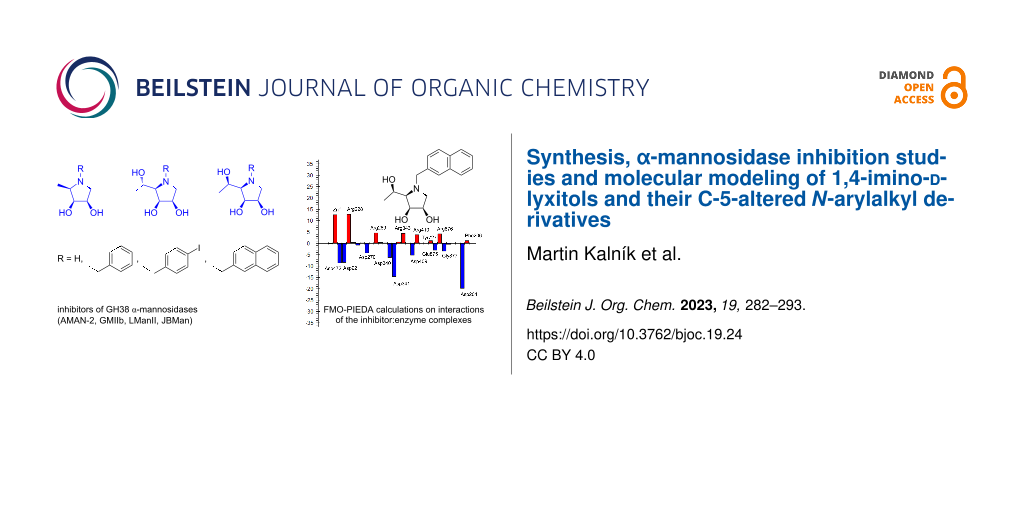
Abstract
A synthesis of 1,4-imino-ᴅ-lyxitols and their N-arylalkyl derivatives altered at C-5 is reported. Their inhibitory activity and selectivity toward four GH38 α-mannosidases (two Golgi types: GMIIb from Drosophila melanogaster and AMAN-2 from Caenorhabditis elegans, and two lysosomal types: LManII from Drosophila melanogaster and JBMan from Canavalia ensiformis) were investigated. 6-Deoxy-DIM was found to be the most potent inhibitor of AMAN-2 (Ki = 0.19 μM), whose amino acid sequence and 3D structure of the active site are almost identical to the human α-mannosidase II (GMII). Although 6-deoxy-DIM was 3.5 times more potent toward AMAN-2 than DIM, their selectivity profiles were almost the same. N-Arylalkylation of 6-deoxy-DIM resulted only in a partial improvement as the selectivity was enhanced at the expense of potency. Structural and physicochemical properties of the corresponding inhibitor:enzyme complexes were analyzed by molecular modeling.

Graphical Abstract
Introduction
Iminosugars are analogs of monosaccharides in which the endocyclic oxygen atom is replaced with a nitrogen atom [1-5]. These compounds have been attracting attention due to their broad spectrum of biological activities [6]. A number of synthetic and naturally occurring iminosugars are able to inhibit various enzymes of medicinal interest including glycosidases, glycosyltransferases and many other carbohydrate processing enzymes that are involved in diseases such as viral infections, diabetes or cancer and lysosomal storage disorders [7-9]. Thus, iminosugar derivatives are promising candidates for pharmaceuticals, and many of them have already been approved for treatments, for example miglitol (type 2 diabetes), miglustat (lysosomal storage disorders, e.g., Gaucher disease) and migalstat (Fabry disease, an orphan drug) [8,10].
Natural iminosugars can be monocyclic or bicyclic compounds since the presence of the nitrogen atom allows for a formation of an additional cycle. In synthetic iminosugar analogs, various structural modifications are possible and many of these compounds inhibit glycoprocessing enzymes [11-14].
One of the best known iminosugars is the natural alkaloid (−)-swainsonine, which is a nanomolar inhibitor of human Golgi α-mannosidase II (GMII, GH38 family, E.C.3.2.1.114). Although such inhibition has been found to suppress metastasis, the potentially positive effect of swainsonine on cancer patients is strongly reduced by its severe side effects [15,16]. These are associated with the accumulation of oligomannoside structures in tissues, serum, and urine, which is caused by the co-inhibition of lysosomal α-mannosidase (LMan, GH38 family, E.C.3.2.1.24) [17] due to structural similarities between the active sites of GMII and LMan. Lysosomal α-mannosidases operate at a lower pH value (pH 4.5) compared to Golgi-type mannosidases (pH 6) and have a significantly broader substrate specificity. Another type of lysosomal α-mannosidase is Jack bean α-mannosidase from Canavalia enciformis (JBMan, GH38 family, E.C.3.2.1.24) which operates in the pH range 4–5. This class II mannosidase is inhibited by swainsonine very effectively (IC50 = 1–5 × 10−7 M) [18] and is frequently used as an acidic α-mannosidase model for structural and mechanistic inhibition studies [19-21]. On the other hand, Caenorhabditis elegans α-mannosidase II (AMAN-2) represents a Golgi-type α-mannosidase (GH38 family, E.C.3.2.1.114) and has the amino acid sequence and predicted 3D structure (based on a built homology model) of the active site almost identical to those of human GMII [22]. In addition, analysis of the available X-ray structures of GH38 enzymes such as dGMII [23], bovine lysosomal α-mannosidase II (bLMan) [17] and JBMan [24] showed that the active sites of Golgi and acidic α-mannosidases are structurally very similar. This explains why potent GMII inhibitors like swainsonine tend to lack significant selectivity. Therefore, the search for potent and selective GMII inhibitors is rather challenging.
Over the last decades, almost all attempts at overcoming the selectivity challenge posed by swainsonine have not been successful [25-27]. The only exception is the latest study by Cheng et al. who reported breakthrough findings in the development of highly potent and selective human GMII inhibitors. A combination of natural product-inspired combinatorial chemistry and computation-guided synthesis provided a nanomolar GMII inhibitor with a 106-fold selectivity over the human LMan while no oligomannose accumulation was observed in animal models [28] (Figure 1).
![[1860-5397-19-24-1]](/bjoc/content/figures/1860-5397-19-24-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Natural iminosugars (1,4-dideoxy-1,4-imino-ᴅ-mannitol (DIM) and swainsonine) and selected examples of synthetic Golgi α-mannosidase II inhibitors with their activity profile toward the Golgi-type α-mannosidases.
Figure 1: Natural iminosugars (1,4-dideoxy-1,4-imino-ᴅ-mannitol (DIM) and swainsonine) and selected examples ...
Another approach to developing selective GMII inhibitors could be based on screening derivatives of 1,4-dideoxy-1,4-imino-ᴅ-mannitol (DIM) as the configurations of all its stereogenic centers match those of swainsonine. Furthermore, DIM is a micromolar GMII inhibitor, which is easily accessible on a large scale from ᴅ-mannose by well-established methods [15,29]. In general, improvements of physicochemical and inhibitory properties of monocyclic iminosugars can be achieved by an alkylation of the endocyclic nitrogen. This reduces their high hydrophilicity which in turn may have a positive impact on the interactions with the hydrophobic pocket of the GMII active site. For example, N-benzylation of DIM afforded a slightly more potent GMII inhibitor than parent DIM [15]. Also, screening of a large library of N-alkyl and N-arylalkyl DIMs revealed that they are less effective inhibitors of JBMan than DIM, indicating that N-alkylation might lead to better selectivity profiles. However, this library has not been assayed for GMII and LM, therefore the compounds’ true inhibitory activity and selectivity toward these relevant GH38 enzymes remain unknown [29].
