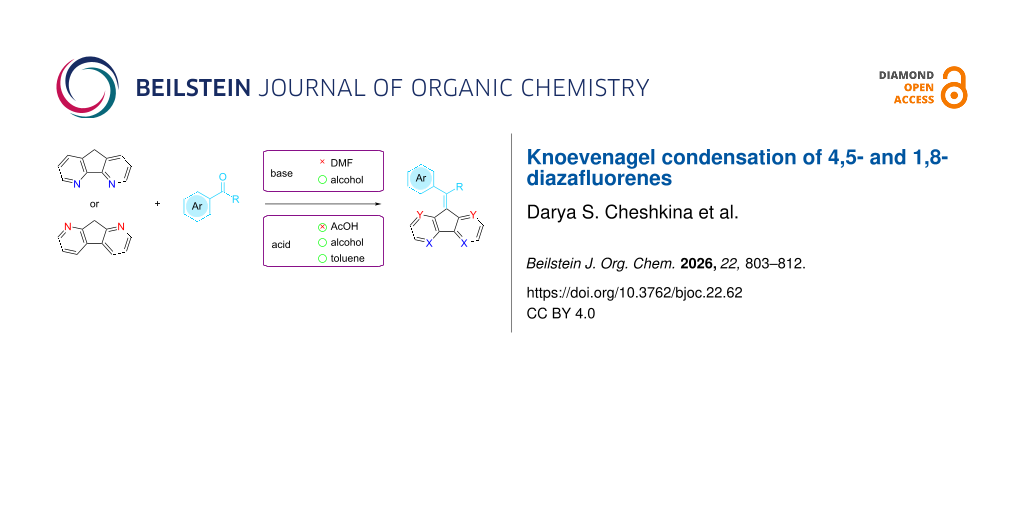
Abstract
Diazafluorenylidenes are potential antitumor agents, ligands, and functional materials. However, there are currently only limited methods for their synthesis, which are complicated and typically based on modifications of 4,5-diazafluorenone. Herein, we systematically studied the Knoevenagel condensation of both 4,5- and 1,8-diazafluorenes with aromatic aldehydes and evaluated the diazafluorenes’ nature and solvent influence on the mechanism and reactivity. We also identified conditions allowing the preparation of both 4,5- and 1,8-diazafluorenylidenes in yields up to 85% minimizing complicated synthetic or experimental workup procedures. Additionally, the condensation of diazafluorenes with ketones resulted in unique di(pyridin-2-yl)methylene)-9H-diazafluorenes being electron-deficient ligands and functional materials.

Graphical Abstract
Introduction
Diazafluorenylidene derivatives were reported to be applied as antitumor agents [1-3], emitters and sensors [4,5]. They have also been extensively used as ligands yielding a wide library of transition-metal complexes [6-8], catalysts [9,10], light-driven molecular motors [11,12], nonlinear optical chromophores [13], and electrochromic materials [14]. Furthermore, diazafluorenylidenes may exhibit polymorphism, high solid-state emission, and decent charge carrier mobility which makes these molecules promising for use in organic optoelectronics [5,15,16].
Current synthetic approaches to diazafluorenylidenes are limited. The typical method for their synthesis involves coupling reactions of diazafluoren-9-one [4,6,7] or diazafluoren-9-diazomethane [17,18] with thiones yielding the products in moderate yields (20–70%). However, these routes require the preliminary synthesis of malodorous thiones and diazomethane derivatives from the corresponding ketones complicating the synthetic protocol. Therefore, the Knoevenagel condensation seemed to be an alternative and preferable approach. Previously reported syntheses of diazafluorenylidenes employed diazafluorenones as the carbonyl component which are reacted with an active methylene moiety like malononitrile or acetonitrile derivatives [14,19,20]. However, the use of diazafluorenes as the methylene component in condensation reactions remains underexplored. These reactions can be carried out under basic [13,21], acidic [3,5] or Lewis acid catalysis [15,22], but the corresponding literature examples are scarce, especially for 1,8-diazafluorene. We have previously reported the condensation of 4,5-diazafluorene (1) with aromatic aldehydes and dialdehydes under ammonium acetate catalysis in acetic acid giving the corresponding products in good yields [5,23]. Remarkably, the only condensation of 1,8-diazafluorene (2) was performed using a TiCl4/pyridine system. However, the reaction yield was very low (11%) [15]. The latter approach was also used [22] for the condensation of 4,5-diazafluorene with ketones being especially challenging due to sterical issues. Moreover, this approach requires the smelly solvent pyridine, moisture-sensitive environmentally aggressive reagent (TiCl4) and subsequent complicated treatment of the resulting reaction mixture making it inapplicable for large-scale high-efficiency production of functional materials. Therefore, analyzing the literature examples we assumed that 4,5- and 1,8-diazafluorenes may have different reactivity in the Knoevenagel condensation and the development of efficient and convenient synthetic routes to diazafluorenylidenes is of considerable importance.
Here, we systematically studied the Knoevenagel condensation of both 4,5- and 1,8-diazafluorenes with aromatic aldehydes and evaluated the influence of the diazafluorene nature and solvent on the reaction mechanisms and reactivity. The reactivity of diazafluorenes under basic conditions was demonstrated to be improved in protic solvents due to solvation of ion pairs by these solvents. We found that acidic conditions allow the preparation of both 4,5- and 1,8-diazafluorenylidenes in yields up to 85%. The basicity of the diazafluorenes was demonstrated to be different and the reaction of 1,8-diazafluorene is impeded because of electrostatic repulsion of the protonated tautomer and the iminium cation. Additionally, we performed the condensation of diazafluorenes with ketones yielding unique di(pyridin-2-yl)methylene)-9H-diazafluorenes.
