-azolyl-1,3-dicarbonyl compounds with hydroxylamine and cyanomethylazoles with hydroxamoyl chlorides are used for the synthesis of a few representatives of 4-(azol-5-yl)isoxazoles [1,4,12-16]. Cycloaddition reactions of azolylacetylenes with nitrile oxides are an alternative method for the synthesis of this type of compounds [14,17] (Scheme 1). Despite of good yields this method has serious limitations for the synthesis of azolylisoxazoles due to the poor availability of the starting materials. Therefore, the search of new regioselective routes to azolylisoxazoles remains a synthetic challenge. We turned our attention to the reaction of enamines with nitrile oxides (or their precursors, hydroxamoyl chlorides). The reaction has been shown to take place regioselectively to form only one of two possible regioisomers [18-26].
![[1860-5397-12-233-i1]](/bjoc/content/inline/1860-5397-12-233-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of azolyl enamines with nitrile oxides.
Scheme 1: Reactions of azolyl enamines with nitrile oxides.
Therefore, this holds some promise for the development of an efficient method based on this reaction for the synthesis of monocyclic, fused and conjugated isoxazoles. At the same time, there are few reports on the systematic study of this reaction [23,24].
A general discussion about the reasons of regioselectivity and stereoselectivity for this reaction is lacking from the literature. To the best of our knowledge there are no examples for the synthesis of 4-isoxazolyl- and imidazolylisoxazoles and imidazolylisoxazolines by this reaction.
We report here the results of experimental and theoretical studies for the reaction of β-azolyl enamines bearing isoxazol-5-yl, imidazol-4-yl and imidazol-5-yl moieties with aryl, pyridyl and cyclohexylhydroxamoyl chlorides, pointing to a concerted mechanism of this reaction.
Results and Discussion
The starting enamines 1a–e bearing imidazole (1a,b) and isoxazole (1c–e) rings were prepared from the corresponding 5-methylazoles by reaction with dimethylformamide dimethyl acetal (DMF-DMA) adapting synthetic procedures published earlier [26-30]. Their structures can be unambiguously assigned as the trans-isomers by observing the coupling constant (J = 13.0–13.6 Hz) for the protons of the vinyl fragment in the 1H NMR spectra.
Hydroxamoyl chlorides are known as masked nitrile oxides and the latter could easily be generated by reaction of the former with a base [31-33]. Nitrile oxides, apart from the expected cycloaddition reaction, could undergo dimerization affording isomeric products with different structures [31-33]. Therefore we could expect, besides isoxazoles, the formation of various byproducts in the reaction of hydroxamoyl chlorides 2a–h with β-azolyl enamines 1a–e (Figure 2). This complicates the base-catalyzed preparation of 4-azolylisoxazoles from enamines and hydroxamoyl chlorides, lowering the yield.
![[1860-5397-12-233-2]](/bjoc/content/figures/1860-5397-12-233-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structures of starting enamines 1 and hydroxamoyl chlorides 2.
Figure 2: Structures of starting enamines 1 and hydroxamoyl chlorides 2.
Fortunately, we have found that the reaction of enamines 1a–e with hydroxamoyl chlorides 2a–h can take place smoothly at room temperature in 1,4-dioxane solution without base to form 4-imidazolyl- and 4-isoxazolylisoxazoles 4a–p as exclusive products in good yields (Scheme 2). Probably the formation of nitrile oxides from hydroxamoyl chlorides occurs catalytically due to formation of dimethylamine as a result of the aromatization reaction with the isoxazoles, in which dimethylamine is formed. One can also propose that enamines 1 or traces of dimethylamine can act as base and facilitate the initial formation of nitrile oxides. Thus, different conjugated heterocycles of type 4 could be prepared starting from different enamines 1a–e and hydroxamoyl chlorides 2a–h. The yields vary from 43 to 90% (Scheme 2, see Supporting Information File 1 for full experimental data), and are mainly in the 65−90% range.
![[1860-5397-12-233-i2]](/bjoc/content/inline/1860-5397-12-233-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 4-azolylisoxazoles 4a–p from enamines 1a–e and hydroxamoyl chlorides 2a–h. Reaction conditions: (i) 1 (1.1 mmol), 2 (1 mmol), 1,4-dioxane (5 mL), rt, 12–48 h, (ii) 3b,c (1 mmol), H2O/HOAc (1:1 mixture) (2 mL), rt, 30 min (3b) or overnight (3c).
Scheme 2: Synthesis of 4-azolylisoxazoles 4a–p from enamines 1a–e and hydroxamoyl chlorides 2a–h. Reaction co...
Imidazolylisoxazoles 4a–h, imidazolylisoxazolines 3a–c and 3-aryl-4-carboxyisoxazol-5-ylisoxazoles 4i–p are novel conjugated heterocycles. There are very few known examples of isoxazol-5-ylisoxazoles, that have been prepared by other methods [14,15]. In contrast to our approach, these methods are not applicable for the regioselective synthesis of compounds containing a 3-aryl-4-carboxyisoxazole fragment, a structural motif in the anticancer pyrazolylisoxazole, and rather the 3,5-disubstituted isoxazole (compound 5, Scheme 2) is expected [12]. The presence of regioisomer 5 was not registered by TLC control and NMR spectra of the crude reaction mixture. A rise of temperature and use of other solvents decrease the product yields due to formation of tar-like products. The presence of a base increases the rate but also considerably decreases the yields of the products due to the formation of a large amount of tar-like products. Enamines 1a–e, bearing imidazole and isoxazole rings and several hydroxamoyl chlorides 2a–h bearing aryls with electron-withdrawing and releasing groups, pyridine and cyclohexane can be used for the synthesis of azolylisoxazoles 4a–p. Reactions of enamines with hydroxamoyl chlorides mainly lead to aromatic isoxazoles via intermediate 4,5-dihydro-5-aminoisoxazoles, which in some cases were isolated [23,24,26]. The latter could be transformed to aromatic isoxazoles by treatment with bases or acids [32]. Reactions of β-azolyl enamines 1a–e with hydroxamoyl chlorides 2a–h mainly afford aromatic isoxazoles 4b,d,f–p and intermediate isoxazolines were not detected by TLC analysis. Conversely, TLC allowed to observe the formation of imidazolylisoxazolines 3 as a result of the reaction of enamines 1a,b with hydroxamoyl chlorides 2. The isoxazolines transform to aromatic isoxazoles 4 during purification. Fortunately, we were able to isolate the products of the reaction of β-imidazolyl enamines 1a,b with hydroxamoyl chlorides 2a,d,g, and the novel imidazolylisoxazolines 3a–c as pure stereoisomers in 67, 65 and 21% yields, respectively. To the best of our knowledge it is the first example of a stereoselective formation of 4-aryl(heteroaryl)isoxazolines in the reaction of acyclic enamines with nitrile oxides. The stereoselective formation of trans-4-alkylisoxazolines in the reaction of β-azolyl enamines with benzonitrile oxides was observed by Pokar et al. [34] in 1980 and diastereoselective formation of fused isoxazolines was recently reported by Jelizi et al. [35]. Their structures are assigned as trans-isomers which are deduced from coupling constants of 4.0 and 4.2 Hz for the C4–H and C5–H in the NMR spectra (see Supporting Information File 2 for NMR spectra descriptions and copies). In turn, isoxazolines 3b,c were easily transformed to aromatic isoxazoles 4c,h in aqueous acetic acid at room temperature. Interestingly, the analogous reaction of hydroxamoyl chlorides 2c,f,g with enamines 1a,b leads directly to aromatic 4-imidazolylisoxazoles 4b,d,e–g. Probably isoxazolines 3a–c are more stable than other compounds of type 3 under column purification conditions. The 3,4-disubstituted isoxazole structures of compounds 4a–p were confirmed by the combination of mass spectrometry, NMR spectroscopy, and X-ray analysis (see Figure 3, Figure 4 and Supporting Information File 2 for details of X-ray study of compounds 4a,o,p).
