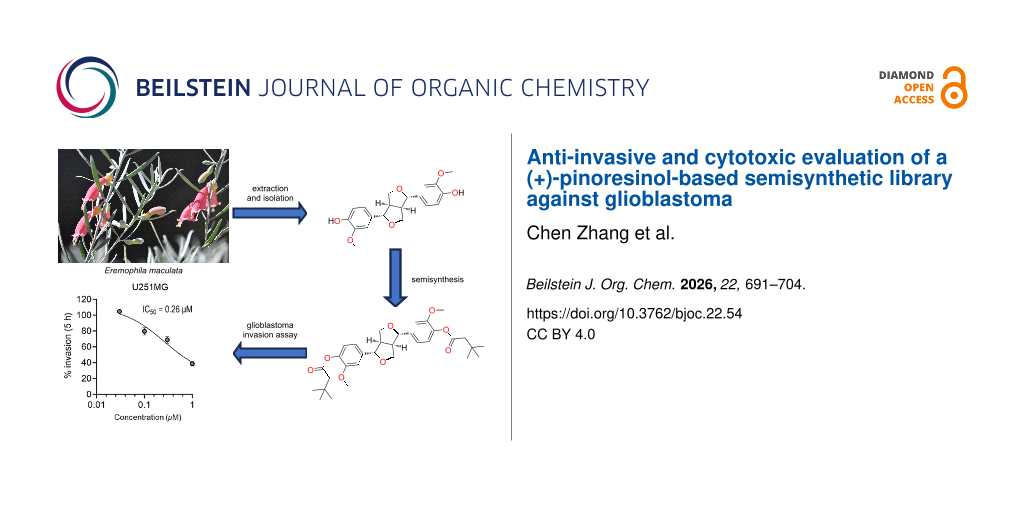
Abstract
The Australian endemic plant genus Eremophila has long been recognized for its unique chemical diversity, with numerous novel and bioactive compounds reported. In this study, we chemically investigated the seeds of Eremophila maculata for the first time, which led to the isolation and characterization of two known plant metabolites, (+)-salicifoliol and (+)-pinoresinol. Due to the reported biological properties of the lignan natural product (+)-pinoresinol and its high abundance from the seeds of E. maculata, we synthesized five analogues for biological evaluations. Preliminary cytotoxicity evaluations of the semisynthetic pinoresinol-based library against two human glioblastoma cell lines, U251MG and KNS42, showed (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol had slight cytotoxicity at 10 µM. A transwell anti-invasive assay on the same compound showed a reduction in the invasion of adult U251MG cells by 50% and pediatric KNS42 cells by 30%, with IC50 values of 0.26 and 0.40 µM, respectively.

Graphical Abstract
Introduction
Since ancient times, natural products have played a crucial role in the development of medicines for various diseases [1-4]. A recent review by Newman et al. has identified that over the past four decades, >30% of therapeutic drugs approved by the FDA are derived from natural products, their derivatives, or synthetic mimetics inspired by natural compounds [5]. The traditional knowledge of native plants for medicinal purposes is invaluable, and there are ongoing investigations by many research groups from around the world that aim to scientifically validate these traditional remedies and explore their potential applications in modern medicine. Australia's native flora, which is renowned for its rich biodiversity, has been used by Australian Aboriginal peoples for thousands of years, and they have long used Eremophila plants such as E. longifolia and E. alternifolia as traditional medicines [6-8]. In recent times, Eremophila plants have been shown to generate impressive chemical diversity, along with a wide range of biological activities from both extracts or isolated natural products, such as antibacterial, antimalarial, and anticancer effects [9-14]. However, many of the 287 species that make up this genus have not yet been chemically investigated, indicating that Eremophila still has the potential to be the source of new bioactive chemicals and/or known compounds with new biological activity. The use of Eremophila-derived specialized metabolites or semisynthetic derivatives has the potential to impact biodiscovery research.
In this study, we investigated the chemistry of the seeds of Eremophila maculata for the first time. Seeds from the Eremophila genus have rarely been chemically investigated due to difficulties in collecting sufficient quantities of this plant part. Owing to access to Griffith University’s NatureBank biota repository, which contains 45 locally collected Eremophila samples, we were able to obtain more than 20 g of E. maculata seeds. Large-scale extraction and isolation studies on this uniquely abundant plant part afforded two known phytochemicals, (+)-salicifoliol (1) [15,16] and (+)-pinoresinol (2) [17]. The abundant natural product 2 was used as a chemical scaffold for semisynthetic studies. At the same time, all derivatives were fully characterized utilizing spectroscopic and spectrometric techniques.
Plants are a vast reservoir of phytochemicals that serve as the source of numerous drugs, including many with anticancer properties [18,19]. These phytochemicals may act on cellular signalling, apoptosis, metabolic pathways, and cell motility in tumor cells. Most anticancer research focuses on antiproliferation or apoptosis of tumor cells. However, tumor cells are highly aggressive and have the capacity to invade and metastasize from a primary tumor to other parts of the body. Strategies that can limit or inhibit the invasiveness of tumor cells without damaging or killing healthy cells represent a powerful approach that can augment current therapies, prevent metastasis, and improve patient quality of life and survival. Cytotoxicity is defined as the ability of a compound to cause cell death, while anti‑invasive properties refer to the ability of compounds to slow down or block cancer cells from moving. In this study, a unique plant-derived lignan‑based library was screened against glioblastoma (brain cancer) cell lines to evaluate anti-invasive and cytotoxicity effects.
Results and Discussion
The air-dried and ground seeds of E. maculata were sequentially extracted with CH2Cl2 and MeOH. Subsequent purifications of these two extracts afforded the two known natural products (+)-salicifoliol (1) [15,16] and (+)-pinoresinol (2, Figure 1) [17]. Their structures were determined by 1D (1H and 13C) and 2D (COSY, HMBC, HSQC, and ROESY) NMR spectroscopy together with MS data analysis. During our comparison of spectroscopic and chiro-optical data for (+)-salicifoliol (1) with reported literature values [16], we noted that an earlier paper [15] reporting the chemical structure of 1 had misdrawn the stereochemistry. The stereochemical assignment for (+)-salicifoliol (1) has been corrected as shown in Figure 1.
![[1860-5397-22-54-1]](/bjoc/content/figures/1860-5397-22-54-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of the two known natural products, (+)-salicifoliol (1) and (+)-pinoresinol (2), that were obtained from the CH2Cl2/MeOH extracts of Eremophila maculata seeds.
Figure 1: Chemical structures of the two known natural products, (+)-salicifoliol (1) and (+)-pinoresinol (2)...
The most abundant metabolite isolated during these chemical studies was (+)-pinoresinol (2), which has been reported to exhibit a variety of bioactivities. These include antifungal activity against the human pathogens Candida albicans (MIC = 12.5 µM), Trichosporon beigelii (MIC = 25 µM), and Malassezia furfur (MIC = 12.5 µM) [20]; antioxidant activity with the inhibition of CuSO4-induced peroxidation of low-density lipoprotein in a concentration-dependent manner from 0.1−10 µM [21], and anti-invasiveness activity on HT115 human colon cancer cells at a concentration ranging from 1.56−25 µM with a reduction of up to 65% invasion in comparison to the control [22]. Owing to the interesting biological activity of 2 and its moderate abundance in the seeds of E. maculata, we decided to generate five semisynthetic analogues 3−7 (Figure 2) and evaluate these compounds for in vitro cytotoxicity and anti-invasive properties using two glioblastoma cancer cell lines [23,24].
![[1860-5397-22-54-2]](/bjoc/content/figures/1860-5397-22-54-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reaction schemes detailing the semisynthesis of (+)-eudesmin (3), (+)-phillygenin (4), (+)-5,5'-dibromopinoresinol (5), (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6), and (+)-4,4'-dipivaloylpinoresinol (7) from (+)-pinoresinol (2).
Figure 2: Reaction schemes detailing the semisynthesis of (+)-eudesmin (3), (+)-phillygenin (4), (+)-5,5'-dib...
During our semisynthetic studies, several reactions including methylations, halogenations, and acylations were performed, leading to the generation of five analogues in varying yield (10−81%) and high purity (>95%). Semisynthetic analogues 3−5 are known compounds [25,26]. However, some of these compounds have not been fully characterized. Regarding compound 5, this molecule had previously been reported as an intermediate in the total synthesis of racemic pinoresinol [25]. Through a bromination reaction using N-bromosuccinimide (NBS) in CH2Cl2 at room temperature (1 h), we produced 5 as a single enantiomer (i.e., (+)-5,5'-dibromopinoresinol). Herein, we report the full spectroscopic and spectrometric characterization of this molecule. Furthermore, analogues 6 and 7 are new semisynthetic molecules and were also fully characterized following NMR, UV–vis, [α]D, ECD, and ESIMS data analyses.