Results and Discussion
Basic conditions
Following the typical protocol of the Knoevenagel condensation, the reactions of 4,5- and 1,8-diazafluorenes with several electron-donor and electron-acceptor-substituted benzaldehydes (viz. 4-methoxybenzaldehyde, 4-bromobenzaldehyde, 3-nitrobenzaldehyde, and unsubstituted benzaldehyde) were first carried out under basic conditions, using t-BuONa in DMF or t-BuOH (Scheme 1 and Table 1). Both diazafluorenes 1 and 2 reacted under such conditions, but only acceptor-substituted aldehydes were able to react in DMF, specifically 4-bromobenzaldehyde and 3-nitrobenzaldehyde. Though, the reaction in tert-butanol was successful for all studied substrates. These observations were attributed to solvation phenomena. Specifically, solvation by aprotic DMF having a high dipole moment promotes the formation of the [Na+–diazafluorenyl−] ion pair, whose anion displays insufficient nucleophilicity for interaction with weakly electrophilic aldehydes (e.g., benzaldehyde and 4-methoxybenzaldehyde). On the other hand, tert-butanol is effectively solvating ions via the formation of hydrogen bonds [24], thus promoting the dissociation of the ion pair and increasing the nucleophilicity of the diazafluorenyl anion, which becomes capable of interacting with less active aldehydes.
![[1860-5397-22-62-i1]](/bjoc/content/inline/1860-5397-22-62-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Knoevenagel reaction of 1 and 2 under basic conditions.
Scheme 1: Knoevenagel reaction of 1 and 2 under basic conditions.
Table 1: Condensation conditions and yields for 4,5- (1) and 1,8-diazafluorenes (2).
| Diazafluorene | 1 | 2 | |||
| R | Product | Yield, % (solvent) | Product | Yield, % (solvent) | |
| 3-NO2 | basic | 3a | 14a (DMF) | 4a | 47a (DMF) |
| 18b (t-BuOH) | 28b (t-BuOH) | ||||
| acidic | 38c,d (AcOH) | 0c (AcOH) | |||
| 67c,d (EtOH) | |||||
| 50c (toluene) | |||||
| 4-Br | basic | 3b | 28a (DMF) | 4b | 32a (DMF) |
| 28b (t-BuOH) | 45b (t-BuOH) | ||||
| acidic | 84c,d (AcOH) | 0c (AcOH) | |||
| 60c,d (EtOH) | |||||
| 27c (toluene) | |||||
| H | basic | 3c | 0a (DMF) | 4c | 0a (DMF) |
| 34b (t-BuOH) | 57b (t-BuOH) | ||||
| acidic | 45c,d (AcOH) | 0c (AcOH) | |||
| 85c,d (EtOH) | |||||
| 41c (toluene) | |||||
| 4-OMe | basic | 3d | 0a (DMF) | 4d | 0a (DMF) |
| 29b (t-BuOH) | 27b (t-BuOH) | ||||
| acidic | 67c,d (AcOH) | 0c (AcOH) | |||
| 39c (EtOH) | |||||
| 52c (toluene) | |||||
Conditions: at-BuONa, solvent, rt, 2 h; bt-BuONa, solvent, 30 °C,12 h; cNH4OAc/AcOH, solvent, 110 °C, 12 h; dthe product precipitated directly from the reaction mixture.
Acidic conditions
As the reaction yields under basic conditions were moderate and the reactivity of aldehydes was insufficient it seemed reasonable to increase their activity by converting them into iminium cations through treatment with ammonium acetate in the presence of acetic acid (Scheme 2). Initially, the Knoevenagel condensation of 1 and 2 was carried out with the available set of aldehydes in glacial acetic acid as a solvent. The reactivity of diazafluorenes 1 and 2 under these conditions turned out to be different: while diazafluorene 1 readily reacted with all aldehydes studied delivering the products with good yields, diazafluorene 2 did not react at all – only starting compounds were quantitatively evaluated from all reactions (Scheme 2, Table 1). To rationalize this observation, we hypothesized that the protonation of nitrogens leads to electrostatic repulsion between the protonated diazafluorene and the iminium cation (vide infra). Therefore, we changed the solvent in the reactions of 1,8-diazafluorene (2) to ethanol or toluene. As a result, condensation products of 2 with various donor- and acceptor-substituted benzaldehydes were obtained in moderate to good yields in both solvents (Table 1). Note, the yields were generally higher in cases when the condensation product precipitated from the reaction mixture (see note d in Table 1), i.e., when the reaction became irreversible.
![[1860-5397-22-62-i2]](/bjoc/content/inline/1860-5397-22-62-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Knoevenagel reaction of 1 and 2 under acidic conditions. Proposed mechanism of condensation and competing protonation equilibrium.
Scheme 2: Knoevenagel reaction of 1 and 2 under acidic conditions. Proposed mechanism of condensation and com...
The proposed mechanism for the condensation reaction in the presence of ammonium acetate is shown in Scheme 2 [25]. Accordingly, the addition of diazafluorene to the iminium cation occurs via the methylene carbon of the corresponding enamine tautomeric form 1B or 2B. Thus, this process is limited by the concentration of this form which is expected to be higher for 1,8-diazafluorene (2) due to the activation by the neighboring nitrogen atoms via the mesomeric effect. On the other hand, diazafluorenes are potentially capable of reversible protonation by acetic acid, which is a competing process decreasing the equilibrium concentration of the enamine form (Scheme 2). Furthermore, an electrostatic repulsion between the protonated diazafluorene (1A/1A’ or 2A/2A’) and the iminium cation can be considered, and this effect should be stronger for 1,8-diazafluorene (2) because the nitrogen atoms are located near the reaction center.
To evaluate the possible protonation effect of 1,8- and 4,5-diazafluorenes we utilized 1H NMR experiments performed immediately after preparation and 6 hours later for solutions of 1 and 2 in pure CD3COOD and in CD3OD with a drop of CD3COOD. Remarkably, 4,5-diazafluorene did not undergo protonation in both experiments – only the signals of the neutral form of 1 were identified (see Supporting Information File 1, Figure S1). Contrary, both the neutral (2) and protonated (2A’) forms of 1,8-diazafluorene were identified in both solvents (see Supporting Information File 1, Figure S2) with relatively high ratio (2 to 2A’ = 1:0.4 in CD3OD and 1:0.9 in CD3COOD). This equilibrium was reached immediately after the addition of the acid and demonstrated higher basicity of the nitrogens in 1,8-diazafluorene as compared with those of 4,5-diazafluorene. Therefore, the protonation (and corresponding repulsion impeding the condensation) effect plays the decisive role solely for compound 2.