![[1860-5397-12-233-3]](/bjoc/content/figures/1860-5397-12-233-3.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 3: Imidazolylisoxazole 4a according to XRD data in the thermal ellipsoids of the 50% probability level.
Figure 3: Imidazolylisoxazole 4a according to XRD data in the thermal ellipsoids of the 50% probability level....
![[1860-5397-12-233-4]](/bjoc/content/figures/1860-5397-12-233-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Isoxazolylisoxazole 4p according to XRD data with thermal ellipsoids of 50% probability level.
Figure 4: Isoxazolylisoxazole 4p according to XRD data with thermal ellipsoids of 50% probability level.
According to the XRD data, the molecules of compound 4a are non-planar with the Ph substituent turned toward the oxazole ring by a 51° angle and the imidazole ring turned toward the oxazole moiety by 23°. Crystals of 4a possess a chiral packing, where not a shortened intermolecular contact is observable. The bond lengths and angles in molecules 4a,o,p are standard. Apparently, hydroxamoyl chlorides transform to nitrile oxides under conditions of the isoxazole synthesis. The formation of isoxazolines 3b,c in the reaction and their transformation to isoxazoles under mild conditions supports a mechanism where the isoxazolines are the intermediates. The exclusive formation of isoxazolines as trans isomers from E-enamines allow us to conclude that the reaction of β-azolyl enamines with hydroxamoyl chlorides proceeds in a regio- and stereospecific manner. In contrast to the reactions of these β-azolyl enamines, similar reactions of β-alkyl enamines as reported by Bujak et al. [24] are regioselective and not stereospecific and therefore not concerted. We could propose stepwise (path 1) and concerted (path 2) reaction mechanisms for the formation of isoxazolines 3a–c as depicted in Scheme 3, in accordance with the proposed reaction mechanisms of heterocyclic enamines proposed earlier by Elliott and co-workers [36]. Path 1 includes the formation of intermediate A as a result of the electrophilic substitution of the β-H atom of the enamines 1a,b after treatment with hydroxamoyl chlorides 2a,d,g.
![[1860-5397-12-233-i3]](/bjoc/content/inline/1860-5397-12-233-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible mechanisms for reaction of hydroxamoyl chlorides 2 with imidazolyl enamines 1a,b.
Scheme 3: Plausible mechanisms for reaction of hydroxamoyl chlorides 2 with imidazolyl enamines 1a,b.
The intermediate A undergoes cyclization to oxazolines 3a–c via addition of its hydroxy group onto the double bond of the enamine fragment. This kind of cyclization was ruled out by Elliott et al. [36] because of a disfavored 5-endo-trig cyclization for the corresponding intermediate oxime.
Path 2 involves the initial transformation of hydroxamoyl chloride to a nitrile oxide, followed by concerted cyclization to triazolines 3a–c. We have found that introduction of electron-withdrawing substituents into the aryl moiety of the hydroxamoyl chloride increases the rate of reaction. Thus, the reaction times of enamine 1e with hydroxamoyl chloride 2a (R = Ph) as determined by TLC analysis is 30 h (product 4p), while those for 2f (R = 6-Cl-2-FC6H3) is 18 h (product 4l) and for 2b (R = 4-MeOC6H4) 32 h (products 4i), respectively. The regio- and stereospecificity and the apparent reaction rate increase by the introduction of electron-withdrawing substituents into the benzonitrile oxide structure, allow to presume that the reaction of enamines with nitrile oxides can be described as 1,3-dipolar cycloadditions with inverse electron demand, similarly to the reaction of enamines with azides [37].
To gain deeper insights into the mechanism of the cycloaddition between nitrile oxides and enamines, quantum chemical calculations were carried out using the Gaussian 09 [38] programs package at B3LYP/def2-TZVP [39-42] theory level. Grimme’s D3BJ dispersion correction [43,44] was applied to improve the long range interactions related calculation accuracy [45]. To the best of our knowledge there is not a high level of theoretical investigations reported on the possible reaction pathways of nitrile oxides with acyclic enamines so far, but only a few works based on a semi-empirical approach and a study of Domingo on reactivity of exocyclic enamines [46-50].
For the theoretical investigation, nitrile oxide 6a and enamine 1a were chosen as the model reactants. The geometries of starting molecules and products can be associated with a local minimum on the potential energy surface (PES) as proven by calculation of the vibrational frequencies among which not an imaginary value was found. Transition state geometries were proven by the presence of the only imaginary frequency appropriate to the reaction’s pathway. Geometry optimization performed on the starting enamine 1a allowed localizing four minimums on the PES with geometries shown in Figure 5.
![[1860-5397-12-233-5]](/bjoc/content/figures/1860-5397-12-233-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Geometries of enamine 1a appropriate to the calculated minima on the PES, and their relative free energies at 298.15 K in gas phase conditions.
Figure 5: Geometries of enamine 1a appropriate to the calculated minima on the PES, and their relative free e...
The transition state between the lowest energy E- and Z-isomers of 1a, 1a_1 and 1a_3, respectively, was calculated by scanning the dihedral angle around the double bond, and found to be 38.09 kcal∙mol−1 above 1a_1 in free energy. Calculations of the suggested mechanisms (Scheme 3) performed for E-isomer 1a_1 and nitrile oxide 6a allowed localizing a concerted transition state for both, observed and not observed regioisomers 3 and 8, respectively. Investigation of the stepwise mechanism’s pathway allowed locating the only transition state (1a_1−7), which is appropriate to the addition of nitrile oxide 2a onto the double bond of enamine 1a_1, and the respective product – oxime 7. However, the intermediate 7 was found to cyclize to isoxazoline 3a through the transition state corresponding to the concerted mechanism, e.g., via TS 1a_1−3a (Scheme 4). Interestingly, isoxazolines 9 and 10 with cis orientation of the Az and NMe2 groups, were found to be the products of transition states 1a_3−9 and 1a_3−10, respectively, both based on 1a_3 geometry of enamine 1a, whereas transition states based on 1a_1 geometry of the enamine lead to isoxazolines 3a and 8 with trans orientation of the Az and NMe2 groups (Scheme 4). Thus, one could conclude, that the configuration around the double bond of the enamine controls the stereoconfiguration of the isoxazoline formed. Apparently, the lower stability of 1a_3 compared to 1a_1, results in an equilibrium strongly shifted towards the latter and, consequently, in the stereoselective formation of isoxazoline 3a.
![[1860-5397-12-233-i4]](/bjoc/content/inline/1860-5397-12-233-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Calculated pathways for the formation of experimentally observed 3a, regioisomer 7 and isoxazoline 8.
Scheme 4: Calculated pathways for the formation of experimentally observed 3a, regioisomer 7 and isoxazoline 8...