An example of our structure elucidation studies is described below, which focuses on the brominated pinoresinol enantiomer (+)-5,5'-dibromopinoresinol (5). HRESIMS data revealed an ion at m/z 536.9519 [M + Na]+ that indicated the molecular formula C20H20Br2O6. The 1H NMR spectrum of 5 in CDCl3 (Table 1) indicated that the symmetry of the reaction starting material (+)-pinoresinol (2) had been retained in the brominated product. This resulted in the presence of two signals corresponding to aromatic protons (δH 7.06 (2H), 6.83 (2H)), as well as two methine (δH 4.75 (2H), 3.11 (2H)), one methylene (δH 4.27/3.90, 4H), and one methoxy signal (δH 3.92, 6H)). The 13C NMR and HSQC spectra of 5 indicated a total of 10 unique carbon signals, including one methoxy (δC 56.6, 2C), two methine (δC 85.4, 53.9, 4C), one methylene (δC 71.8, 2C), and six aromatic carbon signals (δC 147.6, 142.9, 133.3, 122.3, 108.4, 108.0, 12C). Comparison of the 1H and 13C NMR data of 5 and (+)-pinoresinol (2) showed a high degree of homology, with the major differences in NMR data associated with a chemical shift of the aryl resonances. Thus, NMR signals for 5 were assigned following a comparison of the chemical shift with the previously reported natural product 2 and detailed analysis of the 2D NMR data (see Table 1). Key COSY, HMBC, and ROESY correlations for 5 are shown below in Figure 3. Collectively, this data confirmed the planar structure assignment for 5. Moreover, the relative configuration of the semisynthetic derivative 5 was assigned following ROESY data analysis.
Table 1: NMR data for (+)-5,5'-dibromopinoresinol (5) in CDCl3.a
| position | δH (multiplicity, J in Hz) | δC, type | COSY | HMBC | ROESY |
| 1 | — | 133.3, C | — | — | — |
| 2 | 6.83 (d, 1.8) | 108.0, CH | 6,7 | 1,3,4,5,6,7 | 3-OMe,7,8,9a |
| 3 | — | 147.6, C | — | — | — |
| 3-OMe | 3.92 (s) | 56.6, CH3 | — | 3 | 2 |
| 4 | — | 142.9, C | — | — | — |
| 4-OH | —b | — | — | — | — |
| 5 | — | 108.4, C | — | — | — |
| 6 | 7.06 (dd, 1.8; 0.5) | 122.3, CH | 2,7 | 2,3,4,5,7 | 7,8,9a |
| 7 | 4.75 (d, 4.2) | 85.4, CH | 2,6,8 | 1,2,6,8,9 | 2,6,8,9b |
| 8 | 3.11 (m) | 53.9, CH | 7,9a,9b | 1,7,9,7',8',9' | 2,6,7,9a |
| 9a | 4.27 (m) | 71.8, CH2 | 8,9b | 1,7,8,7' | 2,6,8,9b |
| 9b | 3.90 (dd, 9.3; 3.6) | 8,9a | 7,7' | 7 | |
| 1' | — | 133.3, C | — | — | — |
| 2' | 6.83 (d, 1.8) | 108.0, CH | 6',7' | 1',3',4',5',6',7' | 3'-OMe,7',8',9'a |
| 3' | — | 147.6, C | — | — | — |
| 3'-OMe | 3.92 (s) | 56.6, CH3 | — | 3' | 2' |
| 4' | — | 142.9, C | — | — | — |
| 4'-OH | —b | — | — | — | |
| 5' | 108.4, C | — | — | — | |
| 6' | 7.06 (dd, 1.8; 0.5) | 122.3, CH | 2',7' | 2',3',4',5',7' | 7',8',9'a |
| 7' | 4.75 (d, 4.2) | 85.4, CH | 2',6',8' | 1',2',6',8',9' | 2',6',8',9'b |
| 8' | 3.11 (m) | 53.9, CH | 7',9'a,9'b | 1',7',9',7,8,9 | 2',6',7',9'a |
| 9'a | 4.27 (m) | 71.8, CH2 | 8',9'b | 1',7',8',7 | 2',6',8',9'b |
| 9'b | 3.90 (dd, 9.3; 3.6) | 8',9'a | 7',7 | 7' | |
aSpectra recorded at 25 °C (800 MHz for 1H and 200 MHz for 13C). bSignal not observed.
![[1860-5397-22-54-3]](/bjoc/content/figures/1860-5397-22-54-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Key COSY, HMBC, and ROESY correlations for (+)-5,5'-dibromopinoresinol (5).
Figure 3: Key COSY, HMBC, and ROESY correlations for (+)-5,5'-dibromopinoresinol (5).
Following the slow evaporation of a methanolic solution of (+)-eudesmin (3), suitable crystals were obtained for X-ray crystallographic studies (Figure 4), which enabled the absolute configuration of 3 to be assigned as 7S,8R,7'S,8'R. Moreover, this is the first report of a crystal structure of (+)-eudesmin (3). In 2015, Lu et al. reported the purification and full characterization of both enantiomers of eudesmin from Acorus tatarinowii, along with the X-ray crystallographic structure (Cu source) for (−)-eudesmin only, which enabled the absolute configuration of this stereoisomer to be assigned [27]. Our experimental data and stereochemical assignment for (+)-eudesmin (3) are consistent with those reported by Lu et al. [27]. Furthermore, during our studies, ECD data was recorded for (+)-eudesmin (3), (+)-pinoresinol (2), and analogues 4−7 (Figure 5). As expected, similar ECD data was observed for all compounds, and thus the absolute configuration of all new derivatives reported here was assigned as 7S,8R,7'S,8'R [28]. Of note, the ECD data for the two new pinoresinol ester derivatives 6 and 7 showed the biggest differences to the natural products owing to the addition of UV-active acyl moieties.
![[1860-5397-22-54-4]](/bjoc/content/figures/1860-5397-22-54-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP drawing of (+)-eudesmin (3).
Figure 4: ORTEP drawing of (+)-eudesmin (3).
![[1860-5397-22-54-5]](/bjoc/content/figures/1860-5397-22-54-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ECD data for compounds 2–7 in MeOH.
Figure 5: ECD data for compounds 2–7 in MeOH.
Owing to our developing interest in identifying natural products and derivatives that are cytotoxic to human cancer cell lines and/or block invasion or migration of proliferating cancer cells [23,29], all compounds were tested against two human glioblastoma cell lines, U251MG and KNS42 [23,24]. Glioblastoma, which is also known as glioblastoma multiforme (GBM), is a grade IV glioma and one of the deadliest human cancers [30]. Following cytotoxicity evaluations of (+)-pinoresinol (2) and its analogues 3–7 against the U251MG cell line, compound (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) showed slight cytotoxicity (19% cell viability reduction) at a concentration of 10 µM. Also, on the KNS42 cell line, only (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) showed a significant reduction (50%) in cell viability at 10 µM (Figure 6). Interestingly, (+)-4,4'-dipivaloylpinoresinol (7) seemed to increase cell viability in the KNS42 assay at all tested concentrations. As such, further investigations into these data is warranted (Figure 6).
![[1860-5397-22-54-6]](/bjoc/content/figures/1860-5397-22-54-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Percentage of cell viability for (+)-pinoresinol (2) and its analogues (+)-eudesmin (3), (+)-phillygenin (4), (+)-5,5'-dibromopinoresinol (5), (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6), and (+)-4,4'-dipivaloylpinoresinol (7) at different concentrations (0.3, 1, 5, and 10 µM) on cell lines U251MG (left) and KNS42 (right). Cells were seeded in a 96-well plate (Costar #3596) in triplicates at 100 µL/well (i.e., 1,500 cells/well) and incubated for 24 h at 37 °C and 5% CO2. Vehicle control (positive control) was the solvent DMSO, in which all compounds were dissolved, plus cells. The final concentration of DMSO was 0.01%. Medium with only cells was the negative control. All data normalized to vehicle control. n = 3 replicates.