Additionally, we evaluated the stability of the products in acidic media subjecting the condensation products 3b and 4b to heating in glacial acetic acid at 50 °C for 6 hours revealing that compound 3b was stable whereas compound 4b decomposed into the starting reagents (see the TLC data in Supporting Information File 1, Figure S3). Apparently, after the protonation and subsequent addition of a nucleophile (H2O) an intramolecular rearrangement facilitated by the proximity of a neighboring nitrogen atom (in position 1 or 8) occurred (see Scheme 3). Elimination of the aldehyde leads to the formation of 1,8-diazafluorene (2), which can be oxidized into 1,8-diazafluorenone by oxygen present in the reaction mixture. Therefore, the position of nitrogen atoms in diazafluorenes strongly affects their reactivity in acidic media via protonation, electrostatic repulsion, and stability of the condensation products. Therefore, the equilibrium in the reaction of fluorene 2 carried out in acetic acid is shifted toward the starting compounds. Hence, the Knoevenagel condensation of 1,8-diazafluorene (2) should be performed using ammonium acetate, catalytic amounts of acetic acid in a non-acidic solvent.
![[1860-5397-22-62-i3]](/bjoc/content/inline/1860-5397-22-62-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: A proposed mechanism for the 4-to-2 degradation process.
Scheme 3: A proposed mechanism for the 4-to-2 degradation process.
Going beyond the mechanistic and reactivity insights we applied this approach for the synthesis of 1,4-bis((9H-(1,8-diazafluoren)-9-ylidene)methyl)phenylene (4е, Scheme S1 in Supporting Information File 1) which resulted in 65% yield significantly surpassing the previously reported 11% using the TiCl4 system [15]. The method shown here, in contrast to condensation using TiCl4 and pyridine, is cheaper, easier to apply, and the pure target product simply precipitated from the reaction mixture instead of extraction and purification by column chromatography.
Di(pyridin-2-yl)methylene)-9H-diazafluorenes
Furthermore, since ketones are generally considered to be less reactive than aldehydes and may also suffer from steric hindrance during the reaction to further demonstrate the practical potential of Knoevenagel condensation we implemented our acidic conditions and involved 1 and 2 into reaction with di(pyrid-2-yl)ketone with subsequent evaluation of the products’ structure, physicochemical properties and coordination behavior (for 4,5-derivative).
Both 4,5- and 1,8-diazafluorenes reacted with di(pyrid-2-yl)ketone forming di(pyridin-2-yl)methylene)-9H-4,5-diazafluorene (4,5-DPDAF) and 9-(di(pyridin-2-yl)methylene)-9H-1,8-diazafluorene (1,8-DPDAF) in 54% and 34% yields, respectively (Scheme 4). Ammonium acetate and acetic acid in toluene were chosen as conditions providing good solubility of reagents. To the best of our knowledge these reactions are the first examples of a titanium-free condensation of diazafluorenes with ketones. The obtained diazafluorenylidenes were characterized by NMR, HRMS, elemental analysis, cyclic voltammetry (CV), UV–vis spectroscopy (Figure 1) and X-ray data (Figure 2 and Figure S4 in Supporting Information File 1).
![[1860-5397-22-62-i4]](/bjoc/content/inline/1860-5397-22-62-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Condensation of 4,5- (1) and 1,8-diazafluorene (2) with di(pyrid-2-yl)ketone. Reaction yields are given in parentheses.
Scheme 4: Condensation of 4,5- (1) and 1,8-diazafluorene (2) with di(pyrid-2-yl)ketone. Reaction yields are g...
Figure 1a demonstrates the cyclic voltammograms of DPDAFs in CH2Cl2 solution. Both compounds demonstrated pronounced quasi-reversible reduction waves with half-wave potentials E1/2 = −1.74 and −1.68 V (vs Fc/Fc+) for compounds 4,5-DPDAF and 1,8-DPDAF, respectively. The LUMO energies estimated using the onset reduction potentials were −3.12 eV and −3.15 eV for 4,5-DPDAF and 1,8-DPDAF, respectively, indicating a very similar electron-accepting effect of 4,5- and 1,8-diazafluorenes. Figure 1b demonstrates the optical absorption spectra of 4,5-DPDAF and 1,8-DPDAF in THF solution. The spectra have similar absorption maxima at 304 nm and 297 nm for 4,5-DPDAF and 1,8-DPDAF, respectively. However, the absorption spectrum edge demonstrated a bathochromic shift for 1,8-DPDAF (400 nm) as compared to that of 4,5-DPDAF (380 nm) what we assigned to the slightly higher planarity for 1,8-DPDAF due to its lower sterical hindrance. Based on the energies of absorption edges we estimated Eg to be 3.3 eV and 3.1 eV for 4,5-DPDAF and 1,8-DPDAF, respectively. Accordingly, the HOMO energies were −6.38 eV and −6.25 eV for 4,5-DPDAF and 1,8-DPDAF being in accordance with the high electron affinity of diazafluorenes.
![[1860-5397-22-62-1]](/bjoc/content/figures/1860-5397-22-62-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) Cyclic voltammograms of compounds 4,5-DPDAF and 1,8-DPDAF in CH2Cl2 solution and b) optical absorption spectra of 4,5-DPDAF and 1,8-DPDAF in THF.
Figure 1: a) Cyclic voltammograms of compounds 4,5-DPDAF and 1,8-DPDAF in CH2Cl2 solution and b) optical abso...