According to the obtained transition state geometries of the cycloaddition, the concerted mechanism is quite asynchronous, which is indicated by the shorter length of the forming C–C σ-bond compared to C–O (Figure 6). A highly asynchronous transition state was also observed by the Domingo group some years ago for the reaction of exocyclic enamines with benzonitrile oxides leading to spirocyclic isoxazolines [46].
![[1860-5397-12-233-6]](/bjoc/content/figures/1860-5397-12-233-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Structures of the localized transition states. Lengths of the forming bonds are given in Å.
Figure 6: Structures of the localized transition states. Lengths of the forming bonds are given in Å.
It is remarked, that transition states 1a_1−3a and 1a_3−9 have longer lengths of the forming bonds compared to those of transition states 1a_1−8 and 1a_3–10, which indicates an easier attainability of the formers. Calculations of the thermodynamic parameters suggest that transition state 1a_1−3a, leading to observed product 3a, is by 8.9 kcal∙mol−1 more stable than transition state 1a_1−8, leading to the not observed regioisomer 8, and by 5.3 kcal∙mol−1 more stable than state 1a_1−7 of the stepwise mechanism. Isoxazoline 3a in its own is by 7.1 kcal∙mol−1 more stable than regioisomer 8.
These values clearly show a lower energy barrier for transition state 1a_1−3a compared to the others considered here, which explains the observed regioselective formation of isoxazoline 3a (Figure 7). An analysis, applying geometry strain model and molecular orbitals theory [51], reveals that the geometry distortion of nitrile oxide 6a has a major contribution to the activation energy barrier, whereas the geometry distortion energy of the enamine is minor. Also, the analysis shows close values of the orbital interaction energy for all the found transition states (Table 1). It should be noted the close orbital interaction energy values are obtained at longer distances between the reactants in case of transition states 1a_3a and 1a_9, as compared to transition states 1a_8 and 1a_10, respectively. This indicates a longer-range orbital interaction between the reactants in the case of formers, resulting in smaller geometry distortions.
![[1860-5397-12-233-7]](/bjoc/content/figures/1860-5397-12-233-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Summary of the calculated pathways of the cycloaddition reaction between enamine 1a and benzonitrile oxide 6a. The pathway leading to the observed product is highlighted green. Values of the free energies are given in respect to the sum of free energies of enamine 1a_1 and nitrile oxide 6a at the optimized geometries.
Figure 7: Summary of the calculated pathways of the cycloaddition reaction between enamine 1a and benzonitril...
Thus, the lowest electronic activation energy of transition state 1a_1−3a among all calculated transition states is a result of both, an easier attainability of the state governed by longer range orbital interactions, and a smaller value of the total geometry distortion energy (Table 1).
Table 1: Calculated data of the electronic activation energy (∆E), orbital interaction energy (∆Ei), and geometry distortion energies (∆Ed).a
| Entry | TS | ∆Eb | ∆Ei | ∆Ed total | ∆Ed enamine | ∆Ed nitrile oxide |
|---|---|---|---|---|---|---|
| 1 | 1a_1−3a | 8.0384 | −22.8691 | 30.9075 | 7.0448 | 23.8627 |
| 2 | 1a_1−7 | 12.8883 | −23.3032 | 36.1915 | 9.5132 | 26.6783 |
| 3 | 1a_1−8 | 16.9191 | −22.0768 | 38.9959 | 10.4814 | 28.5145 |
| 4 | 1a_3−9 | 8.4848 | −24.1853 | 32.6701 | 7.3005 | 25.3696 |
| 5 | 1a_3−10 | 22.2029 | −23.0625 | 45.2654 | 13.7263 | 31.5391 |
aThe values are given with respect to enamine 1a_1 (1a_3 for TS 1a_3−9 and 1a_3−10) and nitrile oxide 6a; b∆E given in kcal·mol−1.
Plots of respective orbitals at transition state geometries show a better orbital interaction in 1a_1−3a transition state compared to 1a_1−8. The HOMO of 1a_1 is predominantly localized at the β-carbon atom from the amino group whereas the LUMO of 6a has major localization on the carbon atom of the nitrile group (Figure 8). Calculated energy gaps between HOMO of 1a_1 and LUMO of 6a and vice versa at optimized geometries are 3.68 eV and 4.57 eV, respectively. This finding is in agreement with the inverse electron-demand concept for the investigated reaction which explains well the observed reaction rate increase when an electron-withdrawing group is introduced to the structure of nitrile oxide. Thus, according to calculations, reaction of enamines with nitrile oxides leading to isoxazolines, complies to cycloaddition with inverse electron-demand. The observed stereoselectivity of the cycloaddition is probably driven by the higher stability of the E-isomer of the starting enamine, whereas the regioselectivity is controlled by better orbital overlap in the transition state leading to the experimentally observed regioisomer.
![[1860-5397-12-233-8]](/bjoc/content/figures/1860-5397-12-233-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Isosurface plots of the HOMO of enamine 1a_1 (bottom) and the LUMO of nitrile oxide 6 (top) in the geometries of transition states 1a-1−3a (left) and 1a_1−8 (right), visualized at isovalue 0.05 by the use of Chemcraft 1.8 program (http://www.chemcraftprog.com/).
Figure 8: Isosurface plots of the HOMO of enamine 1a_1 (bottom) and the LUMO of nitrile oxide 6 (top) in the ...
Conclusion
β-Azolyl enamines bearing imidazole and isoxazole rings were shown to react regioselectively with aryl, pyridyl and cyclohexylhydroxamoyl chlorides in the absence of a base or a catalyst to afford 4-azolylisoxazoles as the only isomers. The data of combined experimental and theoretical studies allow classifying the reaction of enamines with nitrile oxides as inverse electron-demand 1,3-dipolar cycloaddition. The found stereoselectivity of the cycloaddition is driven by the higher stability of the E-isomer of the starting enamine, whereas the regioselectivity is controlled by better orbital overlap in the transition state leading to the experimentally observed regioisomer. The reason for the lowest electronic activation energy leading to the experimentally observed product among all calculated transition states, is caused by both relatively better stabilizing orbital interactions and relatively smaller geometry distortion energies.
Experimental
1H and 13C NMR spectra were recorded on Bruker Avance II spectrometer in DMSO-d6 or CDCl3 (400 and 100 MHz, respectively) using Me4Si as an internal standard. Mass experiments were performed on Shimadzu GCMS-QP2010 Ultra gas chromatograph operating at an ionization potential of 70 eV (EI). The IR data have been recorded on a Bruker Alpha (NPVO, ZnSe) FTIR spectrometer. Microanalyses were performed on PerkinElmer Series II CHNS/O 2400 elemental analyzer. The melting point was determined on a Stuart SMP 3 apparatus. The progress of the reactions and the purity of the compounds were monitored by TLC on TLC Silica gel 60 F245 Aluminum sheets (Merck KGaA) in EtOAc/hexane (1:2), EtOAc/hexane (1:1), CHCl3/EtOH (1:1), CHCl3/EtOH (20:1), CHCl3/EtOH (50:1) system.
General procedure for the preparation of 4-azolylisoxazoles 4a,b,d–g,i–p
To a solution of the corresponding enamine 1 (1.1 mmol) in 1,4-dioxane (5 mL) an appropriate hydroxamoyl chloride 2 (1 mmol) was added. The reaction mixture was stirred at room temperature for 12–32 h. The solvent was removed under reduced pressure and then 10 mL of C2H5OH/H2O (1:1) mixture was added to the oily precipitate. The formed precipitate was filtered off, washed with EtOH and purified by silica gel (60–120) column chromatography (EtOAc/hexane, CHCl3/EtOH, CH2Cl2) or crystallization from EtOH to afford the desired isoxazole 4.