Figure 6: Percentage of cell viability for (+)-pinoresinol (2) and its analogues (+)-eudesmin (3), (+)-philly...
Based on the glioblastoma cell viability assay results and the previously reported anti-invasive activity of (+)-pinoresinol (2) towards HT115 cancer cells [22], the (+)-pinoresinol-based library 2–7 was also tested using a transwell invasion assay. A preliminary screen was carried out for the U251MG cells at a concentration of 1 µM with a treatment time of 5 h, and only (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) reduced the invasion of the cells (by ≈50%) when compared to the vehicle control. None of the other compounds tested reduced the invasive capability of the cells (Figure 7A).
![[1860-5397-22-54-7]](/bjoc/content/figures/1860-5397-22-54-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: (A) Transwell invasive assay of the (+)-pinoresinol library 2–7 on U251MG cells at a concentration of 1 µM with a treatment time of 5 h. 45,000 cells (100 µL volume) were seeded into the top of each transwell insert (#CLS3422; Sigma-Aldrich) in Dulbecco’s modified Eagle medium (DMEM), with 0% fetal bovine serum (FBS), in triplicates. The bottom wells contained 700 µL of DMEM (10% FBS). This was incubated for 24 h at 37 °C with 5% CO2. Vehicle control (positive control) was the solvent DMSO, in which all compounds were dissolved, plus cells. The final concentration of DMSO was 0.01%. (B) The U251MG cells were treated with different concentrations of (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6, (0.03, 0.1, 0.3, and 1 µM) over 5 h to monitor the effect on the inhibition of invasion. The % invasion of U251MG cells was used to calculate the half-maximal inhibition concentration (IC50) using GraphPad Prism version 9.02. (C) Representative image of transwell for DMSO on U251MG cells taken with a Leica DMi1 10× objective. (D) Representative image of transwell for 1 µM (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) on U251MG cells taken with a Leica DMi1 10× objective. All data normalized to vehicle control. n = 3 replicates.
Figure 7: (A) Transwell invasive assay of the (+)-pinoresinol library 2–7 on U251MG cells at a concentration ...
Based on the results of the transwell invasion assay (Figure 7), (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) was selected for further study. The U251MG cells were treated with different concentrations of 6 (0.03, 0.1, 0.3, and 1 µM) over 5 h and assayed for inhibition of invasion. The IC50 for compound 6 was calculated as 0.26 µM (Figure 7B). The KNS42 cells were treated with different concentrations of (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6, 0.03, 0.1, 0.3, and 1 µM) for 5 h and assayed for inhibition of invasion (Figure 8A). The compound inhibited invasion at 0.3 and 1 µM by up to 30%, and the IC50 for compound 6 was calculated as 0.40 µM (Figure 8).
![[1860-5397-22-54-8]](/bjoc/content/figures/1860-5397-22-54-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: (A) Transwell invasion assay of (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) on KNS42 cells at a concentration range of 0.03–1 µM with a treatment time of 5 h. 45,000 cells (100 µL volume) were seeded into the top of each transwell insert (#CLS3422; Sigma-Aldrich) in DMEM (0% FBS) in triplicates. The bottom wells contained 700 µL of DMEM (10% FBS). This was incubated for 24 h at 37 °C with 5% CO2. Vehicle control (positive control) was the solvent DMSO, in which all compounds were dissolved, plus cells. The final concentration of DMSO was 0.01%. (B) The % invasion of KNS42 cells was used to calculate the half-maximal inhibition concentration (IC50) using GraphPad Prism version 9.02. (C) Representative image of transwell for DMSO on KNS42 cells taken with a Leica DMi1 10× objective. (D) Representative image of transwell for 1 µM (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) on KNS42 cells taken with a Leica DMi1 10× objective. All data normalized to vehicle control. n = 3 replicates.
Figure 8: (A) Transwell invasion assay of (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) on KNS42 cells at ...
Conclusion
In summary, the first chemical investigations of the seeds of E. maculata yielded the known metabolites (+)-salicifoliol (1, 0.014 dry wt %) and (+)-pinoresinol (2, 0.134 dry wt %). The most abundant metabolite 2 was chosen for semisynthetic studies. Five analogues were successfully generated using standard medicinal chemistry methodologies, and all derivatives were fully characterized. The (+)-pinoresinol library 2–7 was evaluated on glioblastoma-derived U251MG and KNS42 cell lines for their cytotoxicity. Only (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6) showed some cytotoxicity. Transwell invasion assays on the library against U251MG cells showed that analogue 6 was the most active compound, as it reduced the invasion of the cells by ≈50%, with an IC50 value of 0.26 µM. Consequently, compound 6 was further tested in the pediatric cell line KNS42 and was shown to reduce cell invasion in the transwell assay, with an IC50 value of 0.40 µM. These findings provide a rationale for more detailed anticancer studies on pinoresinol and its derivatives in the future.
Experimental
General experimental procedures
Melting points were measured using a Cole-Parmer melting point apparatus and are uncorrected. Specific rotations were determined on a JASCO P-2000 polarimeter. UV–vis spectra were recorded using a CAMSPEC M501 UV–vis spectrophotometer. ECD spectra were obtained on a JASCO J-1500 spectropolarimeter and processed using the software SDAR [31]. NMR spectra were recorded at 25 °C on a Bruker AVANCE III HD 500 or HDX 800 MHz NMR spectrometer equipped with a cryoprobe. The 1H and 13C chemical shifts were referenced to solvent peaks for CDCl3 at δH 7.26 and δC 77.2, respectively. UHPLC–LRESIMS analysis was performed using a previously reported method [32], and data was recorded on an Ultimate 3000 RS UHPLC coupled to a Thermo Fisher Scientific MSQ Plus single quadruple ESI mass spectrometer. HRESIMS data were acquired on Bruker maXis II ETD ESI-qTOF. GRACE C18- or C8-bonded silica (35–70 µm, 60 Å) was used for preadsorption work before HPLC separations, and the preadsorbed sample was packed into an Alltech stainless steel guard cartridge (10 × 30 mm). GRACE C8-bonded silica (35–70 µm, 60 Å) was used for the C8 flash column. Isolute Si SPE cartridge (53 µm, 20 g) was used for the separations. A Thermo Fisher Scientific Dionex Ultimate 3000 UHPLC was used for semipreparative HPLC separations. A Thermo BetaSil C18 column (5 μm, 100 Å, 150 × 21.2 mm), Luna C18 HPLC column (5 μm, 100 Å, 150 × 21.2 mm), or a BetaSil phenyl HPLC column (5 μm, 100 Å, 250 × 21.2 mm) was used for semipreparative HPLC separation. Merck silica gel 60 F254 precoated aluminium plates were used for TLC and analyzed under UV light at 254 and 365 nm. For large-scale studies, the plant material was extracted at room temperature using an Edwards Instrument Company Bio-line orbital shaker set to 200 rpm. All chemical reagents used throughout the experiments were purchased from Sigma-Aldrich. All solvents used for chromatography, UV–vis, ECD, [α]D, and MS were Honeywell Burdick & Jackson brand or Lab-Scan HPLC grade. H2O was filtered using a Sartorius Stedium Arium Pro VF ultrapure H2O system.
Plant material
E. maculata seeds used in these studies were obtained from the NatureBank biota library housed at the Institute for Biomedicine and Glycomics, Griffith University, Australia. This seed sample was collected on December 3, 1993 at Mount Crosby, Queensland, Australia. A voucher specimen (AQ600324) has been deposited with the Queensland Herbarium.