As a next step we solved the crystal structures of both compounds. Here we discuss the structure of 4,5-DPDAF and the data for compound 1,8-DPDAF is presented in Supporting Information File 1, note S1. Compound 4,5-DPDAF crystallizes as yellow needles. The conformation of the pyridine fragments is non-planar relative to the diazafluorene fragment due to sterical repulsion of the pyridine moieties and additionally due to C–H···π intramolecular interactions: φ1 = 106.8(2)° (N3–C13–C12–C5) and φ2 = 99.6(2)° (N4–C18–C12–C5) (Figure 2a). The molecules of 4,5-DPDAF are packed into stacks due to π-stacking interactions between diazafluorenes which are stabilized by C–H···N interactions between pyridine and diazafluorene fragments along (a + b) (Figure 2b). The stacks of molecules are linked together via π···π and C–H···π interactions between pyridine fragments (Figure 2c). To further evaluate the complexation behavior of 4,5-DPDAF we co-crystallized the compound with ZnCl2 resulting in a unique complex (Zn–4,5-DPDAF) where Zn is four-coordinated to the nitrogens of the diazafluorene and pyridine rings forming a positively charged cyclic dimer (Figure 2d). The dimers are arranged in a herringbone-type structure via C–H···π, C–Cl···π and π-stacking interactions between diazafluorene and pyridine (Figure 2f). Layers of dimers alternate with layers of ZnCl3(H2O)− anions containing free solvent accessible volume 284 Å3 with removed disordered water molecules (Figure 2g). Figure 2 also shows a schematic representation of the packing difference of the compounds in blue, showing that the dimers in 4,5-DPDAF molecules form stacks, while in the complex the dimers with Zn form a herringbone packing. The complex Zn–4,5-DPDAF represents a unique cyclic dimer structure and highlights a high potential of diazafluorenylidenes for coordination chemistry and materials science.
![[1860-5397-22-62-2]](/bjoc/content/figures/1860-5397-22-62-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure, atom and cycle numbering of 4,5-DPDAF (a) and Zn-4,5-DPDAF (d) with anisotropic displacement ellipsoids drawn at a 50% probability level; crystal structure fragments of 4,5-DPDAF with π-stacking (b) and C–H···π interactions (c); crystal structure fragments of Zn-4,5-DPDAF with π-stacking between dimers (e), their top view (f) and the layered packing of the compound depicting the voids (g). Dashed blue lines represent noncovalent interactions. The arrows indicate the orientation of crystallographic axes. The red line shows the (001) plane. The schematic packing motifs are shown in blue, whereas linking [ZnCl]+ moieties are illustrated by blue dots. Voids are shown as brown contact surfaces created using a probe radius of 1.2 Å.
Figure 2: Molecular structure, atom and cycle numbering of 4,5-DPDAF (a) and Zn-4,5-DPDAF (d) with anisotropi...
Conclusion
In summary, we elaborated efficient Knoevenagel condensation conditions giving 4,5- and 1,8-diazafluorenylidenes in good yields avoiding complicated synthetic or experimental workup procedures. 1,8-Diazafluorene readily condenses with aromatic aldehydes using NH4OAc and AcOH in a non-acidic solvent whereas the reaction for 4,5-diazafluorene may be carried out in pure acetic acid. According to NMR studies, 4,5-diazafluorene has a lower nitrogen basicity as compared to 1,8-diazafluorene. This basicity plays a key role in the reaction of 1,8-diazafluorene condensation because of electrostatic repulsion of the protonated tautomer and the iminium cation. Additionally, the nitrogen positions in 1,8-diazafluorenylidenes facilitate their decomposition in acidic media. Moreover, the reactivity of diazafluorenes under basic conditions also demonstrated to be improved via solvation of the ion pair by a protic solvent. Finally, the unique di(pyridin-2-yl)methylene)-9H-diazafluorenes were obtained by condensation of 4,5- and 1,8-diazafluorenes with di(pyrid-2-yl)ketone highlighting the high synthetic potential of the Knoevenagel reaction on the way toward novel electron-deficient ligands and functional materials.
Experimental
General
4,5-Diazafluorene (1) was synthesized in two steps from 1,10-phenanthroline monohydrate according to literature using a slightly modified procedure [21]. 1,8-Diazafluorene (2) was obtained using the method described in [15]. All reagents and solvents were purchased from commercial sources and used without additional purification. Reaction mixtures were monitored by TLC using Macherey-Nagel pre-coated TLC-sheets Alugram Xtra SIL G/UV254. For column chromatography Macherey-Nagel Kieselgel 60 was used. NMR spectra of products were recorded with Bruker AV 300, Bruker AV 400, and Bruker DRX 500 spectrometers in CDCl3 or (CD3)2CO. NMR samples of the solutions of 4,5- and 1,8-diazafluorenes (6 mg/0.5 mL) for the protonation study were prepared by dissolving 1 and 2 in either pure CD3COOD or CD3OD containing 0.005 mL of CD3COOD. Subsequently, 1H NMR spectra were recorded with a Bruker AV 400 immediately after sample preparation and 6 hours later. HRMS were obtained with a Thermo Scientific Double Focusing System (DFS) high-resolution mass spectrometer. Samples were introduced into the mass spectrometer by direct inlet. Electron ionization with 70 eV energy was used. Measurements of the exact masses of molecular ions were performed with respect to the standard lines of perfluorokerosene (PFK). Elemental analyses were performed for C, H, and N with a CHN-Analyzer Euro EA 300. Melting points were determined with a Thermosystem FP 900 instrument (Mettler Toledo). UV–vis spectra were recorded in diluted (10−5 M) THF in 1 × 1 cm quartz cuvettes using a Varian Cary 5000 UV-VIS-NIR spectrometer. Cyclic voltammetry measurements were performed in CH2Cl2 solution by computer-controlled P-20x potentiostat (SmartStat, Russia) in combination with a three-electrode cell (Gamry); 0.1 M tetrabutylammonium hexafluorophosphate was used as supporting electrolyte. Pt, Pt wire, and Ag/AgCl were used as working, counter, and reference electrodes, respectively. The measurements were standardized by measuring the red/ox potential of ferrocene after each compound analysis. All potentials are reported vs Fc/Fc+ red/ox couple. LUMO energies were estimated using the onset red/ox potentials according to the equation: Elumo= −(Eredonset + 4.8) (eV)