4-(1-Methyl-5-nitro-1H-imidazol-4-yl)-3-phenylisoxazole (4a). Colorless powder, yield 53%, mp 130–132 °С (column, CHCl3/EtOH 20:1); IR (ν/сm−1): 3097, 1611, 1472, 1329, 1134; 1H NMR (DMSO-d6) δ 3.95 (s, 3H, CH3), 7.12–7.62 (m, 5H, CHAr), 8.13 (s, 1H, CHimidaz.), 9.33 (s, 1H, C5−H); 13C NMR (DMSO-d6) δ 35.5, 111.4, 127.6, 128.3, 128.7, 129.8, 132.2, 135.9, 141.7, 160.3, 160.9; EIMS (m/z): 270 [M]+; anal. calcd for C13H10N4O3: C, 57.78; H, 3.73; N, 20.73; found: C, 57.43; H, 3.45; N, 20.99.
4-[4-(1-Methyl-5-nitro-1H-imidazol-4-yl)isoxazol-3-yl]benzonitrile (4b). Pink powder, yield 65%, mp 189–191 °С (column, CH2Cl2); IR (ν/сm−1): 3109, 2227, 1615, 1480, 1354; 1Н NMR (CDCl3) δ 4.02 (s, 3H, CH3), 7.55 (s, 1H, CHimidaz.), 7.67 (s, 4H, CHAr), 8.99 (s, 1H, C5–H); 13С NMR (CDCl3) δ 36.3, 111.4, 113.6, 118.5, 129.3, 132.3, 133.0, 133.6, 135.9, 140.5, 160.0, 160.8; EIMS (m/z): 295 [M]+; anal. calcd for C14H9N5O3: C, 56.95; H, 3.07; N, 23.72; found: C, 56.85; H, 3.01; N, 23.79.
Preparation of isoxazolines 3a–c
Isoxazolines 3a–c were synthesized in the same manner as isoxazoles 4a,b,d–g,i–p (see general procedure). The reaction time is 12 h (3a,b) and 48 h (3c).
N,N-Dimethyl-4-(1-methyl-5-nitro-1H-imidazol-4-yl)-3-phenyl-4,5-dihydroisoxazol-5-amine (3a). Colorless powder, yield 67%, mp 132–134 °С (column, EtOAc/hexane 1:1); 1H NMR (DMSO-d6) δ 2.34 (s, 6H, NMe2), 3.91 (s, 3H, NCH3), 5.28 (d, J = 4.2 Hz, 1Н, СН), 5.30 (d, J = 4.2 Hz, 1Н, СН), 7.29–7.33 (m, 3H, HAr), 7.49–7.58 (m, 3H, HAr), 7.89 (s, 1H, CHimidaz.); 13С NMR (DMSO-d6) δ 35.9, 39.3, 49.1, 104.2, 126.8, 128.1, 129.3, 130.2, 135.5, 142.2, 142.7, 155.2; EIMS (m/z): 315 [M]+; anal. calcd for C15H17N5O3: C, 57.13; H, 5.43; N, 22.21.; found: C, 57.07; H, 5.66; N, 22.10.
3-(4-Chlorophenyl)-N,N-dimethyl-4-(1-methyl-5-nitro-1H-imidazol-4-yl)-4,5-dihydroisoxazol-5-amine (3b). Colorless powder, yield 65%, mp 164–166 °С (EtOH); IR (ν/сm−1): 3120, 1490, 1361, 1306; 1Н NMR (DMSO-d6) δ 2.34 (s, 6H, NMe2), 3.92 (s, 3H, СН3), 5.29 (d, J = 4.0 Hz, 1Н, СН), 5.32 (d, J = 4.0 Hz, 1Н, СН), 7.31 (d, J = 8.8 Hz, 2H, HAr), 7.54 (d, J = 8.8 Hz, 2 H, HAr), 7.89 (s, 1H, CHimidaz.); 13С NMR (DMSO-d6) δ 35.9, 39.3, 49.0, 104.6, 128.3, 128.6, 129.4, 134.8, 135.5, 142.2, 142.4, 154.4; EIMS (m/z): 349 [M]+; anal. calcd for C15H16ClN5O3: C, 51.51; H, 4.61; N, 20.02; found: C, 51.65; H, 4.47; N, 20.28.
Transformation of isoxazolines 3b,c to isoxazoles 4c,h
A solution of corresponding isoxazoline 3b,c (1 mmol) in a mixture of H2O/HOAc (1:1, 2 mL) was stirred at room temperature for 30 minutes (3b) or overnight (3c). The formed precipitate was filtered off, washed with H2O and dried in a desiccator over P2O5 and recrystallized from EtOH (4c) or purified by flash column chromatography (CH2Cl2) (4h).
3-(4-Chlorophenyl)-4-(1-methyl-5-nitro-1H-imidazol-4-yl)isoxazole (4c). Pink powder, yield 70%, mp 129–131 °С (EtOH); 1Н NMR (DMSO-d6) δ 3.95 (s, 3H, СН3), 7.43–7.56 (m, 4H, HAr), 8.13 (s, 1H, CHimidaz.), 9.37 (s, 1H, C5–H); 13С NMR (DMSO-d6) δ 35.5, 111.4, 127.2, 128.8, 129.5, 131.6, 134.7, 135.9, 141.8, 159.4, 161.3; EIMS (m/z): 304 [M]+; anal. calcd for C13H9ClN4O3: C, 51.25; H, 2.98; N, 18.39; found: C, 51.36; H, 2.79; N, 18.52.