In this regard, another promising strategy seems to be a modification of the pyrrolidine core at the C-1 position. For example, attaching an amide moiety directly to C-1 in pyrrolidines possessing the ᴅ-lyxo-configuration and bearing a free endocyclic nitrogen resulted in micromolar GMII inhibitors. In addition, their potency toward GMII was found to be 2.4–3.8 times lower than toward Drosophila melanogaster α-mannosidase dGMII (dGMII) [26]. The incorporation of an N-acyl aminomethyl group onto C-1 further enhanced the potency and led to highly selective nanomolar GMII inhibitors [14] (Figure 1).
Our first investigations were focused on the development of selective GMII inhibitors derived from 1,4-imino-1,4-dideoxy-ʟ-lyxitol. Initially, we modified this core by an alkylation of the endocyclic nitrogen with a benzyl or alkyl unit functionalized either with non-polar hydrocarbons or a polar amine, amidine and guanidine group. The ensuing assay with the model GMIIb enzyme (fruit fly Golgi α-mannosidase II) revealed that N-substitution improved both potency and selectivity, achieving inhibition constants (Ki) up to 4 µM and selectivity indices (SI) up to 350. However, these enhancements were found to be significant only for those N-substituted analogs that bore N-alkyl chains without an additional aryl moiety [30-32] (Figure 1).
Next, we turned our attention to modifications of 1,4-imino-ᴅ-lyxitol which configurationally better resembles swainsonine or DIM. The most promising N-substituents from the previous study were selected and a small library of N-substituted 1,4-imino-ᴅ-lyxitols was prepared [22] (Figure 1). In addition, more relevant Caenorhabditis elegans α-mannosidase II (AMAN-2) (GH38 family, E.C.3.2.1.114) was included in the biochemical assay as its active site is more similar to human GMII than that of GMIIb. The resulting biochemical evaluation revealed that imino-ᴅ-lyxitols with N-substituents possessing a polar basic functional group (amidine or guanidine) were 6–7 times less active toward AMAN-2 than GMIIb and had poorer selectivities (SI below 35). In contrast, imino-ᴅ-lyxitols bearing a non-polar N-arylalkyl chain (benzyl, p-iodobenzyl, 2-naphthylmethyl) showed slightly higher inhibitory activities toward AMAN-2 and good to excellent selectivities [22], indicating that they are more suitable candidates for the next-generation design of potent and selective GMII inhibitors.
Therefore, the current study is dedicated to designing further modifications of these analogs. This contribution deals with the synthesis of 1,4-imino-ᴅ-lyxitols altered at C-5 and substituted at the endocyclic nitrogen by the most successful arylalkyl chains (benzyl, p-iodobenzyl, 2-naphthylmethyl) found in our previous studies [22,30,33]. The biological activity of the novel synthesized compounds was evaluated toward the GH38 family enzymes (AMAN-2, GMIIb, fruit fly lysosomal α-mannosidase II (LManII) and JBMan). Finally, structural and physicochemical properties of inhibitor:enzyme complexes were investigated at the theoretical level using molecular docking, hybrid quantum mechanics/molecular mechanics (QM/MM) calculations and fragmented molecular orbital pair interaction energy decomposition analysis (FMO-PIEDA).
Results and Discussion
Chemistry
The target N-arylalkyl 1,4-imino-ᴅ-lyxitol derivatives modified at C-5 were synthesized by the analogous approach that we had reported previously for their ʟ-enantiomers [33]. First, the key, fully protected N-benzylpyrrolidine 3 was prepared in two steps from known ʟ-ribitol 1 [34] in good overall yield. Next, it was converted to the C-5 deoxygenated N-benzylpyrrolidine 6 via trityl ether cleavage, tosylation of the deprotected OH group, and reduction of the tosylate 5. Hydrogenolysis of the N-benzyl group in 6 followed by a removal of the acetonide and subsequent alkylation of the liberated amine with the corresponding arylalkyl bromides under basic conditions provided the desired N-arylalkyl iminosugars 8 and 9 in moderate yields. In addition, acidic hydrolysis of the acetonide group in 6 afforded the target derivative 7 which after hydrogenolysis and treatment with conc. HCl gave hydrochloride 10 (Scheme 1).
![[1860-5397-19-24-i1]](/bjoc/content/inline/1860-5397-19-24-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of the key pyrrolidine 3 and the target pyrrolidines 7–10. Reagents and conditions: (a) MsCl, Et3N, CH2Cl2, 0 °C–rt, overnight, 80%; (b) BnNH2, 120 °C, 7 h, quant.; (c) PTSA·H2O, CH2Cl2/MeOH 30:1, rt, 24 h, 69%; (d) TsCl, DMAP, CH2Cl2, 0 °C–rt, overnight, 88%; (e) LiBHEt3, THF, 0–40 °C, overnight, 83%; (f) 20% HCl, MeOH, rt, 72 h, 70%; (g) 1. H2, Pd/C, MeOH, rt, 48 h; 2. conc. HCl, 0–40 °C, 2 h, 3. ArCH2Br, K2CO3, DMF, 0 °C–rt, overnight.; (h) 1. H2, Pd/C, MeOH, rt, 2 h; 2. conc. HCl, 0–40 °C, 2 h, 68%.
Scheme 1: Synthesis of the key pyrrolidine 3 and the target pyrrolidines 7–10. Reagents and conditions: (a) M...
The versatile intermediate 3 was further employed in the synthesis of the target compounds 17–20 (Scheme 2) homologated at the C-5 position. The transformation of pyrrolidine 3 to 13 included the replacement of the N-benzyl group with the Cbz group, trityl ether hydrolysis, oxidation of the liberated OH group, and stereoselective addition of MeMgBr to the resulting aldehyde functionality. Hydrogenolysis of the Cbz protecting group in 13 followed by N-alkylation afforded pyrrolidines 14–16 which after acidic hydrolysis of the isopropylidene moiety provided the desired derivatives 17–19. The hydrochloride salt of the free iminosugar 20 was obtained from N-benzyl derivative 17 under the same reaction conditions as described for the hydrochloride 10.
![[1860-5397-19-24-i2]](/bjoc/content/inline/1860-5397-19-24-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of the intermediate 13 and the target pyrrolidines 17–20. Reagents and conditions: (a) 1. H2, Pd/C, MeOH, rt, 48 h; 2. CbzCl, Et3N, CH2Cl2, 0 °C–rt, 2 h, 91% over two steps, (b) PTSA·H2O, CH2Cl2/MeOH 30:1, rt, 20 min, 90%; (c) 1. DMP, CH2Cl2, rt, 1 h; 2. MeMgBr, Et2O, 0 °C–rt, 1 h, 63% over two steps; (d) 1. H2, Pd/C, MeOH, rt, 2 h; 2. ArCH2Br, K2CO3, DMF, 0 °C–rt, overnight; (e) 20% HCl, MeOH, rt, 72 h; (f) H2, Pd/C, MeOH, rt, 5 h, then, conc. HCl, 0 °C, 76%.
Scheme 2: Synthesis of the intermediate 13 and the target pyrrolidines 17–20. Reagents and conditions: (a) 1....