X-ray study. Neat compounds were crystallized by a slow vapor diffusion method from the CHCl3/hexane (4,5-DPDAF) and toluene/iPrOH (1,8-DPDAF) systems. The crystal growth time was about 3 days. The crystals of Zn-4,5-DPDAF (Zn–4,5-DPDAF) were grown by layering a near-saturated ZnCl2 solution in CH3OH over the CHCl3 solution of 4,5-DPDAF. The crystal growth time was about 2 weeks. Single-crystal X-ray diffraction experiments were performed at 296(2) K using a Bruker KAPPA APEX II and Tongda TD-5000 diffractometer with Mo Kα radiation (λ = 0.71073 Å), equipped with a hybrid photon-counting detector (Supporting Information File 1, Table S1). Integration and scaling of the intensity data for Bruker were accomplished with SAINT [26]. Absorption corrections were applied using SADABS [27]. The obtained diffraction data from Tongda were converted into a standard ESPERANTO format [28]. Data reduction, scaling, and absorption correction were carried out using the CrysAlis PRO program package (v. 1.171.42.49). Structure was determined and refinement was performed using SHELXT [29] and SHELXL [30] programs with Olex2 (v. 1.5) [31]. All non-hydrogen atoms were refined anisotropically. The positions of hydrogen atoms were treated using constrained refinement using a riding model [Uiso (H) = 1.2 Ueq (C)]. Structure validation was carried out using CheckCIF service [32]. The structural data of crystals were deposited as CIF files at the Cambridge Crystallographic Database (CCDC No. 2517007 (4,5-DPDAF), 2517008 (1,8-DPDAF), 2517006 (Zn-4,5-DPDAF)) and can be downloaded freely from the following site: https://www.ccdc.cam.ac.uk. Mercury [33], Olex2, and PLATON [32] were used for visualization and analysis of the crystal structure. The free solvent accessible volume in compounds Zn-4,5-DPDAF and 1,8-DPDAF derived from Olex2 “Solvent Mask” routine (PLATON/SQUEEZE procedure) in analysis was found to be 11% (284 Å3, 100 electrons) and 23% (984 Å3, 136 electrons) accordingly. This volume is occupied by highly disordered water molecules (as evidenced from residual electron density peaks) that could not be modeled as a set of discrete atomic sites.
Synthesis
General method (A1) for the condensation in acetic acid
In a manner analogous to [5]. A mixture of diazafluorene (30.0 mg, 0.18 mmol), ammonium acetate (60.5 mg, 0.79 mmol), and aldehyde (0.20 mmol) in glacial acetic acid (5 mL) was heated to 110 °C with stirring for 12 h. The resultant solution was cooled to room temperature and neutralized with aqueous ammonia solution (25%) to pH 6. The precipitate of a product was filtered off and washed sequentially with water, methanol, hexane, then dried in air.
General method (A2) for the condensation in other acidic conditions
A mixture of diazafluorene (30.0 mg, 0.18 mmol), ammonium acetate (60.5 mg, 0.79 mmol), and aldehyde (0.20 mmol), glacial acetic acid (0.02 mL, 0.36 mmol), ethanol or toluene (5 mL) was heated to 110 °C with stirring for 12 h. The resultant solution was cooled to room temperature and neutralized with aqueous ammonia solution (25%) to pH 6. The precipitated compounds (see Table 1) were isolated by filtration followed by washing sequentially with portions of water, methanol, hexane, and dried in air. In the case of absence of any precipitate, the solutions were extracted with chloroform, dried over anhydrous MgSO4 and concentrated under reduced pressure. The products were purified either by column chromatography on silica gel (eluent ethyl acetate) or by addition of diethyl ether to the residue after evaporation followed by filtration of the resulting precipitate, which was then washed with diethyl ether, and dried in air.
General method (B1) for the condensation in basic conditions
In a manner analogous to [34]. A solution of diazafluorene (30.0 mg, 0.18 mmol) in DMF (5 mL) was purged with argon for 20 minutes. Then, t-BuONa (20.6 mg, 0.21 mmol) was added, and the flask was sealed with a septum. The mixture was stirred vigorously for 10 min, and then a solution of an aldehyde (0.20 mmol) in anhydrous DMF (2 mL) was added dropwise. The reaction mixture was stirred at room temperature for 2 h and then poured into water and extracted with chloroform. The extract was dried over MgSO4 and concentrated under reduced pressure. The products were purified by column chromatography on silica gel using ethyl acetate as eluent.
General method (B2) for the condensation in basic conditions
A solution of diazafluorene (30.0 mg, 0.18 mmol) in t-BuOH (5 mL) was purged with argon for 20 minutes. Then, t-BuONa (20.6 mg, 0.21 mmol) was added, and the flask was sealed with a septum. The mixture was stirred vigorously for 10 min, and then a solution of an aldehyde (0.20 mmol) in t-BuOH (2 mL) was added dropwise. The reaction mixture was heated to 30 °C and stirred at this temperature for 12 h, then poured into water and extracted with chloroform. The extract was dried over MgSO4 and concentrated under reduced pressure. The products were purified by column chromatography on silica gel using ethyl acetate as eluent.
The data on the yield, melting point, elemental analysis and spectral characteristics of each compound are provided in Supporting Information File 1 (section 1 and Figures S5–S34).
Synthesis of 1,4-bis((9H-(1,8-diazafluoren)-9-ylidene)methyl)phenylene (4e)
A mixture of 1,8-diazafluorene (30.0 mg, 0.18 mmol), terephthalaldehyde (10.8 mg, 0.08 mmol), ammonium acetate (55.4 mg, 0.72 mmol) and glacial acetic acid (0.02 mL, 0.36 mmol) in toluene (5 mL) was heated to 110 °C with stirring for 12 h. The resultant solution was cooled to room temperature and neutralized with aqueous ammonia solution (25%) to pH 6. The resulting precipitate was filtered, washed successively with water, methanol, hexane and dried in air. Yield: 22.7 mg, 65%. The NMR spectrum corresponds to that described in [15].