X-ray diffraction study
X-ray analyses were accomplished on an Xcalibur 3 diffractometer using the standard procedure (graphite-monochromated Mo K-irradiation, ω-scanning with step 1o, T = 150.00(10) K) (4o,p) or 295(2) K (4a) (See Supporting Information File 2). Using Olex2 [52], the structures were solved with the Superflip [53] structure solution program using Charge Flipping and refined with the ShelXL [54] refinement package using Least Squares minimization. Deposition numbers for compounds 4a (1473400), 4o (1405543) and 4p (1405542) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Supporting Information
Supporting information features copies of 1H and 13C NMR spectra for all compounds synthesized and full experimental and analytical data for isoxazolines 3 and isoxazoles 4.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 269.2 KB | Download |
| Supporting Information File 2: NMR spectra of compounds 3a–c, 4a–p and X-ray study of isoxazoles 4a,o,p. | ||
| Format: PDF | Size: 6.6 MB | Download |
| Supporting Information File 3: Crystallographic information file for compound 4a. | ||
| Format: CIF | Size: 16.1 KB | Download |
| Supporting Information File 4: Crystallographic information file for compound 4o. | ||
| Format: CIF | Size: 15.4 KB | Download |
| Supporting Information File 5: Crystallographic information file for compound 4p. | ||
| Format: CIF | Size: 17.9 KB | Download |
References
-
Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Gehling, V. S.; Hewitt, M. C.; Vaswani, R. G.; Leblanc, Y.; Côté, A.; Nasveschuk, C. G.; Taylor, A. M.; Harmange, J.-C.; Audia, J. E.; Pardo, E.; Joshi, S.; Sandy, P.; Mertz, J. A.; Sims, R. J.; Bergeron, L.; Bryant, B. M.; Bellon, S.; Poy, F.; Jayaram, H.; Sankaranarayanan, R.; Yellapantula, S.; Srinivasamurthy, N. B.; Birudukota, S.; Albrecht, B. K. ACS Med. Chem. Lett. 2013, 4, 835–840. doi:10.1021/ml4001485
Return to citation in text: [1] -
Di Nunno, L.; Vitale, P.; Scilimati, A.; Tacconelli, S.; Patrignani, P. J. Med. Chem. 2004, 47, 4881–4890. doi:10.1021/jm040782x
Return to citation in text: [1] -
Girardin, M.; Dolman, S. J.; Lauzon, S.; Ouellet, S. G.; Hughes, G.; Fernandez, P.; Zhou, G.; O’Shea, P. D. Org. Process Res. Dev. 2011, 15, 1073–1080. doi:10.1021/op200186d
Return to citation in text: [1] [2] [3] -
Brodney, M. A.; Beck, E. M.; Butler, C. R.; Barreiro, G.; Johnson, E. F.; Riddell, D.; Parris, K.; Nolan, C. E.; Fan, Y.; Atchison, K.; Gonzales, C.; Robshaw, A. E.; Doran, S. D.; Bundesmann, M. W.; Buzon, L.; Dutra, J.; Henegar, K.; LaChapelle, E.; Hou, X.; Rogers, B. N.; Pandit, J.; Lira, R.; Martinez-Alsina, L.; Mikochik, P.; Murray, J. C.; Ogilvie, K.; Price, L.; Sakya, S. M.; Yu, A.; Zhang, Y.; O’Neill, B. T. J. Med. Chem. 2015, 58, 3223–3252. doi:10.1021/acs.jmedchem.5b00191
Return to citation in text: [1] -
Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/RCR4503
Return to citation in text: [1] [2] -
Bakulev, V. A.; Tarasov, E. V.; Morzherin, Y. Y.; Luyten, I.; Toppet, S.; Dehaen, W. Tetrahedron 1998, 54, 8501–8514. doi:10.1016/S0040-4020(98)00449-9
Return to citation in text: [1] -
Bakulev, V. A.; Dehaen, W. The Chemistry of 1,2,3-Thiadiazoles; John Wiley & Sons Inc.: Hoboken, 2004. doi:10.1002/0471656992
Return to citation in text: [1] -
Bakulev, V. A.; Mokrushin, V. S. Khim. Geterotsikl. Soedin. 1986, 1011–1028.
Return to citation in text: [1] -
Shafran, Y. M.; Bakulev, V. A.; Mokrushin, V. S.; Alexeev, S. G. Khim. Geterotsikl. Soedin. 1984, 1266–1270.
Return to citation in text: [1] -
Bakulev, V.; Dehaen, W.; Beryozkina, T. Thermal Rearrangements and Transformations of 1,2,3-Triazoles. In Chemistry of 1,2,3-triazoles; Dehaen, W.; Bakulev, V., Eds.; Springer-Verlag: Berlin, Heidelberg, Germany, 2015; pp 1–50.
Return to citation in text: [1] -
Leban, J.; Tasler, S.; Saeb, W.; Chevrier, C. IL17 And IFN-gamma inhibition for the treatment of autoimmune inflammation. Canadian Patent CA 2,825,779, Aug 2, 2012.
Chem. Abstr. 2012, 158, 17532d.
Return to citation in text: [1] [2] [3] -
Buettelmann, B.; Han, B.; Knust, H.; Thomas, A. Aryl-isoxazol-4-yl-imidazole derivatives. WO Patent WO 2007,07,4078, July 5, 2007.
Chem. Abstr. 2007, 147, 143428.
Return to citation in text: [1] [2] [3] -
Thorarensen, A.; Ruble, C. J.; Fisher, J. F.; Romero, D. L.; Beauchamp, T. J.; Northuis, J. M. Antibacterial benzoic acid derivatives. WO Pantent WO 2004,01,8428, March 4, 2004.
Chem. Abstr. 2004, 140, 235498.
Return to citation in text: [1] [2] [3] [4] -
Eiden, F.; Patzelt, G. Arch. Pharm. 1986, 319, 242–251. doi:10.1002/ardp.19863190310
Return to citation in text: [1] [2] -
Lautens, M.; Roy, A. Org. Lett. 2000, 2, 555–557. doi:10.1021/ol005519e
Return to citation in text: [1] -
Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K. B.; Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210–216. doi:10.1021/ja0471525
Return to citation in text: [1] -
Jones, R. C. F.; Bhalay, G.; Carter, P. A. J. Chem. Soc., Perkin Trans. 1 1993, 1715–1716. doi:10.1039/P19930001715
Return to citation in text: [1] -
Jones, R. C. F.; Dunn, S. H.; Duller, K. A. M. J. Chem. Soc., Perkin Trans. 1 1996, 1319–1321. doi:10.1039/P19960001319
Return to citation in text: [1] -
Jones, R. C. F.; Bhalay, G.; Carter, P. A.; Duller, K. A. M.; Dunn, S. H. J. Chem. Soc., Perkin Trans. 1 1999, 765–776. doi:10.1039/a900120d
Return to citation in text: [1] -
Jones, R. C. F.; Duller, K. A. M. ARKIVOC 2002, No. viii, 34–39.
Return to citation in text: [1] -
Sasaki, T.; Yoshioka, T. Bull. Chem. Soc. Jpn. 1968, 41, 2212–2215. doi:10.1246/bcsj.41.2212
Return to citation in text: [1] -
Krompiec, S.; Bujak, P.; Szczepankiewicz, W. Tetrahedron Lett. 2008, 49, 6071–6074. doi:10.1016/j.tetlet.2008.07.176
Return to citation in text: [1] [2] [3] -
Bujak, P.; Krompiec, S.; Malarz, J.; Krompiec, M.; Filapek, M.; Danikiewicz, W.; Kania, M.; Gębarowska, K.; Grudzka, I. Tetrahedron 2010, 66, 5972–5981. doi:10.1016/j.tet.2010.06.040
Return to citation in text: [1] [2] [3] [4] -
Gong, Y.; Wang, Y.; Zhao, W.-T.; Tang, X.-Y. J. Chem. Res. 2013, 37, 499–502. doi:10.3184/174751913X13735527483211
Return to citation in text: [1] -
Bakulev, V. A.; Efimov, I. V.; Belyaev, N. A.; Zhidovinov, S. S.; Rozin, Yu. A.; Volkova, N. N.; Khabarova, A. A.; El'tsov, O. S. Chem. Heterocycl. Compd. 2013, 48, 1880–1882. doi:10.1007/s10593-013-1225-1
Return to citation in text: [1] [2] [3] -
Bakulev, V. A.; Efimov, I. V.; Belyaev, N. A.; Rozin, Y. A.; Volkova, N. N.; El’tsov, O. S. Chem. Heterocycl. Compd. 2012, 47, 1593–1595. doi:10.1007/s10593-012-0952-z
Return to citation in text: [1] -
Beryozkina, T. V.; Zhidovinov, S. S.; Shafran, Y. M.; Eltsov, O. S.; Slepukhin, P. A.; Leban, J.; Marquez, J.; Bakulev, V. A. Tetrahedron 2014, 70, 3915–3923. doi:10.1016/j.tet.2014.04.015
Return to citation in text: [1] -
Shafran, Y.; Rozin, Y.; Beryozkina, T.; Zhidovinov, S.; Eltsov, O.; Subbotina, J.; Leban, J.; Novikova, R.; Bakulev, V. Org. Biomol. Chem. 2012, 10, 5795–5798. doi:10.1039/c2ob25331c
Return to citation in text: [1] -
Hosmane, R. S.; Bhan, A.; Rauser, M. E. J. Org. Chem. 1985, 50, 5892–5895. doi:10.1021/jo00350a099
Return to citation in text: [1] -
Jäger, V.; Colinas, P. A. Nitrile Oxides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons Inc.: New York, 2002; Vol. 59, pp 361–472. doi:10.1002/0471221902.ch6
Return to citation in text: [1] [2] -
Caramella, P.; Grunanger, P. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984; Vol. 1, pp 291–392.