Extraction and isolation
The air-dried and ground seeds of E. maculata (10 g) were extracted with CH2Cl2 (250 mL, 2 h; 250 mL, 16 h) to afford 120.5 mg of crude CH2Cl2 extract. Subsequent extraction with MeOH (250 mL, 2 h; 250 mL, 16 h) afforded 1.43 g of crude MeOH extract. The CH2Cl2 extract (120.5 mg) was subjected to chromatography using a silica flash column (3 × 6 cm) and a 10% stepwise gradient from 100% n–hexane to 100% EtOAc (100 mL washes) followed by a 10% MeOH/90% CH2Cl2 flush (100 mL) to afford 100 fractions that were analyzed by TLC and UHPLC–MS. These data enabled the combining (or discarding) of several fractions, which resulted in 12 final fractions (FA−FL). All 12 samples were analyzed by UHPLC–MS and 1H NMR spectroscopy. Fraction H (22.2 mg) was preadsorbed to C18-bonded silica (≈1 g), packed into a guard cartridge, and attached to a semipreparative Luna C18 HPLC column. A linear gradient from 40% MeOH (0.1% TFA)/60% H2O (0.1% TFA) to 100% MeOH (0.1% TFA) at a flowrate of 4 mL/min over 60 min afforded salicifoliol (1, 1.4 mg, tR 11 min, 0.014 dry wt %) and (+)-pinoresinol (2, 13.4 mg, tR 21–22 min, 0.134 dry wt %). The MeOH extract (1.3 g) was divided into three equal portions and individually preadsorbed to phenyl-bonded silica (≈1 g), packed into a guard cartridge, and subjected to three separate semipreparative BetaSil phenyl HPLC purifications. A linear gradient from 10% MeOH (0.1% TFA)/90% H2O (0.1% TFA) to 100% MeOH (0.1% TFA) at a flowrate of 9 mL/min over 60 min produced semipure (+)-pinoresinol (2, 113.5 mg, tR 41−42 min), which was preadsorbed to C18-bonded silica (≈1 g), packed into a guard cartridge, and subjected to semipreparative C18 HPLC purification. A linear gradient from 40% MeOH (0.1% TFA)/60% H2O (0.1% TFA) to 100% MeOH (0.1% TFA) over 50 min, followed by isocratic conditions of 100% MeOH (0.1% TFA) for 10 min, all at a flow rate of 9 mL/min, gave pure (+)-pinoresinol (2, 19.5 mg, tR 13 min, 0.195 dry wt %). The extraction and isolation protocol described above was repeated on an additional 10 g of seeds to provide a sufficient quantity of 2 for the semisynthetic studies detailed below. The previously described plant compounds salicifoliol (1) [15,16] and (+)-pinoresinol (2) [17] were identified following 1D and 2D NMR (1H, 13C, COSY, HSQC, HMBC, and ROESY), [α]D, MS data analysis, and comparison with literature values.
(+)-Salicifoliol (1): clear gum; [α]D25 +36.4 (c 0.09, MeOH); lit. [α]D25 +56.3 (c 0.06, MeOH) [33]; see Supporting Information File 1 for 1H and 13C NMR data in CDCl3; LRESIMS m/z: [M + H]+ 251, [M − H]− 249.
(+)-Pinoresinol (2): brown gum; [α]D25 +55.5 (c 0.07, MeOH); lit. [α]D25 +20.0 (c 0.75, MeOH) [34]; UV–vis (MeOH) λmax (log ε) 230 (3.94), 279 (3.59) nm; ECD (MeOH) λext (Δε) 208 (+6.25), 256 (−0.08), 282 (+0.26) nm; see Supporting Information File 1 for 1H and 13C NMR data in CDCl3; LRESIMS m/z: [M + Na]+ 381, [M − H]− 357; HRESIMS m/z: [M + Na]+ calcd for C20H22NaO6, 381.1308; found, 381.1307.
Preparation and purification of semisynthetic (+)-pinoresinol derivatives 3–7
Methylation of (+)-pinoresinol (2)
(+)-Pinoresinol (2, 15.8 mg, 0.044 mmol) was dissolved in MeOH/CH2Cl2 1:1 (250 μL) before (trimethylsilyl)diazomethane (2.0 M in diethyl ether, 150 μL) was added dropwise. The reaction mixture was stirred for 20 min at room temperature and then transferred to a separatory funnel containing CH2Cl2 (3 mL) and H2O (3 mL). The H2O layer was washed with CH2Cl2 (3 × 3 mL), and the CH2Cl2-soluble material was dried, preadsorbed to C18-bonded silica (≈1 g), packed into a guard cartridge, and subjected to semipreparative HPLC using a BetaSil C18-bonded silica column. Isocratic conditions of 10% MeOH (0.1% TFA)/90% H2O (0.1% TFA) were applied for 10 min, followed by a linear gradient to 40% MeOH (0.1% TFA)/60% H2O (0.1% TFA) over 5 min, a linear gradient to 100% MeOH (0.1% TFA) over 35 min, and then isocratic conditions of 100% MeOH (0.1% TFA) for 10 min, all at a flow rate of 9 mL/min, to give the dimethylated product (+)-eudesmin (3, 4.4 mg, tR 35−36 min, 26% yield) and the monomethylated product (+)-phillygenin (4, 1.7 mg, tR 32−33 min, 10% yield).
(+)-Eudesmin (3): clear needles (4.4 mg, 26%); mp 94−96 °C (lit. [35] mp 98−100 °C); [α]D24 +110.0 (c 0.4, CHCl3); lit. [α]D +61.0 (c 0.4, CHCl3) [35]; UV–vis (MeOH) λmax (log ε) 230 (4.39), 278 (3.97) nm; ECD (MeOH) λext (Δε) 209 (+9.57), 280 (+0.53) nm; see Supporting Information File 1 for 1H (800 MHz) and 13C NMR (200 MHz) data in CDCl3; LRESIMS m/z: [M + Na]+ 409, [2M + Na]+ 795.
(+)-Phillygenin (4): clear gum (1.7 mg, 10%); [α]D24 +46.8 (c 0.6, CHCl3); lit. [α]D +91.6 (c 0.5, CHCl3) [35]; UV–vis (MeOH) λmax (log ε) 230 (4.01), 279 (3.61) nm; ECD (MeOH) λext (Δε) 206 (+6.69), 280 (+0.22) nm; see Supporting Information File 1 for 1H (800 MHz) and 13C NMR (200 MHz) data in CDCl3; LRESIMS m/z: [M + Na]+ 395.
Bromination of (+)-pinoresinol (2)
(+)-Pinoresinol (2, 10.0 mg, 0.028 mmol) was dissolved in CH2Cl2 (600 μL) before NBS (9.9 mg, 0.056 mmol, 2 equiv) was added. The reaction mixture was stirred for 1 h at room temperature and dried before being preadsorbed to C18-bonded silica (≈1 g). Then, it was packed into a guard cartridge and subjected to semipreparative HPLC using a BetaSil C18-bonded silica column. Isocratic conditions of 10% MeOH (0.1% TFA)/90% H2O (0.1% TFA) were maintained for 10 min, followed by a linear gradient to 40% MeOH (0.1% TFA)/60% H2O (0.1% TFA) over 5 min, a linear gradient to 100% MeOH (0.1% TFA) over 35 min, and then isocratic conditions of 100% MeOH (0.1% TFA) for 10 min, all at a flow rate of 9 mL/min, to give (+)-5,5'-dibromopinoresinol (5, 11.0 mg, tR 37−38 min, 77% yield).
(+)-5,5'-Dibromopinoresinol (5): clear gum (11.0 mg, 77%); [α]D25 +154.7 (c 0.7, MeOH); UV–vis (MeOH) λmax (log ε) 220 (4.28), 286 (3.61) nm; ECD (MeOH) λext (Δε) 213 (+2.61), 284 (+1.27), 340 (0.52) nm; see Supporting Information File 1 for 1H (800 MHz) and 13C NMR (200 MHz) data in CDCl3; LRESIMS m/z: [M (79Br2) − H]− 513, [M (79Br81Br) − H]− 515, [M (81Br2) − H]− 517; HRESIMS m/z: [M + Na]+ calcd for C20H2079Br2NaO6, 536.9519; found, 536.9519 [25].
Acylation of (+)-pinoresinol (2)
(+)-Pinoresinol (2, 13.4 mg, 0.037 mmol) was dissolved in pyridine-d5 (200 μL) before 3,3-dimethylbutyryl chloride (150 μL) was added dropwise. The reaction mixture was stirred for 1 h at room temperature and dried before being preadsorbed to C18-bonded silica (≈1 g). This was then packed into a guard cartridge and subjected to semipreparative HPLC using a BetaSil C18-bonded silica column. Isocratic conditions of 10% MeOH (0.1% TFA)/90% H2O (0.1% TFA) were maintained for 10 min, followed by a linear gradient to 40% MeOH (0.1% TFA)/60% H2O (0.1% TFA) over 5 min, a linear gradient to 100% MeOH (0.1% TFA) over 35 min, and then isocratic conditions of 100% MeOH (0.1% TFA) for 10 min, all at a flow rate of 9 mL/min, to give pure (+)-4,4'-di(3,3-dimethylbutanoyl)pinoresinol (6, 14.6 mg, tR 48−50 min, 70% yield). (+)-Pinoresinol (2, 8.0 mg, 0.022 mmol) was dissolved in pyridine-d5 (200 μL) before pivaloyl chloride (100 μL) was added dropwise. The reaction mixture was stirred for 1 h at room temperature and purified using the same method described above to give (+)-4,4'-dipivaloylpinoresinol (7, 9.5 mg, tR 46−50 min, 81% yield).