Next, the configuration at the C-5 stereocenter in 13 was inverted via cyclic carbamate 21. Aminoalcohol 22 obtained after a basic hydrolysis of 21 was N-alkylated with the corresponding arylalkyl bromides to furnish derivatives 23–25. These were subjected to an acidic hydrolysis of the acetonide moiety to give the target compounds 26–28. Iminosugar 29 (6-deoxy-DIM) was obtained from N-benzylpyrrolidine 26 by the same procedure as described for the preparation of hydrochlorides 10 and 20 (Scheme 3). It should be noted that the hydrochlorides 20 and 29 were synthesized via derivatives 17 and 26 because the alternative approach through acetonides 13 and 22 involved tedious purifications of the final hydrochlorides.
![[1860-5397-19-24-i3]](/bjoc/content/inline/1860-5397-19-24-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of the target pyrrolidines 26–29. Reagents and conditions: (a) Tf2O, pyridine, CH2Cl2, 0 °C, 1.5 h, 64%; (b) 10% aq NaOH, EtOH, reflux, 24 h, 67%; (c) ArCH2Br, K2CO3, DMF, 0 °C–rt, overnight; (d) 20% HCl, MeOH, rt, 72 h; (e) H2, Pd/C, MeOH, rt, 5 h, then, conc. HCl, 0 °C, 86%.
Scheme 3: Synthesis of the target pyrrolidines 26–29. Reagents and conditions: (a) Tf2O, pyridine, CH2Cl2, 0 ...
Enzyme assay
The potency and selectivity of the synthesized iminosugar hydrochlorides 10, 20, 29 and their N-arylalkyl analogs 7–9, 17–19 and 26–28 were evaluated toward the class II GH38 α-mannosidases, and DIM and swainsonine were used as standards. The enzymes screened included two Golgi types (GMIIb and AMAN-2) and two lysosomal types (LManII and JBMan) (Table 1). As for the Golgi-type mannosidases, AMAN-2 is a more relevant enzyme because its amino acid sequence and the 3D structure of its active site are almost identical to those of human GMII [22].
Table 1: Inhibition (IC50, Ki values and selectivity index, SI) of class II GH38 α-mannosidases (GMIIb, AMAN-2, LManII and JBMan) by the synthesized iminosugar derivatives.
| compound | IC50 [Ki] (µM) | SIa | |||
| GMIIb | AMAN-2 | LManII | JBMan | ||
| 7 | 245 ± 35 | 975 ± 25 | >4000 | >4000 | n.d. |
| 8 | 205 ± 35 | 385 ± 15 | >4000 | >4000 | n.d. |
| 9 | 310 ± 22 | 605 ± 35 | >4000 | >4000 | n.d. |
| 10 | 525 ± 25 | 415 ± 15 | >4000 | 2875 ± 275 | n.d. |
| 17 | 335 ± 37 | 990 ± 10 | >4000 | >4000 | n.d. |
| 18 | 170 ± 10 | 205 ± 5 | >4000 | >4000 | n.d. |
| 19 | 860 ± 140 | 1675 ± 75 | >4000 | >4000 | n.d. |
| 20 | 30 ± 5 | 48 ± 6 | 295 ± 20 | 105 ± 25 | 2.2b |
| 26 |
33 ± 3.7
[16.2 ± 4.4 ] |
115 ± 18
[58 ± 8] |
505 ± 57
[270 ± 27] |
298 ± 30
[80 ± 6] |
1.4 |
| 27 |
13 ± 5.5
[4.2 ± 1.1] |
66 ± 10
[34 ± 7] |
410 ± 80
[263 ± 18] |
164 ± 28
[73 ± 3] |
2.1 |
| 28 |
13.5 ± 5.5
[5.2 ± 1.4] |
22 ± 4
[18 ± 3] |
118 ± 10
[98 ± 11] |
78 ± 16
[44 ± 8] |
2.4 |
| 29 |
0.12 ± 0.02
[0.065 ± 0.01] |
0.24 ± 0.05
[0.19 ± 0.02] |
0.82 ± 0.19
[0.38 ± 0.04] |
0.32 ± 0.11
[0.12 ± 0.01]c |
0.6 |
| DIM |
0.19 ± 0.04
[0.13 ± 0.02] |
0.81 ± 0.03
[0.68 ± 0.03] |
2.55 ± 0.10
[1.95 ± 0.35] |
0.72 ± 0.03
[0.38 ± 0.03] |
0.6 |
| swainsonine |
0.004
[0.0027]d |
0.004 ± 0.02
0.01e |
0.012
[0.0071]d |
0.20f
[n.d.] |
20 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-24-i4.svg?max-width=637&scale=1.0)
30 |
3.9 ± 0.1g | 2.3 ± 0.1g | 30.5 ± 3g | 10.5 ± 1.3g | 5b |
![[Graphic 2]](/bjoc/content/inline/1860-5397-19-24-i5.svg?max-width=637&scale=1.0)
31 |
7.6 ± 1.1g | 2.4 ± 0.1g | 845 ± 170g | 1950 ± 250g | 812b |
aSelectivity index, SI [Ki (JBMan)/Ki (AMAN-2)], n.d.: not determined; bSI [IC50 (JBMan)/IC50 (AMAN-2)]; cKi = 0.5 µM measured by Eis [37]; dIC50 and Ki measured by Nemčovičová et al. [35]; eIC50 estimated from the inhibition assays measured by Paschinger et al. [36] who reported 45% inhibition of AMAN-2 by 10 nM concentration of swainsonine; fIC50 measured by Poláková et al. [38]; gIC50 measuared by Kóňa et al. [22].
Iminosugars 7–10 deoxygenated at C-5 were the least effective inhibitors toward the Golgi enzymes (IC50 in the range of 205–975 µM) while the lysosomal enzymes were virtually unaffected. As for the C-5 homologated imino-ᴅ-lyxitols, compounds 20 and 26–29 showed much higher potencies than analogues 17–19 toward the GH38 enzymes tested. Thus, it appears that N-substitution of the free iminosugar 20 considerably reduces the inhibitory activity. Among imino-ᴅ-lyxitols 26–29, 6-deoxy-DIM 29 was found to be the most potent derivative (Ki = 0.065 μM and 0.19 μM for GMIIb and AMAN-2, respectively), being almost 4 times more active toward AMAN-2 than DIM. However, it also exhibited undesirably strong inhibition of LManII and JBMan, giving a low selectivity index. N-Arylalkylation of 6-deoxy-DIM 29 slightly enhanced the selectivity but significantly reduced the potency. Out of the N-arylalkylated iminosugars 26–28, the naphthyl derivative 28 showed the strongest activity against AMAN-2 (Ki = 18 μM) and a selectivity similar to the p-iodobenzyl analog 27. These findings suggest that both the C-5 hydroxy group and R-configuration at the corresponding carbon are necessary for retaining the inhibitory potential of the investigated imino-ᴅ-lyxitol derivatives.