Return to citation in text: [1] [2] [3] -
Grundmann, C.; Grünanger, P. The Nitrile Oxides; Springer-Verlag: Berlin, 1971; pp 47–58. doi:10.1007/978-3-642-99991-8
Return to citation in text: [1] [2] -
Pokar, D.; Rossi, L. M.; Trimarco, P.; Vago, L. J. Heterocycl. Chem. 1980, 17, 881–885. doi:10.1002/jhet.5570170508
Return to citation in text: [1] -
Jelizi, H.; Wannassi, N.; Rammah, M. E. B.; Ciamala, K.; Knorr, M.; Rousselin, Y.; Kubicki, M. M.; Strohmann, C.; Enescu, M. J. Heterocycl. Chem. 2014, 51, 383–391. doi:10.1002/jhet.1588
Return to citation in text: [1] -
Altuğ, C.; Dürüst, Y.; Elliott, M. C.; Kariuki, B. M.; Rorstad, T.; Zaal, M. Org. Biomol. Chem. 2010, 8, 4978–4986. doi:10.1039/c0ob00286k
Return to citation in text: [1] [2] -
Lopez, A. S.; Munk, M. E.; Houk, K. N. J. Org. Chem. 2013, 78, 1576–1582. doi:10.1021/jo302695n
Return to citation in text: [1] -
Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.
Return to citation in text: [1] -
Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a
Return to citation in text: [1] -
Weigend, F. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. doi:10.1039/b515623h
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344
Return to citation in text: [1] -
Grimme, S.; Ehrlich, S.; Goerigk, L. J. Comput. Chem. 2011, 32, 1456–1465. doi:10.1002/jcc.21759
Return to citation in text: [1] -
Armstrong, A.; Boto, R. A.; Dingwall, P.; Contreras-García, J.; Harvey, M. J.; Mason, N. J.; Rzepa, H. S. Chem. Sci. 2014, 5, 2057–2071. doi:10.1039/c3sc53416b
Return to citation in text: [1] -
Houk, K. N.; Sims, J.; Duke, R. E., Jr.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287–7301. doi:10.1021/ja00803a017
Return to citation in text: [1] [2] -
Caramella, P.; Corsico, A. C.; Corsaro, A.; Del Monto, D.; Albini, F. M. Tetrahedron 1982, 38, 173–182. doi:10.1016/0040-4020(82)85063-1
Return to citation in text: [1] -
Tsoleridis, C. A.; Dimtsas, J.; Hatzimimikou, D.; Stephanidou-Stephanatou, J. Tetrahedron 2006, 62, 4232–4242. doi:10.1016/j.tet.2006.01.064
Return to citation in text: [1] -
Domingo, L. R.; Picher, M. T.; Arroyo, P.; Saez, J. A. J. Org. Chem. 2006, 71, 9319–9330. doi:10.1021/jo0613986
Return to citation in text: [1] -
Ess, D. H.; Houk, K. N. J. Am. Chem. Soc. 2007, 129, 10646–10647. doi:10.1021/ja0734086
Return to citation in text: [1] -
Fernández, I.; Bickelhaupt, F. M. Chem. Soc. Rev. 2014, 43, 4953–4967. doi:10.1039/c4cs00055b
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/S0021889808042726
Return to citation in text: [1] -
Palatinus, L.; Chapuis, G. J. Appl. Crystallogr. 2007, 40, 786–790. doi:10.1107/S0021889807029238
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1]
| 35. | Jelizi, H.; Wannassi, N.; Rammah, M. E. B.; Ciamala, K.; Knorr, M.; Rousselin, Y.; Kubicki, M. M.; Strohmann, C.; Enescu, M. J. Heterocycl. Chem. 2014, 51, 383–391. doi:10.1002/jhet.1588 |
| 24. | Bujak, P.; Krompiec, S.; Malarz, J.; Krompiec, M.; Filapek, M.; Danikiewicz, W.; Kania, M.; Gębarowska, K.; Grudzka, I. Tetrahedron 2010, 66, 5972–5981. doi:10.1016/j.tet.2010.06.040 |
| 36. | Altuğ, C.; Dürüst, Y.; Elliott, M. C.; Kariuki, B. M.; Rorstad, T.; Zaal, M. Org. Biomol. Chem. 2010, 8, 4978–4986. doi:10.1039/c0ob00286k |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 3. | Di Nunno, L.; Vitale, P.; Scilimati, A.; Tacconelli, S.; Patrignani, P. J. Med. Chem. 2004, 47, 4881–4890. doi:10.1021/jm040782x |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 13. |
Buettelmann, B.; Han, B.; Knust, H.; Thomas, A. Aryl-isoxazol-4-yl-imidazole derivatives. WO Patent WO 2007,07,4078, July 5, 2007.
Chem. Abstr. 2007, 147, 143428. |
| 14. |
Thorarensen, A.; Ruble, C. J.; Fisher, J. F.; Romero, D. L.; Beauchamp, T. J.; Northuis, J. M. Antibacterial benzoic acid derivatives. WO Pantent WO 2004,01,8428, March 4, 2004.
Chem. Abstr. 2004, 140, 235498. |
| 46. | Houk, K. N.; Sims, J.; Duke, R. E., Jr.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287–7301. doi:10.1021/ja00803a017 |
| 47. | Caramella, P.; Corsico, A. C.; Corsaro, A.; Del Monto, D.; Albini, F. M. Tetrahedron 1982, 38, 173–182. doi:10.1016/0040-4020(82)85063-1 |
| 48. | Tsoleridis, C. A.; Dimtsas, J.; Hatzimimikou, D.; Stephanidou-Stephanatou, J. Tetrahedron 2006, 62, 4232–4242. doi:10.1016/j.tet.2006.01.064 |
| 49. | Domingo, L. R.; Picher, M. T.; Arroyo, P.; Saez, J. A. J. Org. Chem. 2006, 71, 9319–9330. doi:10.1021/jo0613986 |
| 50. | Ess, D. H.; Houk, K. N. J. Am. Chem. Soc. 2007, 129, 10646–10647. doi:10.1021/ja0734086 |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 4. | Girardin, M.; Dolman, S. J.; Lauzon, S.; Ouellet, S. G.; Hughes, G.; Fernandez, P.; Zhou, G.; O’Shea, P. D. Org. Process Res. Dev. 2011, 15, 1073–1080. doi:10.1021/op200186d |
| 12. |
Leban, J.; Tasler, S.; Saeb, W.; Chevrier, C. IL17 And IFN-gamma inhibition for the treatment of autoimmune inflammation. Canadian Patent CA 2,825,779, Aug 2, 2012.