(+)-4,4'-Di(3,3-dimethylbutanoyl)pinoresinol (6): clear gum (14.6 mg, 70%); [α]D25 +96.2 (c 0.9, MeOH); UV–vis (MeOH) λmax (log ε) 220 (4.27), 272 (3.78) nm; ECD (MeOH) λext (Δε) 223 (−2.49), 292 (−1.88) nm; see Supporting Information File 1 for 1H (800 MHz) and 13C NMR (200 MHz) data in CDCl3; LRESIMS m/z: [M − H]− 553, [M + Na]+ 577; HRESIMS m/z: [M + Na]+ calcd for C32H42NaO8, 577.2772; found, 577.2766.
(+)-4,4'-Dipivaloylpinoresinol (7): clear gum (9.5 mg, 81%); [α]D25 +30.0 (c 0.05, MeOH); UV–vis (MeOH) λmax (log ε) 220 (4.27), 273 (3.76) nm; ECD (MeOH) λext (Δε) 224 (2.71), 291 (−2.11); see Supporting Information File 1 for 1H (800 MHz) and 13C NMR (200 MHz) data in CDCl3; LRESIMS m/z: [M + Na]+ 549; HRESIMS m/z: [M + Na]+ calcd for C30H38NaO8, 549.2459; found, 549.2458.
X-ray crystallography analysis of (+)-eudesmin (3)
Intensity data were collected on a Rigaku XtaLAB synergy diffractometer using Cu Kα radiation at 100.0(1) K using an Oxford Cryostream cooling device. The structure was solved by direct methods and difference Fourier synthesis [36]. Hydrogen atoms bound to the carbon atom were placed at their idealized positions and included in subsequent refinement cycles. Thermal ellipsoid plots were generated using the program Mercury [37], integrated within the WINGX suite of programs [38]. Crystallographic data for (+)-eudesmin (3) have been deposited with the Cambridge Crystallographic Data Centre and assigned CCDC Number 2366804. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Crystal data for (+)-eudesmin (3)
C22H26O6, M = 386.43, T = 100.0 K, 1.54184 Å, monoclinic, space group P21, a = 11.42420(10) Å, b = 32.78820(10) Å, c = 11.53130(10) Å, β = 116.7680(10)°, V = 3856.50(6) Å3, Z = 8, Z’ = 4, Dc = 1.331 mg⋅M−3, μ(Cu Kα) = 0.792 mm−1, F(000) = 1648, crystal size 0.6 × 0.4 × 0.24 mm3, 136064 reflections measured, θmax = 79.97°, 16547 independent reflections (Rint = 0.056), final R = 0.0334 (I > 2σ(I), 16091 data), wR(F2) = 0.0886 (all data), GOF = 1.041, absolute structure parameter = 0.02(3).
Cancer cell lines and growth
Two glioblastoma cell lines U251MG (#203170) and KNS42 (#IF050356) were obtained from Cell Bank Australia and maintained in the laboratory in either DMEM with 10% FBS and 100 U/mL penicillin-streptomycin (Pen-Strep) (Figure 9). Cells were grown to 80% confluence in T75 flasks with appropriate medium. The cells were then detached form the surface of the flask by incubating in 0.5% trypsin-EDTA for 3 min, followed by neutralization with 7 mL of DMEM complete medium.
![[1860-5397-22-54-9]](/bjoc/content/figures/1860-5397-22-54-9.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: The U251 cell line was derived from a grade III–IV malignant tumor, specifically an astrocytoma, obtained from a 75-year-old male patient. The cell line exhibits a fibroblastic pattern of growth and has a doubling time of 23–24 h. KNS42 cells are derived from a 16-year-old male patient with GBM, and cells appear as polygonal and flat clusters, with a doubling time of approximately 14 h.
Figure 9: The U251 cell line was derived from a grade III–IV malignant tumor, specifically an astrocytoma, ob...
Cytotoxicity or percentage of cell viability
An aliquot (10 µL) of the harvested cells was counted in a haemocytometer after the addition of 10 µL of trypan blue to determine the cell count. The remaining cells were centrifuged at 1,100 rpm for 5 min. Based on the cell count, the spun cells were resuspended in the appropriate volume of complete medium to achieve a cell number of 15,000 cells per mL. Cells were seeded in a 96-well plate (Costar #3596) in triplicates at 100 µL/well (i.e., 1,500 cells/well) and incubated for 24 h at 37 °C and 5% CO2. Vehicle control (positive control) was the solvent DMSO, in which all compounds were dissolved, plus cells. The final concentration of DMSO was 0.01%. Medium with only cells was the negative control. After incubation, the medium was removed, and 100 µL/well of fresh medium with four different concentrations (0.3, 1, 5, and 10 µM) of the compounds were added. Plates were placed in the incubator for 24 h at 37 °C with 5% CO2. After 24 h, the medium was replaced with 100 µL/well of fresh medium with alamarBlue (1:10 dilution, Thermo Fisher #DAL1100) and incubated at 37 °C and 5% CO2. AlamarBlue is a redox indicator used for measuring cell viability [39], and the reaction is based on the live cells converting resazurin to resorufin, i.e., blue to pink /red in color [40].
Changes in cell viability were detected using fluorescence (excitation at 530–560 and emission at 590 nm) or absorbance (detected at 570 and 600 nm) in a plate reader (FLUOstar Omega, BMG Labtech). In this study, cell viability was quantitated by measuring changes in fluorescence after 45 and 90 min of incubation at 37 °C and 5% CO2. The % cell viability was calculated using the following formula:
![[1860-5397-22-54-i1]](/bjoc/content/inline/1860-5397-22-54-i1.svg?max-width=590&scale=1.18182)
Transwell invasion assay
This assay is used to study the invasive capabilities of tumor cells to pharmacological inhibitors. It involves the addition of an extracellular matrix, such as Matrigel, on top of a porous membrane that allows cells to invade in response to the chemotactic gradient. The transwell assay protocol was adapted from the method outlined by Justus et al. [41]. Corning 6.5 mm transwell assay plates (24 wells) with polycarbonate inserts (24) containing 8.0 µm pores (Sigma-Aldrich, #CLS3422) were used in this study.
Cell starvation
U251MG and KNS42 cells were grown in DMEM complete medium in a T75 flask to 50–60% confluency. Cells were washed once with 1× phosphate-buffered saline (PBS), and PBS was replaced with 15 mL of DMEM containing 2% FBS and 100 U/mL Pen-Strep. The flask was placed at 37 °C and 5% CO2 for 24 h of starvation.
Insert preparation
40 µL of Matrigel (1:40 dilution) was added to each insert, followed by fixing for 5 min under UV light in a biosafety cabinet. The inserts were left to dry overnight in the biosafety cabinet. After 24 h, 50 µL of FBS-free DMEM with 100 U/mL Pen-Strep was added to each insert and placed in the incubator at 37 °C and 5% CO2 to rehydrate for 1 h.
Cell preparation
Medium from the starved cells was aspirated, and the cells were washed with 10 mL of 1× PBS. The PBS was replaced with 3 mL of warm 1× trypsin-EDTA solution, and the flask was placed in an incubator at 37 °C and 5% CO2 to detach the cells. The trypsin-EDTA medium was neutralized with 7 mL of DMEM complete medium, and the cell suspension was transferred to a 15 mL centrifuge tube. An aliquot (10 µL) was taken to count the number of viable cells after the addition of an equal volume of trypan blue. Depending on the cell count, the cell pellet in the tube was resuspended in a volume of FBS-free medium to give 450,000 cells/mL, such that each insert would contain 45,000 cells/100 µL.
Assay setup
An aliquot of 700 µL each of DMEM complete medium containing 10% FBS and Pen-Strep and the appropriate test compound (1 µM) was added to the wells of the plate. Positive control was the vehicle control, i.e., cell suspension with DMSO. To each prepared insert, 100 µL of cell suspension (prepared above) with the appropriate test compound was added, and the inserts were placed into the wells with corresponding medium plus compound. The plates were incubated for 5 h in an incubator at 37 °C and 5% CO2.