Molecular modeling
In order to better understand the results of our inhibition study, the inhibitor:enzyme interactions were analyzed by molecular modeling. Structures of the inhibitors were docked into an X-ray structure of dGMII and geometries of the resulting inhibitor:dGMII complexes (for 10, 20, 28–30 and DIM) were optimized at the hybrid QM/MM level (BP86/LACVP*:OPLS2005). Based on the previous pKa calculations [22] of DIM, 30 and 31 bound at the active site of dGMII (their pKa = 4.9–5.4 at pH 6 of Golgi), all imino-ᴅ-lyxitol derivatives in this study were modeled in the neutral form (despite their pKa values in aqueous solution may be higher than 7). Superimposing the binding pose of the most potent inhibitor 29 in the active site of dGMII on the bound swainsonine in the X-ray complex (PDB ID: 3BLB) [23,39] (Figure 2) showed that 29 and swainsonine bind to GMII in a similar manner. The pyrrolidine ring of 29 interacts with the Zn2+ ion cofactor, amino acid residues Asp92, Asp204 (catalytic nucleophile), Asp341 (catalytic acid), Asp472, and Trp 95. The (R)-1-hydroxyethyl group at the C-5 position of the ring forms hydrogen bonds with the side chains of Tyr727 and Asp472 [d(C5-OH···Tyr727-OH) = 1.48 Å, and d(C5-OH···Asp472-COO−) = 1.55 Å, BP86/LACVP*:OPLS2005] and interacts with the hydrophobic pocket created by Tyr727, Phe206 and Trp415. Also, the binding position of this side chain is in a good overlap with the hydroxyethylene part of the piperidine ring of swainsonine. This indicates that the (R)-1-hydroxyethyl group of 29 could mimic the interactions of the hydroxypiperidinyl moiety of swainsonine in the active site of dGMII.
![[1860-5397-19-24-2]](/bjoc/content/figures/1860-5397-19-24-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Superposition of the inhibitor 29 (green), docked into dGMII, with X-ray complexes of swainsonine (pink) with dGMII (PDB ID: 3BLB) [39]. The hydroxyethyl moiety at C-5 of 29 is placed at the same position as the hydroxypiperidine moiety of swainsonine.
Figure 2: Superposition of the inhibitor 29 (green), docked into dGMII, with X-ray complexes of swainsonine (...
To evaluate the inhibitory effect of the (R)-1-hydroxyethyl substituent of 29 and other derivatives synthesized in this work, pair interaction energies between the bound inhibitors (29, 10, 20, 28, 30 and DIM) and amino acid residues of dGMII were calculated at the quantum mechanics level (FMO-PIEDA-MP2/6-31G*) in an active-site model of the inhibitor:enzyme complexes optimized at the hybrid QM/MM level (BP86/LACVP*:OPLS2005). The FMO-PIEDA results are compiled in Table 2 and visualized in Figure 3. Firstly, the results for the complex 29:dGMII were analyzed: the overall interaction energy (ΔEI-E) between 29 and the enzyme is −563.6 kcal mol−1, towards which the interaction energy between the (R)-1-hydroxyethyl group of 29 and the enzyme (ΔElinker-E = −49.3 kcal mol−1) contributes only 9%. As can be seen in Figure 3, the main ΔElinker-E contributors are Tyr727 (−26.7 kcal mol−1) and Asp472 (−26.3 kcal mol−1). Both of these amino acid residues use their side chains to interact with the hydroxy group of the (R)-1-hydroxyethyl moiety of 29. The interaction with Trp95 is also significant but to a lesser extent (−5.8 kcal mol−1), and the overall interaction of 29 with the Phe206-Trp415 hydrophobic pocket is insignificant (less than −1.2 kcal mol−1). Therefore, the main contributors remain Tyr727 and Asp472 (two hydrogen bonds). A similar conclusion can also be made for the other calculated inhibitors with one (20, 28, 30) or two (DIM) hydroxy groups at C-5. This explains why the inhibitory activity of 29 [Ki(AMAN-2) = 0.19 µM] is only slightly better than the previously synthesized derivative 30 (with a hydroxymethyl moiety at C-5) [Ki(AMAN-2) = 2.3 µM] [22].
Table 2: Interaction energies (ΔEI-E, in kcal mol−1) for complexes (inhibitor:dGMII) calculated at the MP2//BP86 level. Also interaction energies between the enzyme and the inhibitor fragments [the pyrrolidine core (ΔEring-E) or the structural moiety at C-5 (ΔElinker-E)] are also compiled.
| C-5 linker | N-substitution | ring conform. | ΔEI-E | ΔEring-E | ΔElinker-E | |
| 10 | -CH3 | -H | E1/2E | −512.09 | −516.22 | 4.13 |
| 20 | (1S)-CH2(OH)-CH3 | -H | 2E/E1 | −551.18 | −508.17 | −43.01 |
| 30 | -CH2-OH | -H | E1 | −569.30 | −518.31 | −50.99 |
| 29 | (1R)-CH2(OH)-CH3 | -H | E1 | −563.60 | −514.35 | −49.25 |
| 28 | (1R)-CH2(OH)-CH3 | N-2-naphthylmethyl | E1 | −558.78 | −502.70 | −56.07 |
| DIM | -CH2(OH)-CH2-OH | -H | E1/2E | −578.74 | −514.58 | −64.16 |
![[1860-5397-19-24-3]](/bjoc/content/figures/1860-5397-19-24-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: FMO-PIEDA total pair interaction energies (ΔElinker-E) (in kcal mol−1) between the structural moiety at C-5 of the inhibitor (10, 30, 29, 28, 20 and DIM) and the active-site amino acid residues of dGMII. The most significant ΔElinker-E are marked.
Figure 3: FMO-PIEDA total pair interaction energies (ΔElinker-E) (in kcal mol−1) between the structural moiet...
To evaluate the inhibitory effect of the structural moiety at C-5 of the 1,4-imino-ᴅ-lyxitols, FMO-PIEDA interaction energies were calculated for another five derivatives (10, 20, 30, 28 and DIM). The inhibitory activities of these derivatives increased in the following order: 10 [IC50(AMAN-2) = 415 µM] < 20 [IC50(AMAN-2) = 48 µM] < 28 [IC50(AMAN-2) = 22 µM] < 30 [IC50(AMAN-2) = 2.3 µM] < DIM [IC50(AMAN-2) = 0.81 µM] < 29 [IC50(AMAN-2) = 0.24 µM]. Interestingly, almost the same trend was found for the overall FMO-PIEDA interaction energies (ΔEI-E) and the energies (ΔElinker-E) between the side chain at C-5 and the active site (Table 2). ΔEI-E and ΔElinker-E increase in the following order: 10 (−512.09 kcal mol−1) < 20 (−551.18 kcal mol−1) < 28 (−558.78 kcal mol−1) < 29 (−563.60 kcal mol−1) < 30 (−569.30 kcal mol−1) < DIM (−578.74 kcal mol−1); and 10 (+4.13 kcal mol−1) < 20 (−43.01 kcal mol−1) < 29 (−49.25 kcal mol−1) < 30 (−50.99 kcal mol−1) < 28 (−56.07 kcal mol−1) < DIM (−64.16 kcal mol−1), respectively. The calculations predict that the functional group attached to C-5 of the inhibitor must bear one or two hydroxy groups (with R-configuration in case of 28, 29 and DIM). The methyl group itself (in 10) contributes repulsively and decreases the overall interaction energy. Thus, only hydroxymethyl, (R)-1-hydroxyethyl and (R)-1,2-dihydroxyethyl are suitable substituents at the C-5 position of 1,4-imino-ᴅ-lyxitols. The calculations further predict that alkylation of the nitrogen atom may further increase the interactions of the (R)-1-hydroxyethyl group at C-5 with the enzyme (N-2-naphthylmethyl in 28). However, this alkylation also decreases the interactions of the lyxitol core with the enzyme (ΔEring-E = −502.70 kcal mol−1 of 28 is weaker than ΔEring-E = −514.35 kcal mol−1 of 29) and the overall ΔEI-E for 28 became lower than for 29, which is in agreement with the measured inhibitory activities of these compounds. This is a surprising result because N-alkylation of 30 (structure 31 in Table 1) did not decrease inhibition of Golgi-type α-mannosidases [22]. It seems that the slightly bulkier (R)-1-hydroxyethyl group at C-5 of 29 compared to the hydroxymethyl at C-5 of 30 allows for a different conformation of the N-2-naphthylmethyl group in the active site of α-mannosidases (Figure 4). This would induce a shift in the binding pose of the lyxitol core in 28 compared to 29 and a subsequent weakening in ΔEring-E (from −514.35 kcal mol−1 in 29 to −502.70 kcal mol−1 in 28), which is the major component of the overall interaction energy between the inhibitor and the enzyme. This assumption was further supported by additional FMO-PIEDA calculations for 31. In both 31 and 28, the 2-naphthylmethyl group is attached to the ring nitrogen of the inhibitor. The interaction energy between the N-2-naphthylmethyl moiety of the inhibitors and the enzyme was almost the same (−30.67 kcal mol−1 for 31 and −30.99 kcal mol−1 for 28, Figure 5), confirming that this substituent is suitable for interacting with the enzyme, but at the same time it worsened the interactions of the lyxitol core of 28 (in connection to the mentioned (R)-1-hydroxyethyl group at C-5).