Chem. Abstr. 2012, 158, 17532d. |
| 13. |
Buettelmann, B.; Han, B.; Knust, H.; Thomas, A. Aryl-isoxazol-4-yl-imidazole derivatives. WO Patent WO 2007,07,4078, July 5, 2007.
Chem. Abstr. 2007, 147, 143428. |
| 14. |
Thorarensen, A.; Ruble, C. J.; Fisher, J. F.; Romero, D. L.; Beauchamp, T. J.; Northuis, J. M. Antibacterial benzoic acid derivatives. WO Pantent WO 2004,01,8428, March 4, 2004.
Chem. Abstr. 2004, 140, 235498. |
| 15. | Eiden, F.; Patzelt, G. Arch. Pharm. 1986, 319, 242–251. doi:10.1002/ardp.19863190310 |
| 16. | Lautens, M.; Roy, A. Org. Lett. 2000, 2, 555–557. doi:10.1021/ol005519e |
| 46. | Houk, K. N.; Sims, J.; Duke, R. E., Jr.; Strozier, R. W.; George, J. K. J. Am. Chem. Soc. 1973, 95, 7287–7301. doi:10.1021/ja00803a017 |
| 2. | Gehling, V. S.; Hewitt, M. C.; Vaswani, R. G.; Leblanc, Y.; Côté, A.; Nasveschuk, C. G.; Taylor, A. M.; Harmange, J.-C.; Audia, J. E.; Pardo, E.; Joshi, S.; Sandy, P.; Mertz, J. A.; Sims, R. J.; Bergeron, L.; Bryant, B. M.; Bellon, S.; Poy, F.; Jayaram, H.; Sankaranarayanan, R.; Yellapantula, S.; Srinivasamurthy, N. B.; Birudukota, S.; Albrecht, B. K. ACS Med. Chem. Lett. 2013, 4, 835–840. doi:10.1021/ml4001485 |
| 13. |
Buettelmann, B.; Han, B.; Knust, H.; Thomas, A. Aryl-isoxazol-4-yl-imidazole derivatives. WO Patent WO 2007,07,4078, July 5, 2007.
Chem. Abstr. 2007, 147, 143428. |
| 43. | Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344 |
| 44. | Grimme, S.; Ehrlich, S.; Goerigk, L. J. Comput. Chem. 2011, 32, 1456–1465. doi:10.1002/jcc.21759 |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 4. | Girardin, M.; Dolman, S. J.; Lauzon, S.; Ouellet, S. G.; Hughes, G.; Fernandez, P.; Zhou, G.; O’Shea, P. D. Org. Process Res. Dev. 2011, 15, 1073–1080. doi:10.1021/op200186d |
| 45. | Armstrong, A.; Boto, R. A.; Dingwall, P.; Contreras-García, J.; Harvey, M. J.; Mason, N. J.; Rzepa, H. S. Chem. Sci. 2014, 5, 2057–2071. doi:10.1039/c3sc53416b |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 6. | Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/RCR4503 |
| 6. | Galenko, A. V.; Khlebnikov, A. F.; Novikov, M. S.; Pakalnis, V. V.; Rostovskii, N. V. Russ. Chem. Rev. 2015, 84, 335–377. doi:10.1070/RCR4503 |
| 12. |
Leban, J.; Tasler, S.; Saeb, W.; Chevrier, C. IL17 And IFN-gamma inhibition for the treatment of autoimmune inflammation. Canadian Patent CA 2,825,779, Aug 2, 2012.
Chem. Abstr. 2012, 158, 17532d. |
| 39. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 40. | Weigend, F. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. doi:10.1039/b515623h |
| 41. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 42. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 5. | Brodney, M. A.; Beck, E. M.; Butler, C. R.; Barreiro, G.; Johnson, E. F.; Riddell, D.; Parris, K.; Nolan, C. E.; Fan, Y.; Atchison, K.; Gonzales, C.; Robshaw, A. E.; Doran, S. D.; Bundesmann, M. W.; Buzon, L.; Dutra, J.; Henegar, K.; LaChapelle, E.; Hou, X.; Rogers, B. N.; Pandit, J.; Lira, R.; Martinez-Alsina, L.; Mikochik, P.; Murray, J. C.; Ogilvie, K.; Price, L.; Sakya, S. M.; Yu, A.; Zhang, Y.; O’Neill, B. T. J. Med. Chem. 2015, 58, 3223–3252. doi:10.1021/acs.jmedchem.5b00191 |
| 36. | Altuğ, C.; Dürüst, Y.; Elliott, M. C.; Kariuki, B. M.; Rorstad, T.; Zaal, M. Org. Biomol. Chem. 2010, 8, 4978–4986. doi:10.1039/c0ob00286k |
| 4. | Girardin, M.; Dolman, S. J.; Lauzon, S.; Ouellet, S. G.; Hughes, G.; Fernandez, P.; Zhou, G.; O’Shea, P. D. Org. Process Res. Dev. 2011, 15, 1073–1080. doi:10.1021/op200186d |
| 1. | Giomi, D.; Cordero, F. M.; Machetti, F. Isoxazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: U. K., 2008; Vol. 4, pp 365–485. |
| 7. | Bakulev, V. A.; Tarasov, E. V.; Morzherin, Y. Y.; Luyten, I.; Toppet, S.; Dehaen, W. Tetrahedron 1998, 54, 8501–8514. doi:10.1016/S0040-4020(98)00449-9 |
| 8. | Bakulev, V. A.; Dehaen, W. The Chemistry of 1,2,3-Thiadiazoles; John Wiley & Sons Inc.: Hoboken, 2004. doi:10.1002/0471656992 |
| 9. | Bakulev, V. A.; Mokrushin, V. S. Khim. Geterotsikl. Soedin. 1986, 1011–1028. |
| 10. | Shafran, Y. M.; Bakulev, V. A.; Mokrushin, V. S.; Alexeev, S. G. Khim. Geterotsikl. Soedin. 1984, 1266–1270. |
| 11. | Bakulev, V.; Dehaen, W.; Beryozkina, T. Thermal Rearrangements and Transformations of 1,2,3-Triazoles. In Chemistry of 1,2,3-triazoles; Dehaen, W.; Bakulev, V., Eds.; Springer-Verlag: Berlin, Heidelberg, Germany, 2015; pp 1–50. |
| 37. | Lopez, A. S.; Munk, M. E.; Houk, K. N. J. Org. Chem. 2013, 78, 1576–1582. doi:10.1021/jo302695n |
| 23. | Krompiec, S.; Bujak, P.; Szczepankiewicz, W. Tetrahedron Lett. 2008, 49, 6071–6074. doi:10.1016/j.tetlet.2008.07.176 |
| 24. | Bujak, P.; Krompiec, S.; Malarz, J.; Krompiec, M.; Filapek, M.; Danikiewicz, W.; Kania, M.; Gębarowska, K.; Grudzka, I. Tetrahedron 2010, 66, 5972–5981. doi:10.1016/j.tet.2010.06.040 |
| 14. |
Thorarensen, A.; Ruble, C. J.; Fisher, J. F.; Romero, D. L.; Beauchamp, T. J.; Northuis, J. M. Antibacterial benzoic acid derivatives. WO Pantent WO 2004,01,8428, March 4, 2004.