Supporting Information
| Supporting Information File 1: Analytical data, protonation data, thin-layer chromatography data, X-ray data, spectra of synthesized compounds. | ||
| Format: PDF | Size: 2.8 MB | Download |
| Supporting Information File 2: Crystallographic information files for compounds 1,8-DPDAF, 4,5-DPDAF and Zn–4,5-DPDAF. | ||
| Format: ZIP | Size: 546.0 KB | Download |
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Lattmann, E.; Begum, A.; Plater, M. J. Drug Des. Discovery 1999, 16, 195–201.
Return to citation in text: [1] -
Yin, Q.; Awadasseid, A.; Zhou, Y.; Wang, M.; Xiong, X.; Wu, Y.; Zhang, W. Med. Chem. Res. 2022, 31, 1476–1487. doi:10.1007/s00044-022-02928-5
Return to citation in text: [1] -
Zhou, K.; Liu, J.; Xiong, X.; Cheng, M.; Hu, X.; Narva, S.; Zhao, X.; Wu, Y.; Zhang, W. Eur. J. Med. Chem. 2019, 178, 484–499. doi:10.1016/j.ejmech.2019.06.012
Return to citation in text: [1] [2] -
Sako, K.; Kakehi, T.; Nakano, S.; Oku, H.; Shen, X. F.; Iwanaga, T.; Yoshikawa, M.; Sugahara, K.; Toyota, S.; Takemura, H.; Shinmyozu, T.; Shiotsuka, M.; Tatemitsu, H. Tetrahedron Lett. 2014, 55, 749–752. doi:10.1016/j.tetlet.2013.12.013
Return to citation in text: [1] [2] -
Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Kazantsev, M. S. Dyes Pigm. 2024, 229, 112261. doi:10.1016/j.dyepig.2024.112261
Return to citation in text: [1] [2] [3] [4] [5] -
Katoh, K.; Sasamori, T.; Tokitoh, N.; Sato, N. Chem. Lett. 2007, 36, 1122–1123. doi:10.1246/cl.2007.1122
Return to citation in text: [1] [2] -
Katoh, K.; Sato, N. Chem. Lett. 2008, 37, 618–619. doi:10.1246/cl.2008.618
Return to citation in text: [1] [2] -
Riklin, M.; von Zelewsky, A.; Bashall, A.; McPartlin, M.; Baysal, A.; Connor, J. A.; Wallis, J. D. Helv. Chim. Acta 1999, 82, 1666–1680. doi:10.1002/(sici)1522-2675(19991006)82:10<1666::aid-hlca1666>3.0.co;2-k
Return to citation in text: [1] -
Fomenko, I. S.; Afewerki, M.; Gongola, M. I.; Vasilyev, E. S.; Shul’pina, L. S.; Ikonnikov, N. S.; Shul’pin, G. B.; Samsonenko, D. G.; Yanshole, V. V.; Nadolinny, V. A.; Lavrov, A. N.; Tkachev, A. V.; Gushchin, A. L. Molecules 2022, 27, 4072. doi:10.3390/molecules27134072
Return to citation in text: [1] -
Baran, M. F.; Durap, F.; Aydemir, M.; Baysal, A. Appl. Organomet. Chem. 2016, 30, 1030–1035. doi:10.1002/aoc.3538
Return to citation in text: [1] -
Wezenberg, S. J.; Chen, K.-Y.; Feringa, B. L. Angew. Chem., Int. Ed. 2015, 54, 11457–11461. doi:10.1002/anie.201505781
Return to citation in text: [1] -
Faulkner, A.; van Leeuwen, T.; Feringa, B. L.; Wezenberg, S. J. J. Am. Chem. Soc. 2016, 138, 13597–13603. doi:10.1021/jacs.6b06467
Return to citation in text: [1] -
Colombo, A.; Dragonetti, C.; Righetto, S.; Roberto, D.; Valore, A.; Benincori, T.; Colombo, F.; Sannicolò, F. J. Mater. Chem. 2012, 22, 19761–19766. doi:10.1039/c2jm33509c
Return to citation in text: [1] [2] -
Sako, K.; Mugishima, Y.; Iwanaga, T.; Toyota, S.; Takemura, H.; Watanabe, M.; Shinmyozu, T.; Shiotsuka, M.; Tatemitsu, H. Tetrahedron Lett. 2011, 52, 5865–5868. doi:10.1016/j.tetlet.2011.08.164
Return to citation in text: [1] [2] -
Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Trul, A. A.; Koskin, I. P.; Trukhanov, V. A.; Shundrina, I. K.; Paraschuk, D. Y.; Kazantsev, M. S. ACS Appl. Mater. Interfaces 2025, 17, 52403–52415. doi:10.1021/acsami.5c11469
Return to citation in text: [1] -
Levy, A.; Cohen, S.; Agranat, I. Org. Biomol. Chem. 2003, 1, 2755–2763. doi:10.1039/b303041e
Return to citation in text: [1] -
Querol, M.; Stoekli-Evans, H.; Belser, P. Org. Lett. 2002, 4, 1067–1070. doi:10.1021/ol017130c
Return to citation in text: [1] -
Zhao, J.-F.; Chen, L.; Sun, P.-J.; Hou, X.-Y.; Zhao, X.-H.; Li, W.-J.; Xie, L.-H.; Qian, Y.; Shi, N.-E.; Lai, W.-Y.; Fan, Q.-L.; Huang, W. Tetrahedron 2011, 67, 1977–1982. doi:10.1016/j.tet.2010.12.065
Return to citation in text: [1] -
Wang, D.; Lee, M. M. S.; Xu, W.; Shan, G.; Zheng, X.; Kwok, R. T. K.; Lam, J. W. Y.; Hu, X.; Tang, B. Z. Angew. Chem., Int. Ed. 2019, 58, 5628–5632. doi:10.1002/anie.201900366
Return to citation in text: [1] -
Plater, M. J.; Kemp, S.; Lattmann, E. J. Chem. Soc., Perkin Trans. 1 2000, 971–979. doi:10.1039/a909109b
Return to citation in text: [1] [2] -
Vasilyev, E. S.; Bizyaev, S. N.; Komarov, V. Y.; Tkachev, A. V. Mendeleev Commun. 2019, 29, 584–586. doi:10.1016/j.mencom.2019.09.036
Return to citation in text: [1] [2] -
Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Kazantsev, M. S. Chem. Sustainable Dev. 2024, 32, 548–555. doi:10.15372/csd2024589
Return to citation in text: [1] -
Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527632220
Return to citation in text: [1] -
Kanakaraju, S.; Prasanna, B.; Basavoju, S.; Chandramouli, G. V. P. Arabian J. Chem. 2017, 10, S2705–S2713. doi:10.1016/j.arabjc.2013.10.014
Return to citation in text: [1] -
SAINT Data Reduction and Frame Integration Program for the CCD Area-Detector System; Bruker Analytical X-ray Systems: Madison, Wisconsin, USA, 2006.