![[1860-5397-19-24-4]](/bjoc/content/figures/1860-5397-19-24-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: QM/MM optimized complexes 28:dGMII (top) and 31:dGMII (bottom) [22]. N-2-naphthylmethyl group (grey) and (R)-1-hydroxyethyl group (green) of 28 and the hydroxymethyl group (green) of 31 are visualized by van der Waals spheres to highlight the unfavorable interaction (marked with a black arrow) between the N-2-naphthylmethyl group and the methyl component of (R)-1-hydroxyethyl group at C-5 of 28. In case of 31, the smaller hydroxymethyl group at C-5 allows for a different binding conformation of the N-2-naphthylmethyl group.
Figure 4: QM/MM optimized complexes 28:dGMII (top) and 31:dGMII (bottom) [22]. N-2-naphthylmethyl group (grey) an...
![[1860-5397-19-24-5]](/bjoc/content/figures/1860-5397-19-24-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: FMO-PIEDA total pair interaction energies (ΔElinker-E) (in kcal mol−1) between the N-2-naphthylmethyl moiety of the inhibitors 28 and 31 and the active-site amino acid residues of dGMII. The most significant ΔElinker-E are marked.
Figure 5: FMO-PIEDA total pair interaction energies (ΔElinker-E) (in kcal mol−1) between the N-2-naphthylmeth...
Conclusion
1,4-Imino-ᴅ-lyxitols and their C-5 altered N-arylalkyl derivatives were synthesized for α-mannosidase inhibition studies. Their evaluation revealed that deoxygenation at C-5 (derivatives 7–10) provided the least effective inhibitors of the target Golgi enzymes. The comparison between iminosugars 17–20 and their C-5 epimers 26–29 showed that the hydroxy group must adopt an R-configuration in order to exhibit a strong inhibition profile. Also, unsubstituted 6-deoxy-DIM 29 turned out to be the best inhibitor out of all the analogues synthesized in this study as its N-arylalkylation significantly reduced potency and barely improved selectivity. This is in sharp contrast to the previously synthesized N-2-naphthylmethyl 1,4-imino-ᴅ-lyxitol which showed a high selectivity toward Golgi-type α-mannosidases [22]. The FMO-PIEDA calculations revealed that N-2-naphthylmethyl is too bulky for the 6-deoxy-DIM core and causes weaker binding of the inhibitor ring to the enzyme. Therefore, the next efforts should be focused on the identification of a novel N-substitution pattern of the DIM skeleton that would have more beneficial effects on the inhibition profile.
Supporting Information
| Supporting Information File 1: Experimental (synthesis, enzyme assay, molecular modelling). | ||
| Format: PDF | Size: 997.5 KB | Download |
| Supporting Information File 2: Copies of NMR spectra. | ||
| Format: PDF | Size: 1.8 MB | Download |
| Supporting Information File 3: Optimized QM/MM complexes (inhibitor:enzyme). | ||
| Format: MAE | Size: 20.8 MB | Download |
Acknowledgements
We would also like to thank Prof. I. B. H. Wilson from the University of Natural Resources and Life Sciences in Vienna for donating a clone of Pichia pastoris expressing AMAN-2 mannosidase. Peter Gabko, MSci, is gratefully acknowledged for our insightful discussions.
Funding
This work was supported by the Scientific Grant Agency of the Ministry of Education of Slovak Republic and Slovak Academy of Sciences (the projects VEGA-2/0010/23 and VEGA-2/0061/23), SAS Taiwan project (SAS-MOST/JRP/2019/882/GM-INHIB), the project implementation CEMBAM (Centre for Medical Bio-Additive Manufacturing and Research, ITMS2014+: 313011V358 supported by the Operational Programme Integrated Infrastructure funded by the European Regional Development Fund) and the Slovak Research and Development Agency (the project APVV-19-0376).
References
-
Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/s0957-4166(00)00113-0
Return to citation in text: [1] -
Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048
Return to citation in text: [1] -
D'Alonzo, D.; Guaragna, A.; Palumbo, G. Curr. Med. Chem. 2009, 16, 473–505. doi:10.2174/092986709787315540
Return to citation in text: [1] -
Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. H.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017
Return to citation in text: [1] -
Compain, P. Synlett 2014, 25, 1215–1240. doi:10.1055/s-0033-1340822
Return to citation in text: [1] -
Horne, G.; Wilson, F. X. Prog. Med. Chem. 2011, 50, 135–176. doi:10.1016/b978-0-12-381290-2.00004-5
Return to citation in text: [1] -
Stütz, A. E.; Wrodnigg, T. M. Adv. Carbohydr. Chem. Biochem. 2011, 66, 187–298. doi:10.1016/b978-0-12-385518-3.00004-3
Return to citation in text: [1] -
Conforti, I.; Marra, A. Org. Biomol. Chem. 2021, 19, 5439–5475. doi:10.1039/d1ob00382h
Return to citation in text: [1] [2] -
Kiefel, M. J. Glycomimetics as inhibitors in anti-infection therapy. In Microbial Glycobiology; Holst, O.; Brennan, P. J.; Itzstein, M. V.; Moran, A. P., Eds.; Academic Press: San Diego, CA, USA, 2010; pp 915–932. doi:10.1016/b978-0-12-374546-0.00047-x
Return to citation in text: [1] -
Pereira, D. M.; Valentão, P.; Andrade, P. B. Chem. Sci. 2018, 9, 1740–1752. doi:10.1039/c7sc04712f
Return to citation in text: [1] -
Liang, P.-H.; Cheng, W.-C.; Lee, Y.-L.; Yu, H.-P.; Wu, Y.-T.; Lin, Y.-L.; Wong, C.-H. ChemBioChem 2006, 7, 165–173. doi:10.1002/cbic.200500321
Return to citation in text: [1] -
Calveras, J.; Egido-Gabás, M.; Gómez, L.; Casas, J.; Parella, T.; Joglar, J.; Bujons, J.; Clapés, P. Chem. – Eur. J. 2009, 15, 7310–7328. doi:10.1002/chem.200900838
Return to citation in text: [1] -
Stocker, B. L.; Jongkees, S. A. K.; Win-Mason, A. L.; Dangerfield, E. M.; Withers, S. G.; Timmer, M. S. M. Carbohydr. Res. 2013, 367, 29–32. doi:10.1016/j.carres.2012.11.011
Return to citation in text: [1] -
Cheng, T.-J. R.; Chan, T.-H.; Tsou, E.-L.; Chang, S.-Y.; Yun, W.-Y.; Yang, P.-J.; Wu, Y.-T.; Cheng, W.-C. Chem. – Asian J. 2013, 8, 2600–2604. doi:10.1002/asia.201300680
Return to citation in text: [1] [2] -
Winchester, B.; al Daher, S.; Carpenter, N. C.; Cenci di Bello, I.; Choi, S. S.; Fairbanks, A. J.; Fleet, G. W. J. Biochem. J. 1993, 290, 743–749. doi:10.1042/bj2900743
Return to citation in text: [1] [2] [3] -
Goss, P. E.; Baker, M. A.; Carver, J. P.; Dennis, J. W. Clin. Cancer Res. 1995, 1, 935–944.