Cell fixing and staining
After 5 h of incubation, 700 µL of 80% ethanol was added to the empty wells of the plate. The inserts were gently lifted, and a cotton-tipped applicator was used to remove the medium and uninvaded cells from the top of the inserts before being placed into ethanol to fix the cells (7−10 min). The inserts were lifted, any excess ethanol was removed with a cotton-tipped applicator, and the inserts were placed upside down to dry. Ethanol was removed from the wells, and 700 µL of 0.2% w/v crystal violet solution was added to each well. The dried inserts were placed into this solution for 10 min to stain the cells. After 10 min, the inserts were gently lifted and rinsed in autoclaved water 2−3 times to remove excess stain. The inside of the inserts was cleaned with cotton-tipped applicators to remove excess water and stain, and the inserts were placed upside down to dry overnight in a biosafety cabinet.
Cell imaging and counting
A Leica DMi1 microscope camera with a 10× objective was used to take three images per insert (different fields of view) using the Leica Application Suite (version 3.7.2.22383). Each image was processed in Fiji (ImageJ). A grid was applied to each image by selecting ‘Analyze’>’Tools’>’Grid’, and the cells were counted, grid by grid, using a manual cell counter. The cell counts from the three images per insert were then averaged to obtain the average number of invaded cells per insert.
Data analysis
All raw data collected from cell counting was processed in Microsoft Excel. This included the average cell number for each replicate and test compound and the average invasion percentage for each sample extract. The % invasion was calculated using the following formula:
![[1860-5397-22-54-i2]](/bjoc/content/inline/1860-5397-22-54-i2.svg?max-width=590&scale=1.18182)
The data was exported into Prism (GraphPad 9.02), where a one-way analysis of variance (ANOVA) was performed to determine whether the results were significant.
Dose-response curve
The compound that showed significant inhibition of invasion of the cells was selected for this assay. The test compound was diluted to provide a concentration range (0.03, 0.1, 0.3, and 1 µM). These dilutions were used in the assay as detailed in the sections above.
Supporting Information
| Supporting Information File 1: Compound characterization data. | ||
| Format: PDF | Size: 4.9 MB | Download |
Acknowledgements
We acknowledge the Benefit Sharing Agreement between the Queensland Government and Griffith University under the Biodiscovery Act 2004 (QLD), enabling equitable benefit sharing from biodiscovery using Queensland-sourced native biological material.
We thank NatureBank (https://www.griffith.edu.au/research/institute-biomedicine-glycomics/facilities/naturebank) for supplying the Eremophila seeds used in these studies.
Funding
The authors acknowledge the Australian Research Council (ARC) for support towards NMR, MS, and HPLC equipment (grants LE0668477, LE140100119, LE0237908, and LE230100128). S. A. R would like to acknowledge funding from Channel 7 Children’s Foundation grant. S. A. R. and R. A. D. both thank the NeuroSurgical Research Foundation for grant funding. C. Z. would like to thank Griffith University for Ph. D. scholarships (GUPRS and GUIPRS).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Villa, F. A.; Gerwick, L. Immunopharmacol. Immunotoxicol. 2010, 32, 228–237. doi:10.3109/08923970903296136
Return to citation in text: [1] -
Balunas, M. J.; Kinghorn, A. D. Life Sci. 2005, 78, 431–441. doi:10.1016/j.lfs.2005.09.012
Return to citation in text: [1] -
Achan, J.; Talisuna, A. O.; Erhart, A.; Yeka, A.; Tibenderana, J. K.; Baliraine, F. N.; Rosenthal, P. J.; D'Alessandro, U. Malar. J. 2011, 10, 144. doi:10.1186/1475-2875-10-144
Return to citation in text: [1] -
Kardos, N.; Demain, A. L. Appl. Microbiol. Biotechnol. 2011, 92, 677–687. doi:10.1007/s00253-011-3587-6
Return to citation in text: [1] -
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2020, 83, 770–803. doi:10.1021/acs.jnatprod.9b01285
Return to citation in text: [1] -
Quinn, R. J. Drug Dev. Res. 1999, 46, 250–254. doi:10.1002/(sici)1098-2299(199903/04)46:3/4<250::aid-ddr9>3.0.co;2-8
Return to citation in text: [1] -
Chinnock, R. J. Eremophila and allied genera: a monograph of the plant family Myoporaceae; Rosenberg Publishing: Dural, Australia, 2007.
Return to citation in text: [1] -
Richmond, G. S. J. Adel. Bot. Gard. 1993, 15, 101–107.
Return to citation in text: [1] -
Sadgrove, N. J.; Jones, G. L. J. Ethnopharmacol. 2013, 147, 638–644. doi:10.1016/j.jep.2013.03.060
Return to citation in text: [1] -
Barnes, E. C.; Kavanagh, A. M.; Ramu, S.; Blaskovich, M. A.; Cooper, M. A.; Davis, R. A. Phytochemistry 2013, 93, 162–169. doi:10.1016/j.phytochem.2013.02.021
Return to citation in text: [1] -
Kumar, R.; Duffy, S.; Avery, V. M.; Carroll, A. R.; Davis, R. A. J. Nat. Prod. 2018, 81, 1079–1083. doi:10.1021/acs.jnatprod.7b00992
Return to citation in text: [1] -
Pedersen, H. A.; Ndi, C.; Semple, S. J.; Buirchell, B.; Møller, B. L.; Staerk, D. J. Nat. Prod. 2020, 83, 1598–1610. doi:10.1021/acs.jnatprod.0c00070
Return to citation in text: [1] -
Zhao, Y.; Li, T.; Kjaerulff, L.; Venter, H.; Coriani, S.; Møller, B. L.; Semple, S.; Staerk, D. J. Nat. Prod. 2023, 86, 2638–2650. doi:10.1021/acs.jnatprod.3c00655
Return to citation in text: [1] -
Petersen, M. J.; Liang, C.; Kjaerulff, L.; Ndi, C.; Semple, S.; Buirchell, B.; Coriani, S.; Møller, B. L.; Staerk, D. Phytochemistry 2022, 196, 113072. doi:10.1016/j.phytochem.2021.113072
Return to citation in text: [1] -
Hye Jin, K.; Kye Jung, S.; Hyun Soo, K.; Donghyn, K.; Hyung Jun, K.; Young Deog, H.; Kee Dong, Y. Nat. Prod. Sci. 2023, 29, 268–283. doi:10.20307/nps.2023.29.4.268
Return to citation in text: [1] [2] [3] [4] -
Yamauchi, S.; Ina, T.; Kirikihira, T.; Masuda, T. Biosci., Biotechnol., Biochem. 2004, 68, 183–192. doi:10.1271/bbb.68.183
Return to citation in text: [1] [2] [3] [4] -
Umezawa, T.; Kuroda, H.; Isohata, T.; Higuchi, T.; Shimada, M. Biosci., Biotechnol., Biochem. 1994, 58, 230–234. doi:10.1271/bbb.58.230