Return to citation in text: [1] -
SADABS; Program for area detector adsorption correction; Sheldrick, G. M.; Institute for Inorganic Chemistry, University of Goettingen: Goettingen, Germany, 1996.
Return to citation in text: [1] -
Rothkirch, A.; Gatta, G. D.; Meyer, M.; Merkel, S.; Merlini, M.; Liermann, H.-P. J. Synchrotron Radiat. 2013, 20, 711–720. doi:10.1107/s0909049513018621
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x
Return to citation in text: [1] [2] -
Macrae, C. F.; Sovago, I.; Cottrell, S. J.; Galek, P. T. A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G. P.; Stevens, J. S.; Towler, M.; Wood, P. A. J. Appl. Crystallogr. 2020, 53, 226–235. doi:10.1107/s1600576719014092
Return to citation in text: [1] -
Sonina, A. A.; Becker, C. S.; Kuimov, A. D.; Shundrina, I. K.; Komarov, V. Y.; Kazantsev, M. S. CrystEngComm 2021, 23, 2654–2664. doi:10.1039/d0ce01794a
Return to citation in text: [1]
| 31. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 32. | Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x |
| 33. | Macrae, C. F.; Sovago, I.; Cottrell, S. J.; Galek, P. T. A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G. P.; Stevens, J. S.; Towler, M.; Wood, P. A. J. Appl. Crystallogr. 2020, 53, 226–235. doi:10.1107/s1600576719014092 |
| 1. | Lattmann, E.; Begum, A.; Plater, M. J. Drug Des. Discovery 1999, 16, 195–201. |
| 2. | Yin, Q.; Awadasseid, A.; Zhou, Y.; Wang, M.; Xiong, X.; Wu, Y.; Zhang, W. Med. Chem. Res. 2022, 31, 1476–1487. doi:10.1007/s00044-022-02928-5 |
| 3. | Zhou, K.; Liu, J.; Xiong, X.; Cheng, M.; Hu, X.; Narva, S.; Zhao, X.; Wu, Y.; Zhang, W. Eur. J. Med. Chem. 2019, 178, 484–499. doi:10.1016/j.ejmech.2019.06.012 |
| 11. | Wezenberg, S. J.; Chen, K.-Y.; Feringa, B. L. Angew. Chem., Int. Ed. 2015, 54, 11457–11461. doi:10.1002/anie.201505781 |
| 12. | Faulkner, A.; van Leeuwen, T.; Feringa, B. L.; Wezenberg, S. J. J. Am. Chem. Soc. 2016, 138, 13597–13603. doi:10.1021/jacs.6b06467 |
| 5. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Kazantsev, M. S. Dyes Pigm. 2024, 229, 112261. doi:10.1016/j.dyepig.2024.112261 |
| 23. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Kazantsev, M. S. Chem. Sustainable Dev. 2024, 32, 548–555. doi:10.15372/csd2024589 |
| 9. | Fomenko, I. S.; Afewerki, M.; Gongola, M. I.; Vasilyev, E. S.; Shul’pina, L. S.; Ikonnikov, N. S.; Shul’pin, G. B.; Samsonenko, D. G.; Yanshole, V. V.; Nadolinny, V. A.; Lavrov, A. N.; Tkachev, A. V.; Gushchin, A. L. Molecules 2022, 27, 4072. doi:10.3390/molecules27134072 |
| 10. | Baran, M. F.; Durap, F.; Aydemir, M.; Baysal, A. Appl. Organomet. Chem. 2016, 30, 1030–1035. doi:10.1002/aoc.3538 |
| 15. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297 |
| 6. | Katoh, K.; Sasamori, T.; Tokitoh, N.; Sato, N. Chem. Lett. 2007, 36, 1122–1123. doi:10.1246/cl.2007.1122 |
| 7. | Katoh, K.; Sato, N. Chem. Lett. 2008, 37, 618–619. doi:10.1246/cl.2008.618 |
| 8. | Riklin, M.; von Zelewsky, A.; Bashall, A.; McPartlin, M.; Baysal, A.; Connor, J. A.; Wallis, J. D. Helv. Chim. Acta 1999, 82, 1666–1680. doi:10.1002/(sici)1522-2675(19991006)82:10<1666::aid-hlca1666>3.0.co;2-k |
| 3. | Zhou, K.; Liu, J.; Xiong, X.; Cheng, M.; Hu, X.; Narva, S.; Zhao, X.; Wu, Y.; Zhang, W. Eur. J. Med. Chem. 2019, 178, 484–499. doi:10.1016/j.ejmech.2019.06.012 |
| 5. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Kazantsev, M. S. Dyes Pigm. 2024, 229, 112261. doi:10.1016/j.dyepig.2024.112261 |
| 4. | Sako, K.; Kakehi, T.; Nakano, S.; Oku, H.; Shen, X. F.; Iwanaga, T.; Yoshikawa, M.; Sugahara, K.; Toyota, S.; Takemura, H.; Shinmyozu, T.; Shiotsuka, M.; Tatemitsu, H. Tetrahedron Lett. 2014, 55, 749–752. doi:10.1016/j.tetlet.2013.12.013 |
| 5. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Kazantsev, M. S. Dyes Pigm. 2024, 229, 112261. doi:10.1016/j.dyepig.2024.112261 |
| 15. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297 |
| 22. | Vasilyev, E. S.; Bizyaev, S. N.; Komarov, V. Y.; Tkachev, A. V. Mendeleev Commun. 2019, 29, 584–586. doi:10.1016/j.mencom.2019.09.036 |