Return to citation in text: [1] -
Heikinheimo, P.; Helland, R.; Leiros, H.-K. S.; Leiros, I.; Karlsen, S.; Evjen, G.; Ravelli, R.; Schoehn, G.; Ruigrok, R.; Tollersrud, O.-K.; McSweeney, S.; Hough, E. J. Mol. Biol. 2003, 327, 631–644. doi:10.1016/s0022-2836(03)00172-4
Return to citation in text: [1] [2] -
Kang, M. S.; Elbein, A. D. Plant Physiol. 1983, 71, 551–554. doi:10.1104/pp.71.3.551
Return to citation in text: [1] -
Fiaux, H.; Kuntz, D. A.; Hoffman, D.; Janzer, R. C.; Gerber-Lemaire, S.; Rose, D. R.; Juillerat-Jeanneret, L. Bioorg. Med. Chem. 2008, 16, 7337–7346. doi:10.1016/j.bmc.2008.06.021
Return to citation in text: [1] -
Mane, R. S.; Ghosh, S.; Singh, S.; Chopade, B. A.; Dhavale, D. D. Bioorg. Med. Chem. 2011, 19, 6720–6725. doi:10.1016/j.bmc.2011.09.046
Return to citation in text: [1] -
Mirabella, S.; D'Adamio, G.; Matassini, C.; Goti, A.; Delgado, S.; Gimeno, A.; Robina, I.; Moreno-Vargas, A. J.; Šesták, S.; Jiménez-Barbero, J.; Cardona, F. Chem. – Eur. J. 2017, 23, 14585–14596. doi:10.1002/chem.201703011
Return to citation in text: [1] -
Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
van den Elsen, J. M. H.; Kuntz, D. A.; Rose, D. R. EMBO J. 2001, 20, 3008–3017. doi:10.1093/emboj/20.12.3008
Return to citation in text: [1] [2] -
Howard, E.; Cousido‐Siah, A.; Lepage, M. L.; Schneider, J. P.; Bodlenner, A.; Mitschler, A.; Meli, A.; Izzo, I.; Alvarez, H. A.; Podjarny, A.; Compain, P. Angew. Chem., Int. Ed. 2018, 57, 8002–8006. doi:10.1002/anie.201801202
Return to citation in text: [1] -
Lee, Z. Y.; Loo, J. S. E.; Wibowo, A.; Mohammat, M. F.; Foo, J. B. Carbohydr. Res. 2021, 508, 108395. doi:10.1016/j.carres.2021.108395
Return to citation in text: [1] -
Armstrong, Z.; Kuo, C.-L.; Lahav, D.; Liu, B.; Johnson, R.; Beenakker, T. J. M.; de Boer, C.; Wong, C.-S.; van Rijssel, E. R.; Debets, M. F.; Florea, B. I.; Hissink, C.; Boot, R. G.; Geurink, P. P.; Ovaa, H.; van der Stelt, M.; van der Marel, G. M.; Codée, J. D. C.; Aerts, J. M. F. G.; Wu, L.; Overkleeft, H. S.; Davies, G. J. J. Am. Chem. Soc. 2020, 142, 13021–13029. doi:10.1021/jacs.0c03880
Return to citation in text: [1] [2] -
Chen, W.-A.; Sayyad, A.; Chen, C.-W.; Chen, Y.-H.; Cheng, T.-J. R.; Cheng, W.-C. Asian J. Org. Chem. 2019, 8, 2233–2242. doi:10.1002/ajoc.201900637
Return to citation in text: [1] -
Chen, W.-A.; Chen, Y.-H.; Hsieh, C.-Y.; Hung, P.-F.; Chen, C.-W.; Chen, C.-H.; Lin, J.-L.; Cheng, T.-J. R.; Hsu, T.-L.; Wu, Y.-T.; Shen, C.-N.; Cheng, W.-C. Chem. Sci. 2022, 13, 6233–6243. doi:10.1039/d1sc05894k
Return to citation in text: [1] -
Yang, L.-F.; Shimadate, Y.; Kato, A.; Li, Y.-X.; Jia, Y.-M.; Fleet, G. W. J.; Yu, C.-Y. Org. Biomol. Chem. 2020, 18, 999–1011. doi:10.1039/c9ob02029b
Return to citation in text: [1] [2] -
Šesták, S.; Bella, M.; Klunda, T.; Gurská, S.; Džubák, P.; Wöls, F.; Wilson, I. B. H.; Sladek, V.; Hajdúch, M.; Poláková, M.; Kóňa, J. ChemMedChem 2018, 13, 373–383. doi:10.1002/cmdc.201700607
Return to citation in text: [1] [2] -
Klunda, T.; Šesták, S.; Kóňa, J.; Poláková, M. Bioorg. Chem. 2019, 83, 424–431. doi:10.1016/j.bioorg.2018.10.066
Return to citation in text: [1] -
Klunda, T.; Hricovíni, M.; Šesták, S.; Kóňa, J.; Poláková, M. New J. Chem. 2021, 45, 10940–10951. doi:10.1039/d1nj01176f
Return to citation in text: [1] -
Bella, M.; Šesták, S.; Moncoľ, J.; Koóš, M.; Poláková, M. Beilstein J. Org. Chem. 2018, 14, 2156–2162. doi:10.3762/bjoc.14.189
Return to citation in text: [1] [2] -
An, S.; Kim, G.; Kim, H. J.; Ahn, S.; Kim, H. Y.; Ko, H.; Hyun, Y. E.; Nguyen, M.; Jeong, J.; Liu, Z.; Han, J.; Choi, H.; Yu, J.; Kim, J. W.; Lee, H. W.; Jacobson, K. A.; Cho, W. J.; Kim, Y.-M.; Kang, K. W.; Noh, M.; Jeong, L. S. J. Med. Chem. 2020, 63, 16012–16027. doi:10.1021/acs.jmedchem.0c01874
Return to citation in text: [1] -
Nemčovičová, I.; Šesták, S.; Rendić, D.; Plšková, M.; Mucha, J.; Wilson, I. B. H. Glycoconjugate J. 2013, 30, 899–909. doi:10.1007/s10719-013-9495-5
Return to citation in text: [1] -
Paschinger, K.; Hackl, M.; Gutternigg, M.; Kretschmer-Lubich, D.; Stemmer, U.; Jantsch, V.; Lochnit, G.; Wilson, I. B. H. J. Biol. Chem. 2006, 281, 28265–28277. doi:10.1074/jbc.m602878200
Return to citation in text: [1] -
Eis, M. J.; Rule, C. J.; Wurzburg, B. A.; Ganem, B. Tetrahedron Lett. 1985, 26, 5397–5398. doi:10.1016/s0040-4039(00)98217-0
Return to citation in text: [1] -
Poláková, M.; Stanton, R.; Wilson, I. B. H.; Holková, I.; Šesták, S.; Machová, E.; Jandová, Z.; Kóňa, J. Carbohydr. Res. 2015, 406, 34–40. doi:10.1016/j.carres.2015.01.004
Return to citation in text: [1] -
Kuntz, D. A.; Rose, D. R. PDB Protein Data Bank: Crystal structure of Golgi Mannosidase II in complex with swainsonine at 1.3 Angstrom resolution; wwPDB Foundation: Piscataway, NJ, USA, 2007; PDB ID: 3BLB. doi:10.2210/pdb3blb/pdb