Return to citation in text: [1] [2] [3] -
Yuan, M.; Zhang, G.; Bai, W.; Han, X.; Li, C.; Bian, S. Oxid. Med. Cell. Longevity 2022, 1429869. doi:10.1155/2022/1429869
Return to citation in text: [1] -
Yool, A. J.; Ramesh, S. Front. Pharmacol. 2020, 11, 358. doi:10.3389/fphar.2020.00358
Return to citation in text: [1] -
Hwang, B.; Lee, J.; Liu, Q.-H.; Woo, E.-R.; Lee, D. G. Molecules 2010, 15, 3507–3516. doi:10.3390/molecules15053507
Return to citation in text: [1] -
Kang, M.-H.; Naito, M.; Sakai, K.; Uchida, K.; Osawa, T. Life Sci. 1999, 66, 161–171. doi:10.1016/s0024-3205(99)00574-3
Return to citation in text: [1] -
Hashim, Y. Z. H.-Y.; Rowland, I. R.; McGlynn, H.; Servili, M.; Selvaggini, R.; Taticchi, A.; Esposto, S.; Montedoro, G.; Kaisalo, L.; Wähälä, K.; Gill, C. I. R. Int. J. Cancer 2008, 122, 495–500. doi:10.1002/ijc.23148
Return to citation in text: [1] [2] -
Varricchio, A.; Khan, S.; Price, Z. K.; Davis, R. A.; Ramesh, S. A.; Yool, A. J. Cancers 2023, 15, 1027. doi:10.3390/cancers15041027
Return to citation in text: [1] [2] [3] -
Chen, S.; Deng, X.; Sheng, H.; Rong, Y.; Zheng, Y.; Zhang, Y.; Lin, J. Mol. Ther.–Nucleic Acids 2021, 26, 417–431. doi:10.1016/j.omtn.2021.07.024
Return to citation in text: [1] [2] -
Yue, F.; Lan, W.; Zhang, L.; Lu, F.; Sun, R.; Ralph, J. Front. Energy Res. 2021, 9, 640337. doi:10.3389/fenrg.2021.640337
Return to citation in text: [1] [2] [3] -
Miyazawa, M.; Kasahara, H.; Kameoka, H. Phytochemistry 1993, 34, 1501–1507. doi:10.1016/s0031-9422(00)90836-7
Return to citation in text: [1] -
Lu, Y.; Xue, Y.; Liu, J.; Yao, G.; Li, D.; Sun, B.; Zhang, J.; Liu, Y.; Qi, C.; Xiang, M.; Luo, Z.; Du, G.; Zhang, Y. J. Nat. Prod. 2015, 78, 2205–2214. doi:10.1021/acs.jnatprod.5b00328
Return to citation in text: [1] [2] -
Stomberg, R.; Langer, V.; Li, S.; Lundquist, K. Acta Crystallogr., Sect. E: Struct. Rep. Online 2001, 57, 692–694. doi:10.1107/s1600536801011084
Return to citation in text: [1] -
Tietjen, I.; Cassel, J.; Register, E. T.; Zhou, X. Y.; Messick, T. E.; Keeney, F.; Lu, L. D.; Beattie, K. D.; Rali, T.; Tebas, P.; Ertl, H. C. J.; Salvino, J. M.; Davis, R. A.; Montaner, L. J. Antimicrob. Agents Chemother. 2021, 65, e00772-21. doi:10.1128/aac.00772-21
Return to citation in text: [1] -
Wu, W.; Klockow, J. L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H. E. Pharmacol. Res. 2021, 171, 105780. doi:10.1016/j.phrs.2021.105780
Return to citation in text: [1] -
Weeratunga, S.; Hu, N.-J.; Simon, A.; Hofmann, A. BMC Bioinf. 2012, 13, 201. doi:10.1186/1471-2105-13-201
Return to citation in text: [1] -
Hayes, S.; Taki, A. C.; Lum, K. Y.; Byrne, J. J.; Ekins, M. G.; Gasser, R. B.; Davis, R. A. Beilstein J. Org. Chem. 2022, 18, 1544–1552. doi:10.3762/bjoc.18.164
Return to citation in text: [1] -
González, A. G.; Estévez-Reyes, R.; Mato, C. J. Nat. Prod. 1989, 52, 1139–1142. doi:10.1021/np50065a037
Return to citation in text: [1] -
Páska, C.; Innocenti, G.; Ferlin, M.; Kunvári, M.; László, M. Nat. Prod. Lett. 2002, 16, 359–363. doi:10.1080/1057530290033123
Return to citation in text: [1] -
Iida, T.; Nakano, M.; Ito, K. Phytochemistry 1982, 21, 673–675. doi:10.1016/0031-9422(82)83163-4
Return to citation in text: [1] [2] [3] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466–470. doi:10.1107/s0021889807067908
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/s0021889899006020
Return to citation in text: [1] -
Ligasová, A.; Koberna, K. Molecules 2021, 26, 5515. doi:10.3390/molecules26185515
Return to citation in text: [1] -
O'Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Eur. J. Biochem. 2000, 267, 5421–5426. doi:10.1046/j.1432-1327.2000.01606.x
Return to citation in text: [1] -
Justus, C. R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L. V. J. Visualized Exp. 2014, No. 88, e51046. doi:10.3791/51046
Return to citation in text: [1]
| 17. | Umezawa, T.; Kuroda, H.; Isohata, T.; Higuchi, T.; Shimada, M. Biosci., Biotechnol., Biochem. 1994, 58, 230–234. doi:10.1271/bbb.58.230 |
| 33. | González, A. G.; Estévez-Reyes, R.; Mato, C. J. Nat. Prod. 1989, 52, 1139–1142. doi:10.1021/np50065a037 |
| 34. | Páska, C.; Innocenti, G.; Ferlin, M.; Kunvári, M.; László, M. Nat. Prod. Lett. 2002, 16, 359–363. doi:10.1080/1057530290033123 |
| 1. | Villa, F. A.; Gerwick, L. Immunopharmacol. Immunotoxicol. 2010, 32, 228–237. doi:10.3109/08923970903296136 |
| 2. | Balunas, M. J.; Kinghorn, A. D. Life Sci. 2005, 78, 431–441. doi:10.1016/j.lfs.2005.09.012 |
| 3. | Achan, J.; Talisuna, A. O.; Erhart, A.; Yeka, A.; Tibenderana, J. K.; Baliraine, F. N.; Rosenthal, P. J.; D'Alessandro, U. Malar. J. 2011, 10, 144. doi:10.1186/1475-2875-10-144 |
| 4. | Kardos, N.; Demain, A. L. Appl. Microbiol. Biotechnol. 2011, 92, 677–687. doi:10.1007/s00253-011-3587-6 |
| 15. | Hye Jin, K.; Kye Jung, S.; Hyun Soo, K.; Donghyn, K.; Hyung Jun, K.; Young Deog, H.; Kee Dong, Y. Nat. Prod. Sci. 2023, 29, 268–283. doi:10.20307/nps.2023.29.4.268 |
| 16. | Yamauchi, S.; Ina, T.; Kirikihira, T.; Masuda, T. Biosci., Biotechnol., Biochem. 2004, 68, 183–192. doi:10.1271/bbb.68.183 |
| 23. | Varricchio, A.; Khan, S.; Price, Z. K.; Davis, R. A.; Ramesh, S. A.; Yool, A. J. Cancers 2023, 15, 1027. doi:10.3390/cancers15041027 |
| 24. | Chen, S.; Deng, X.; Sheng, H.; Rong, Y.; Zheng, Y.; Zhang, Y.; Lin, J. Mol. Ther.–Nucleic Acids 2021, 26, 417–431. doi:10.1016/j.omtn.2021.07.024 |
| 38. | Farrugia, L. J. J. Appl. Crystallogr. 1999, 32, 837–838. doi:10.1107/s0021889899006020 |
| 9. | Sadgrove, N. J.; Jones, G. L. J. Ethnopharmacol. 2013, 147, 638–644. doi:10.1016/j.jep.2013.03.060 |
| 10. | Barnes, E. C.; Kavanagh, A. M.; Ramu, S.; Blaskovich, M. A.; Cooper, M. A.; Davis, R. A. Phytochemistry 2013, 93, 162–169. doi:10.1016/j.phytochem.2013.02.021 |
| 11. | Kumar, R.; Duffy, S.; Avery, V. M.; Carroll, A. R.; Davis, R. A. J. Nat. Prod. 2018, 81, 1079–1083. doi:10.1021/acs.jnatprod.7b00992 |
| 12. | Pedersen, H. A.; Ndi, C.; Semple, S. J.; Buirchell, B.; Møller, B. L.; Staerk, D. J. Nat. Prod. 2020, 83, 1598–1610. doi:10.1021/acs.jnatprod.0c00070 |