| 4. | Sako, K.; Kakehi, T.; Nakano, S.; Oku, H.; Shen, X. F.; Iwanaga, T.; Yoshikawa, M.; Sugahara, K.; Toyota, S.; Takemura, H.; Shinmyozu, T.; Shiotsuka, M.; Tatemitsu, H. Tetrahedron Lett. 2014, 55, 749–752. doi:10.1016/j.tetlet.2013.12.013 |
| 6. | Katoh, K.; Sasamori, T.; Tokitoh, N.; Sato, N. Chem. Lett. 2007, 36, 1122–1123. doi:10.1246/cl.2007.1122 |
| 7. | Katoh, K.; Sato, N. Chem. Lett. 2008, 37, 618–619. doi:10.1246/cl.2008.618 |
| 14. | Sako, K.; Mugishima, Y.; Iwanaga, T.; Toyota, S.; Takemura, H.; Watanabe, M.; Shinmyozu, T.; Shiotsuka, M.; Tatemitsu, H. Tetrahedron Lett. 2011, 52, 5865–5868. doi:10.1016/j.tetlet.2011.08.164 |
| 19. | Zhao, J.-F.; Chen, L.; Sun, P.-J.; Hou, X.-Y.; Zhao, X.-H.; Li, W.-J.; Xie, L.-H.; Qian, Y.; Shi, N.-E.; Lai, W.-Y.; Fan, Q.-L.; Huang, W. Tetrahedron 2011, 67, 1977–1982. doi:10.1016/j.tet.2010.12.065 |
| 20. | Wang, D.; Lee, M. M. S.; Xu, W.; Shan, G.; Zheng, X.; Kwok, R. T. K.; Lam, J. W. Y.; Hu, X.; Tang, B. Z. Angew. Chem., Int. Ed. 2019, 58, 5628–5632. doi:10.1002/anie.201900366 |
| 34. | Sonina, A. A.; Becker, C. S.; Kuimov, A. D.; Shundrina, I. K.; Komarov, V. Y.; Kazantsev, M. S. CrystEngComm 2021, 23, 2654–2664. doi:10.1039/d0ce01794a |
| 5. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Kazantsev, M. S. Dyes Pigm. 2024, 229, 112261. doi:10.1016/j.dyepig.2024.112261 |
| 15. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297 |
| 16. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Trul, A. A.; Koskin, I. P.; Trukhanov, V. A.; Shundrina, I. K.; Paraschuk, D. Y.; Kazantsev, M. S. ACS Appl. Mater. Interfaces 2025, 17, 52403–52415. doi:10.1021/acsami.5c11469 |
| 13. | Colombo, A.; Dragonetti, C.; Righetto, S.; Roberto, D.; Valore, A.; Benincori, T.; Colombo, F.; Sannicolò, F. J. Mater. Chem. 2012, 22, 19761–19766. doi:10.1039/c2jm33509c |
| 21. | Plater, M. J.; Kemp, S.; Lattmann, E. J. Chem. Soc., Perkin Trans. 1 2000, 971–979. doi:10.1039/a909109b |
| 15. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297 |
| 14. | Sako, K.; Mugishima, Y.; Iwanaga, T.; Toyota, S.; Takemura, H.; Watanabe, M.; Shinmyozu, T.; Shiotsuka, M.; Tatemitsu, H. Tetrahedron Lett. 2011, 52, 5865–5868. doi:10.1016/j.tetlet.2011.08.164 |
| 32. | Spek, A. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 148–155. doi:10.1107/s090744490804362x |
| 13. | Colombo, A.; Dragonetti, C.; Righetto, S.; Roberto, D.; Valore, A.; Benincori, T.; Colombo, F.; Sannicolò, F. J. Mater. Chem. 2012, 22, 19761–19766. doi:10.1039/c2jm33509c |
| 17. | Levy, A.; Cohen, S.; Agranat, I. Org. Biomol. Chem. 2003, 1, 2755–2763. doi:10.1039/b303041e |
| 18. | Querol, M.; Stoekli-Evans, H.; Belser, P. Org. Lett. 2002, 4, 1067–1070. doi:10.1021/ol017130c |
| 5. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Kazantsev, M. S. Dyes Pigm. 2024, 229, 112261. doi:10.1016/j.dyepig.2024.112261 |
| 25. | Kanakaraju, S.; Prasanna, B.; Basavoju, S.; Chandramouli, G. V. P. Arabian J. Chem. 2017, 10, S2705–S2713. doi:10.1016/j.arabjc.2013.10.014 |
| 22. | Vasilyev, E. S.; Bizyaev, S. N.; Komarov, V. Y.; Tkachev, A. V. Mendeleev Commun. 2019, 29, 584–586. doi:10.1016/j.mencom.2019.09.036 |
| 24. | Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527632220 |
| 29. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 30. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 27. | SADABS; Program for area detector adsorption correction; Sheldrick, G. M.; Institute for Inorganic Chemistry, University of Goettingen: Goettingen, Germany, 1996. |
| 28. | Rothkirch, A.; Gatta, G. D.; Meyer, M.; Merkel, S.; Merlini, M.; Liermann, H.-P. J. Synchrotron Radiat. 2013, 20, 711–720. doi:10.1107/s0909049513018621 |
| 15. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297 |
| 26. | SAINT Data Reduction and Frame Integration Program for the CCD Area-Detector System; Bruker Analytical X-ray Systems: Madison, Wisconsin, USA, 2006. |
| 15. | Cheshkina, D. S.; Becker, C. S.; Sonina, A. A.; Koskin, I. P.; Shundrina, I. K.; Mostovich, E. A.; Kazantsev, M. S. J. Phys. Chem. C 2024, 128, 15070–15081. doi:10.1021/acs.jpcc.4c04297 |
| 21. | Plater, M. J.; Kemp, S.; Lattmann, E. J. Chem. Soc., Perkin Trans. 1 2000, 971–979. doi:10.1039/a909109b |
© 2026 Cheshkina et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.