Chem. Abstr. 2004, 140, 235498. |
| 17. | Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V. V.; Noodleman, L.; Sharpless, K. B.; Fokin, V. V. J. Am. Chem. Soc. 2005, 127, 210–216. doi:10.1021/ja0471525 |
| 51. | Fernández, I.; Bickelhaupt, F. M. Chem. Soc. Rev. 2014, 43, 4953–4967. doi:10.1039/c4cs00055b |
| 18. | Jones, R. C. F.; Bhalay, G.; Carter, P. A. J. Chem. Soc., Perkin Trans. 1 1993, 1715–1716. doi:10.1039/P19930001715 |
| 19. | Jones, R. C. F.; Dunn, S. H.; Duller, K. A. M. J. Chem. Soc., Perkin Trans. 1 1996, 1319–1321. doi:10.1039/P19960001319 |
| 20. | Jones, R. C. F.; Bhalay, G.; Carter, P. A.; Duller, K. A. M.; Dunn, S. H. J. Chem. Soc., Perkin Trans. 1 1999, 765–776. doi:10.1039/a900120d |
| 21. | Jones, R. C. F.; Duller, K. A. M. ARKIVOC 2002, No. viii, 34–39. |
| 22. | Sasaki, T.; Yoshioka, T. Bull. Chem. Soc. Jpn. 1968, 41, 2212–2215. doi:10.1246/bcsj.41.2212 |
| 23. | Krompiec, S.; Bujak, P.; Szczepankiewicz, W. Tetrahedron Lett. 2008, 49, 6071–6074. doi:10.1016/j.tetlet.2008.07.176 |
| 24. | Bujak, P.; Krompiec, S.; Malarz, J.; Krompiec, M.; Filapek, M.; Danikiewicz, W.; Kania, M.; Gębarowska, K.; Grudzka, I. Tetrahedron 2010, 66, 5972–5981. doi:10.1016/j.tet.2010.06.040 |
| 25. | Gong, Y.; Wang, Y.; Zhao, W.-T.; Tang, X.-Y. J. Chem. Res. 2013, 37, 499–502. doi:10.3184/174751913X13735527483211 |
| 26. | Bakulev, V. A.; Efimov, I. V.; Belyaev, N. A.; Zhidovinov, S. S.; Rozin, Yu. A.; Volkova, N. N.; Khabarova, A. A.; El'tsov, O. S. Chem. Heterocycl. Compd. 2013, 48, 1880–1882. doi:10.1007/s10593-013-1225-1 |
| 52. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/S0021889808042726 |
| 53. | Palatinus, L.; Chapuis, G. J. Appl. Crystallogr. 2007, 40, 786–790. doi:10.1107/S0021889807029238 |
| 32. | Caramella, P.; Grunanger, P. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984; Vol. 1, pp 291–392. |
| 34. | Pokar, D.; Rossi, L. M.; Trimarco, P.; Vago, L. J. Heterocycl. Chem. 1980, 17, 881–885. doi:10.1002/jhet.5570170508 |
| 12. |
Leban, J.; Tasler, S.; Saeb, W.; Chevrier, C. IL17 And IFN-gamma inhibition for the treatment of autoimmune inflammation. Canadian Patent CA 2,825,779, Aug 2, 2012.
Chem. Abstr. 2012, 158, 17532d. |
| 23. | Krompiec, S.; Bujak, P.; Szczepankiewicz, W. Tetrahedron Lett. 2008, 49, 6071–6074. doi:10.1016/j.tetlet.2008.07.176 |
| 24. | Bujak, P.; Krompiec, S.; Malarz, J.; Krompiec, M.; Filapek, M.; Danikiewicz, W.; Kania, M.; Gębarowska, K.; Grudzka, I. Tetrahedron 2010, 66, 5972–5981. doi:10.1016/j.tet.2010.06.040 |
| 26. | Bakulev, V. A.; Efimov, I. V.; Belyaev, N. A.; Zhidovinov, S. S.; Rozin, Yu. A.; Volkova, N. N.; Khabarova, A. A.; El'tsov, O. S. Chem. Heterocycl. Compd. 2013, 48, 1880–1882. doi:10.1007/s10593-013-1225-1 |
| 31. | Jäger, V.; Colinas, P. A. Nitrile Oxides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons Inc.: New York, 2002; Vol. 59, pp 361–472. doi:10.1002/0471221902.ch6 |
| 32. | Caramella, P.; Grunanger, P. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984; Vol. 1, pp 291–392. |
| 33. | Grundmann, C.; Grünanger, P. The Nitrile Oxides; Springer-Verlag: Berlin, 1971; pp 47–58. doi:10.1007/978-3-642-99991-8 |
| 14. |
Thorarensen, A.; Ruble, C. J.; Fisher, J. F.; Romero, D. L.; Beauchamp, T. J.; Northuis, J. M. Antibacterial benzoic acid derivatives. WO Pantent WO 2004,01,8428, March 4, 2004.
Chem. Abstr. 2004, 140, 235498. |
| 15. | Eiden, F.; Patzelt, G. Arch. Pharm. 1986, 319, 242–251. doi:10.1002/ardp.19863190310 |
| 26. | Bakulev, V. A.; Efimov, I. V.; Belyaev, N. A.; Zhidovinov, S. S.; Rozin, Yu. A.; Volkova, N. N.; Khabarova, A. A.; El'tsov, O. S. Chem. Heterocycl. Compd. 2013, 48, 1880–1882. doi:10.1007/s10593-013-1225-1 |
| 27. | Bakulev, V. A.; Efimov, I. V.; Belyaev, N. A.; Rozin, Y. A.; Volkova, N. N.; El’tsov, O. S. Chem. Heterocycl. Compd. 2012, 47, 1593–1595. doi:10.1007/s10593-012-0952-z |
| 28. | Beryozkina, T. V.; Zhidovinov, S. S.; Shafran, Y. M.; Eltsov, O. S.; Slepukhin, P. A.; Leban, J.; Marquez, J.; Bakulev, V. A. Tetrahedron 2014, 70, 3915–3923. doi:10.1016/j.tet.2014.04.015 |
| 29. | Shafran, Y.; Rozin, Y.; Beryozkina, T.; Zhidovinov, S.; Eltsov, O.; Subbotina, J.; Leban, J.; Novikova, R.; Bakulev, V. Org. Biomol. Chem. 2012, 10, 5795–5798. doi:10.1039/c2ob25331c |
| 30. | Hosmane, R. S.; Bhan, A.; Rauser, M. E. J. Org. Chem. 1985, 50, 5892–5895. doi:10.1021/jo00350a099 |
| 54. | Sheldrick, G. M. Acta Crystallogr., Sect. A 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 31. | Jäger, V.; Colinas, P. A. Nitrile Oxides. In Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H., Eds.; John Wiley & Sons Inc.: New York, 2002; Vol. 59, pp 361–472. doi:10.1002/0471221902.ch6 |
| 32. | Caramella, P.; Grunanger, P. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; John Wiley & Sons: New York, 1984; Vol. 1, pp 291–392. |
| 33. | Grundmann, C.; Grünanger, P. The Nitrile Oxides; Springer-Verlag: Berlin, 1971; pp 47–58. doi:10.1007/978-3-642-99991-8 |
© 2016 Efimov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)