Return to citation in text: [1] [2]
| 37. | Eis, M. J.; Rule, C. J.; Wurzburg, B. A.; Ganem, B. Tetrahedron Lett. 1985, 26, 5397–5398. doi:10.1016/s0040-4039(00)98217-0 |
| 35. | Nemčovičová, I.; Šesták, S.; Rendić, D.; Plšková, M.; Mucha, J.; Wilson, I. B. H. Glycoconjugate J. 2013, 30, 899–909. doi:10.1007/s10719-013-9495-5 |
| 36. | Paschinger, K.; Hackl, M.; Gutternigg, M.; Kretschmer-Lubich, D.; Stemmer, U.; Jantsch, V.; Lochnit, G.; Wilson, I. B. H. J. Biol. Chem. 2006, 281, 28265–28277. doi:10.1074/jbc.m602878200 |
| 1. | Asano, N.; Nash, R. J.; Molyneux, R. J.; Fleet, G. W. J. Tetrahedron: Asymmetry 2000, 11, 1645–1680. doi:10.1016/s0957-4166(00)00113-0 |
| 2. | Winchester, B. G. Tetrahedron: Asymmetry 2009, 20, 645–651. doi:10.1016/j.tetasy.2009.02.048 |
| 3. | D'Alonzo, D.; Guaragna, A.; Palumbo, G. Curr. Med. Chem. 2009, 16, 473–505. doi:10.2174/092986709787315540 |
| 4. | Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. H.; Storer, R. Drug Discovery Today 2011, 16, 107–118. doi:10.1016/j.drudis.2010.08.017 |
| 5. | Compain, P. Synlett 2014, 25, 1215–1240. doi:10.1055/s-0033-1340822 |
| 11. | Liang, P.-H.; Cheng, W.-C.; Lee, Y.-L.; Yu, H.-P.; Wu, Y.-T.; Lin, Y.-L.; Wong, C.-H. ChemBioChem 2006, 7, 165–173. doi:10.1002/cbic.200500321 |
| 12. | Calveras, J.; Egido-Gabás, M.; Gómez, L.; Casas, J.; Parella, T.; Joglar, J.; Bujons, J.; Clapés, P. Chem. – Eur. J. 2009, 15, 7310–7328. doi:10.1002/chem.200900838 |
| 13. | Stocker, B. L.; Jongkees, S. A. K.; Win-Mason, A. L.; Dangerfield, E. M.; Withers, S. G.; Timmer, M. S. M. Carbohydr. Res. 2013, 367, 29–32. doi:10.1016/j.carres.2012.11.011 |
| 14. | Cheng, T.-J. R.; Chan, T.-H.; Tsou, E.-L.; Chang, S.-Y.; Yun, W.-Y.; Yang, P.-J.; Wu, Y.-T.; Cheng, W.-C. Chem. – Asian J. 2013, 8, 2600–2604. doi:10.1002/asia.201300680 |
| 28. | Chen, W.-A.; Chen, Y.-H.; Hsieh, C.-Y.; Hung, P.-F.; Chen, C.-W.; Chen, C.-H.; Lin, J.-L.; Cheng, T.-J. R.; Hsu, T.-L.; Wu, Y.-T.; Shen, C.-N.; Cheng, W.-C. Chem. Sci. 2022, 13, 6233–6243. doi:10.1039/d1sc05894k |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 8. | Conforti, I.; Marra, A. Org. Biomol. Chem. 2021, 19, 5439–5475. doi:10.1039/d1ob00382h |
| 10. | Pereira, D. M.; Valentão, P.; Andrade, P. B. Chem. Sci. 2018, 9, 1740–1752. doi:10.1039/c7sc04712f |
| 15. | Winchester, B.; al Daher, S.; Carpenter, N. C.; Cenci di Bello, I.; Choi, S. S.; Fairbanks, A. J.; Fleet, G. W. J. Biochem. J. 1993, 290, 743–749. doi:10.1042/bj2900743 |
| 29. | Yang, L.-F.; Shimadate, Y.; Kato, A.; Li, Y.-X.; Jia, Y.-M.; Fleet, G. W. J.; Yu, C.-Y. Org. Biomol. Chem. 2020, 18, 999–1011. doi:10.1039/c9ob02029b |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 7. | Stütz, A. E.; Wrodnigg, T. M. Adv. Carbohydr. Chem. Biochem. 2011, 66, 187–298. doi:10.1016/b978-0-12-385518-3.00004-3 |
| 8. | Conforti, I.; Marra, A. Org. Biomol. Chem. 2021, 19, 5439–5475. doi:10.1039/d1ob00382h |
| 9. | Kiefel, M. J. Glycomimetics as inhibitors in anti-infection therapy. In Microbial Glycobiology; Holst, O.; Brennan, P. J.; Itzstein, M. V.; Moran, A. P., Eds.; Academic Press: San Diego, CA, USA, 2010; pp 915–932. doi:10.1016/b978-0-12-374546-0.00047-x |
| 24. | Howard, E.; Cousido‐Siah, A.; Lepage, M. L.; Schneider, J. P.; Bodlenner, A.; Mitschler, A.; Meli, A.; Izzo, I.; Alvarez, H. A.; Podjarny, A.; Compain, P. Angew. Chem., Int. Ed. 2018, 57, 8002–8006. doi:10.1002/anie.201801202 |
| 39. | Kuntz, D. A.; Rose, D. R. PDB Protein Data Bank: Crystal structure of Golgi Mannosidase II in complex with swainsonine at 1.3 Angstrom resolution; wwPDB Foundation: Piscataway, NJ, USA, 2007; PDB ID: 3BLB. doi:10.2210/pdb3blb/pdb |
| 6. | Horne, G.; Wilson, F. X. Prog. Med. Chem. 2011, 50, 135–176. doi:10.1016/b978-0-12-381290-2.00004-5 |
| 25. | Lee, Z. Y.; Loo, J. S. E.; Wibowo, A.; Mohammat, M. F.; Foo, J. B. Carbohydr. Res. 2021, 508, 108395. doi:10.1016/j.carres.2021.108395 |
| 26. | Armstrong, Z.; Kuo, C.-L.; Lahav, D.; Liu, B.; Johnson, R.; Beenakker, T. J. M.; de Boer, C.; Wong, C.-S.; van Rijssel, E. R.; Debets, M. F.; Florea, B. I.; Hissink, C.; Boot, R. G.; Geurink, P. P.; Ovaa, H.; van der Stelt, M.; van der Marel, G. M.; Codée, J. D. C.; Aerts, J. M. F. G.; Wu, L.; Overkleeft, H. S.; Davies, G. J. J. Am. Chem. Soc. 2020, 142, 13021–13029. doi:10.1021/jacs.0c03880 |
| 27. | Chen, W.-A.; Sayyad, A.; Chen, C.-W.; Chen, Y.-H.; Cheng, T.-J. R.; Cheng, W.-C. Asian J. Org. Chem. 2019, 8, 2233–2242. doi:10.1002/ajoc.201900637 |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 19. | Fiaux, H.; Kuntz, D. A.; Hoffman, D.; Janzer, R. C.; Gerber-Lemaire, S.; Rose, D. R.; Juillerat-Jeanneret, L. Bioorg. Med. Chem. 2008, 16, 7337–7346. doi:10.1016/j.bmc.2008.06.021 |