| 13. | Zhao, Y.; Li, T.; Kjaerulff, L.; Venter, H.; Coriani, S.; Møller, B. L.; Semple, S.; Staerk, D. J. Nat. Prod. 2023, 86, 2638–2650. doi:10.1021/acs.jnatprod.3c00655 |
| 14. | Petersen, M. J.; Liang, C.; Kjaerulff, L.; Ndi, C.; Semple, S.; Buirchell, B.; Coriani, S.; Møller, B. L.; Staerk, D. Phytochemistry 2022, 196, 113072. doi:10.1016/j.phytochem.2021.113072 |
| 25. | Yue, F.; Lan, W.; Zhang, L.; Lu, F.; Sun, R.; Ralph, J. Front. Energy Res. 2021, 9, 640337. doi:10.3389/fenrg.2021.640337 |
| 26. | Miyazawa, M.; Kasahara, H.; Kameoka, H. Phytochemistry 1993, 34, 1501–1507. doi:10.1016/s0031-9422(00)90836-7 |
| 39. | Ligasová, A.; Koberna, K. Molecules 2021, 26, 5515. doi:10.3390/molecules26185515 |
| 6. | Quinn, R. J. Drug Dev. Res. 1999, 46, 250–254. doi:10.1002/(sici)1098-2299(199903/04)46:3/4<250::aid-ddr9>3.0.co;2-8 |
| 7. | Chinnock, R. J. Eremophila and allied genera: a monograph of the plant family Myoporaceae; Rosenberg Publishing: Dural, Australia, 2007. |
| 8. | Richmond, G. S. J. Adel. Bot. Gard. 1993, 15, 101–107. |
| 21. | Kang, M.-H.; Naito, M.; Sakai, K.; Uchida, K.; Osawa, T. Life Sci. 1999, 66, 161–171. doi:10.1016/s0024-3205(99)00574-3 |
| 36. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 5. | Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2020, 83, 770–803. doi:10.1021/acs.jnatprod.9b01285 |
| 22. | Hashim, Y. Z. H.-Y.; Rowland, I. R.; McGlynn, H.; Servili, M.; Selvaggini, R.; Taticchi, A.; Esposto, S.; Montedoro, G.; Kaisalo, L.; Wähälä, K.; Gill, C. I. R. Int. J. Cancer 2008, 122, 495–500. doi:10.1002/ijc.23148 |
| 37. | Macrae, C. F.; Bruno, I. J.; Chisholm, J. A.; Edgington, P. R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P. A. J. Appl. Crystallogr. 2008, 41, 466–470. doi:10.1107/s0021889807067908 |
| 17. | Umezawa, T.; Kuroda, H.; Isohata, T.; Higuchi, T.; Shimada, M. Biosci., Biotechnol., Biochem. 1994, 58, 230–234. doi:10.1271/bbb.58.230 |
| 15. | Hye Jin, K.; Kye Jung, S.; Hyun Soo, K.; Donghyn, K.; Hyung Jun, K.; Young Deog, H.; Kee Dong, Y. Nat. Prod. Sci. 2023, 29, 268–283. doi:10.20307/nps.2023.29.4.268 |
| 35. | Iida, T.; Nakano, M.; Ito, K. Phytochemistry 1982, 21, 673–675. doi:10.1016/0031-9422(82)83163-4 |
| 15. | Hye Jin, K.; Kye Jung, S.; Hyun Soo, K.; Donghyn, K.; Hyung Jun, K.; Young Deog, H.; Kee Dong, Y. Nat. Prod. Sci. 2023, 29, 268–283. doi:10.20307/nps.2023.29.4.268 |
| 16. | Yamauchi, S.; Ina, T.; Kirikihira, T.; Masuda, T. Biosci., Biotechnol., Biochem. 2004, 68, 183–192. doi:10.1271/bbb.68.183 |
| 20. | Hwang, B.; Lee, J.; Liu, Q.-H.; Woo, E.-R.; Lee, D. G. Molecules 2010, 15, 3507–3516. doi:10.3390/molecules15053507 |
| 25. | Yue, F.; Lan, W.; Zhang, L.; Lu, F.; Sun, R.; Ralph, J. Front. Energy Res. 2021, 9, 640337. doi:10.3389/fenrg.2021.640337 |
| 18. | Yuan, M.; Zhang, G.; Bai, W.; Han, X.; Li, C.; Bian, S. Oxid. Med. Cell. Longevity 2022, 1429869. doi:10.1155/2022/1429869 |
| 19. | Yool, A. J.; Ramesh, S. Front. Pharmacol. 2020, 11, 358. doi:10.3389/fphar.2020.00358 |
| 35. | Iida, T.; Nakano, M.; Ito, K. Phytochemistry 1982, 21, 673–675. doi:10.1016/0031-9422(82)83163-4 |
| 17. | Umezawa, T.; Kuroda, H.; Isohata, T.; Higuchi, T.; Shimada, M. Biosci., Biotechnol., Biochem. 1994, 58, 230–234. doi:10.1271/bbb.58.230 |
| 16. | Yamauchi, S.; Ina, T.; Kirikihira, T.; Masuda, T. Biosci., Biotechnol., Biochem. 2004, 68, 183–192. doi:10.1271/bbb.68.183 |
| 35. | Iida, T.; Nakano, M.; Ito, K. Phytochemistry 1982, 21, 673–675. doi:10.1016/0031-9422(82)83163-4 |
| 27. | Lu, Y.; Xue, Y.; Liu, J.; Yao, G.; Li, D.; Sun, B.; Zhang, J.; Liu, Y.; Qi, C.; Xiang, M.; Luo, Z.; Du, G.; Zhang, Y. J. Nat. Prod. 2015, 78, 2205–2214. doi:10.1021/acs.jnatprod.5b00328 |
| 25. | Yue, F.; Lan, W.; Zhang, L.; Lu, F.; Sun, R.; Ralph, J. Front. Energy Res. 2021, 9, 640337. doi:10.3389/fenrg.2021.640337 |
| 40. | O'Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Eur. J. Biochem. 2000, 267, 5421–5426. doi:10.1046/j.1432-1327.2000.01606.x |
| 27. | Lu, Y.; Xue, Y.; Liu, J.; Yao, G.; Li, D.; Sun, B.; Zhang, J.; Liu, Y.; Qi, C.; Xiang, M.; Luo, Z.; Du, G.; Zhang, Y. J. Nat. Prod. 2015, 78, 2205–2214. doi:10.1021/acs.jnatprod.5b00328 |
| 41. | Justus, C. R.; Leffler, N.; Ruiz-Echevarria, M.; Yang, L. V. J. Visualized Exp. 2014, No. 88, e51046. doi:10.3791/51046 |
| 32. | Hayes, S.; Taki, A. C.; Lum, K. Y.; Byrne, J. J.; Ekins, M. G.; Gasser, R. B.; Davis, R. A. Beilstein J. Org. Chem. 2022, 18, 1544–1552. doi:10.3762/bjoc.18.164 |
| 15. | Hye Jin, K.; Kye Jung, S.; Hyun Soo, K.; Donghyn, K.; Hyung Jun, K.; Young Deog, H.; Kee Dong, Y. Nat. Prod. Sci. 2023, 29, 268–283. doi:10.20307/nps.2023.29.4.268 |
| 16. | Yamauchi, S.; Ina, T.; Kirikihira, T.; Masuda, T. Biosci., Biotechnol., Biochem. 2004, 68, 183–192. doi:10.1271/bbb.68.183 |
| 22. | Hashim, Y. Z. H.-Y.; Rowland, I. R.; McGlynn, H.; Servili, M.; Selvaggini, R.; Taticchi, A.; Esposto, S.; Montedoro, G.; Kaisalo, L.; Wähälä, K.; Gill, C. I. R. Int. J. Cancer 2008, 122, 495–500. doi:10.1002/ijc.23148 |
| 31. | Weeratunga, S.; Hu, N.-J.; Simon, A.; Hofmann, A. BMC Bioinf. 2012, 13, 201. doi:10.1186/1471-2105-13-201 |
| 23. | Varricchio, A.; Khan, S.; Price, Z. K.; Davis, R. A.; Ramesh, S. A.; Yool, A. J. Cancers 2023, 15, 1027. doi:10.3390/cancers15041027 |
| 24. | Chen, S.; Deng, X.; Sheng, H.; Rong, Y.; Zheng, Y.; Zhang, Y.; Lin, J. Mol. Ther.–Nucleic Acids 2021, 26, 417–431. doi:10.1016/j.omtn.2021.07.024 |
| 30. | Wu, W.; Klockow, J. L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H. E. Pharmacol. Res. 2021, 171, 105780. doi:10.1016/j.phrs.2021.105780 |
| 28. | Stomberg, R.; Langer, V.; Li, S.; Lundquist, K. Acta Crystallogr., Sect. E: Struct. Rep. Online 2001, 57, 692–694. doi:10.1107/s1600536801011084 |
| 23. | Varricchio, A.; Khan, S.; Price, Z. K.; Davis, R. A.; Ramesh, S. A.; Yool, A. J. Cancers 2023, 15, 1027. doi:10.3390/cancers15041027 |
| 29. | Tietjen, I.; Cassel, J.; Register, E. T.; Zhou, X. Y.; Messick, T. E.; Keeney, F.; Lu, L. D.; Beattie, K. D.; Rali, T.; Tebas, P.; Ertl, H. C. J.; Salvino, J. M.; Davis, R. A.; Montaner, L. J. Antimicrob. Agents Chemother. 2021, 65, e00772-21. doi:10.1128/aac.00772-21 |
© 2026 Zhang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.