| 20. | Mane, R. S.; Ghosh, S.; Singh, S.; Chopade, B. A.; Dhavale, D. D. Bioorg. Med. Chem. 2011, 19, 6720–6725. doi:10.1016/j.bmc.2011.09.046 |
| 21. | Mirabella, S.; D'Adamio, G.; Matassini, C.; Goti, A.; Delgado, S.; Gimeno, A.; Robina, I.; Moreno-Vargas, A. J.; Šesták, S.; Jiménez-Barbero, J.; Cardona, F. Chem. – Eur. J. 2017, 23, 14585–14596. doi:10.1002/chem.201703011 |
| 23. | van den Elsen, J. M. H.; Kuntz, D. A.; Rose, D. R. EMBO J. 2001, 20, 3008–3017. doi:10.1093/emboj/20.12.3008 |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 18. | Kang, M. S.; Elbein, A. D. Plant Physiol. 1983, 71, 551–554. doi:10.1104/pp.71.3.551 |
| 17. | Heikinheimo, P.; Helland, R.; Leiros, H.-K. S.; Leiros, I.; Karlsen, S.; Evjen, G.; Ravelli, R.; Schoehn, G.; Ruigrok, R.; Tollersrud, O.-K.; McSweeney, S.; Hough, E. J. Mol. Biol. 2003, 327, 631–644. doi:10.1016/s0022-2836(03)00172-4 |
| 23. | van den Elsen, J. M. H.; Kuntz, D. A.; Rose, D. R. EMBO J. 2001, 20, 3008–3017. doi:10.1093/emboj/20.12.3008 |
| 39. | Kuntz, D. A.; Rose, D. R. PDB Protein Data Bank: Crystal structure of Golgi Mannosidase II in complex with swainsonine at 1.3 Angstrom resolution; wwPDB Foundation: Piscataway, NJ, USA, 2007; PDB ID: 3BLB. doi:10.2210/pdb3blb/pdb |
| 17. | Heikinheimo, P.; Helland, R.; Leiros, H.-K. S.; Leiros, I.; Karlsen, S.; Evjen, G.; Ravelli, R.; Schoehn, G.; Ruigrok, R.; Tollersrud, O.-K.; McSweeney, S.; Hough, E. J. Mol. Biol. 2003, 327, 631–644. doi:10.1016/s0022-2836(03)00172-4 |
| 38. | Poláková, M.; Stanton, R.; Wilson, I. B. H.; Holková, I.; Šesták, S.; Machová, E.; Jandová, Z.; Kóňa, J. Carbohydr. Res. 2015, 406, 34–40. doi:10.1016/j.carres.2015.01.004 |
| 15. | Winchester, B.; al Daher, S.; Carpenter, N. C.; Cenci di Bello, I.; Choi, S. S.; Fairbanks, A. J.; Fleet, G. W. J. Biochem. J. 1993, 290, 743–749. doi:10.1042/bj2900743 |
| 16. | Goss, P. E.; Baker, M. A.; Carver, J. P.; Dennis, J. W. Clin. Cancer Res. 1995, 1, 935–944. |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 26. | Armstrong, Z.; Kuo, C.-L.; Lahav, D.; Liu, B.; Johnson, R.; Beenakker, T. J. M.; de Boer, C.; Wong, C.-S.; van Rijssel, E. R.; Debets, M. F.; Florea, B. I.; Hissink, C.; Boot, R. G.; Geurink, P. P.; Ovaa, H.; van der Stelt, M.; van der Marel, G. M.; Codée, J. D. C.; Aerts, J. M. F. G.; Wu, L.; Overkleeft, H. S.; Davies, G. J. J. Am. Chem. Soc. 2020, 142, 13021–13029. doi:10.1021/jacs.0c03880 |
| 15. | Winchester, B.; al Daher, S.; Carpenter, N. C.; Cenci di Bello, I.; Choi, S. S.; Fairbanks, A. J.; Fleet, G. W. J. Biochem. J. 1993, 290, 743–749. doi:10.1042/bj2900743 |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 29. | Yang, L.-F.; Shimadate, Y.; Kato, A.; Li, Y.-X.; Jia, Y.-M.; Fleet, G. W. J.; Yu, C.-Y. Org. Biomol. Chem. 2020, 18, 999–1011. doi:10.1039/c9ob02029b |
| 34. | An, S.; Kim, G.; Kim, H. J.; Ahn, S.; Kim, H. Y.; Ko, H.; Hyun, Y. E.; Nguyen, M.; Jeong, J.; Liu, Z.; Han, J.; Choi, H.; Yu, J.; Kim, J. W.; Lee, H. W.; Jacobson, K. A.; Cho, W. J.; Kim, Y.-M.; Kang, K. W.; Noh, M.; Jeong, L. S. J. Med. Chem. 2020, 63, 16012–16027. doi:10.1021/acs.jmedchem.0c01874 |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 30. | Šesták, S.; Bella, M.; Klunda, T.; Gurská, S.; Džubák, P.; Wöls, F.; Wilson, I. B. H.; Sladek, V.; Hajdúch, M.; Poláková, M.; Kóňa, J. ChemMedChem 2018, 13, 373–383. doi:10.1002/cmdc.201700607 |
| 33. | Bella, M.; Šesták, S.; Moncoľ, J.; Koóš, M.; Poláková, M. Beilstein J. Org. Chem. 2018, 14, 2156–2162. doi:10.3762/bjoc.14.189 |
| 33. | Bella, M.; Šesták, S.; Moncoľ, J.; Koóš, M.; Poláková, M. Beilstein J. Org. Chem. 2018, 14, 2156–2162. doi:10.3762/bjoc.14.189 |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 22. | Kóňa, J.; Šesták, S.; Wilson, I. B. H.; Poláková, M. Org. Biomol. Chem. 2022, 20, 8932–8943. doi:10.1039/d2ob01545e |
| 14. | Cheng, T.-J. R.; Chan, T.-H.; Tsou, E.-L.; Chang, S.-Y.; Yun, W.-Y.; Yang, P.-J.; Wu, Y.-T.; Cheng, W.-C. Chem. – Asian J. 2013, 8, 2600–2604. doi:10.1002/asia.201300680 |
| 30. | Šesták, S.; Bella, M.; Klunda, T.; Gurská, S.; Džubák, P.; Wöls, F.; Wilson, I. B. H.; Sladek, V.; Hajdúch, M.; Poláková, M.; Kóňa, J. ChemMedChem 2018, 13, 373–383. doi:10.1002/cmdc.201700607 |
| 31. | Klunda, T.; Šesták, S.; Kóňa, J.; Poláková, M. Bioorg. Chem. 2019, 83, 424–431. doi:10.1016/j.bioorg.2018.10.066 |
| 32. | Klunda, T.; Hricovíni, M.; Šesták, S.; Kóňa, J.; Poláková, M. New J. Chem. 2021, 45, 10940–10951. doi:10.1039/d1nj01176f |
© 2023 Kalník et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.