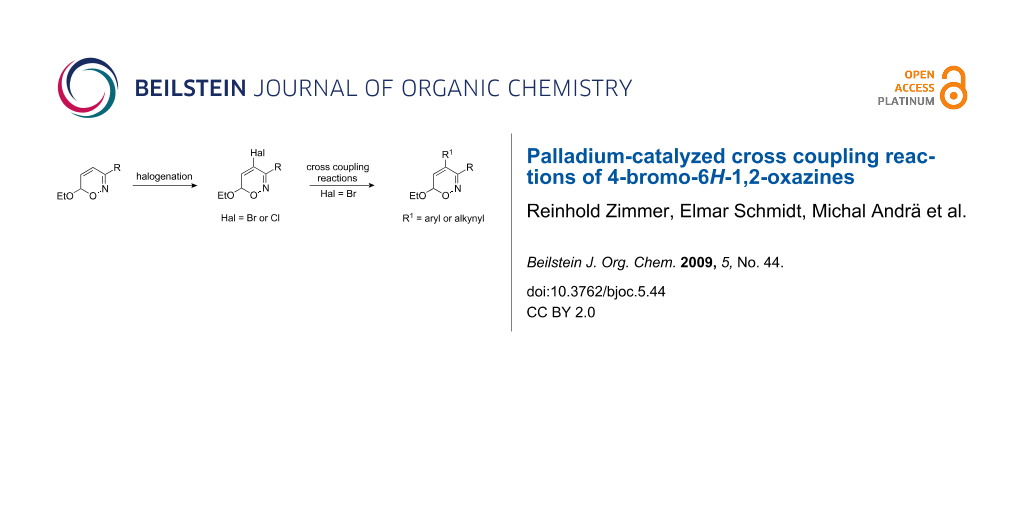
Abstract
A number of 4-aryl- and 4-alkynyl-substituted 6H-1,2-oxazines 8 and 9 have been prepared in good yields via cross coupling reactions of halogenated precursors 2, which in turn are easily accessible by bromination of 6H-1,2-oxazines 1. Lewis-acid promoted reaction of 1,2-oxazine 9c with 1-hexyne provided alkynyl-substituted pyridine derivative 12 thus demonstrating the potential of this approach for the synthesis of pyridines.

Graphical Abstract
Introduction
A broad range of synthetic applications demonstrates that 1,2-oxazine derivatives constitute a versatile class of N,O heterocycles [1-13]. Considerable attention has been paid to 6H-1,2-oxazines 1 bearing a C-4,C-5-double bond [14-18], which are useful intermediates in the synthesis of γ-lactams [19], γ-amino acids [20], amino alcohols [20], aziridines [21], pyrrolizidines [22], and pyrrolidine derivatives [15,23,24]. In the context of our ongoing exploration of the synthetic potential of these heterocycles we were interested to modify the substitution pattern of the C-4,C-5 double bond of 6H-1,2-oxazines [25-27]. Herein, we describe our results dealing with the halogenation of 6H-1,2-oxazines 1 and the use of the resulting products as precursors in palladium-catalyzed cross coupling reactions.
Results and Discussion
Not much is known about halogenated 6H-1,2-oxazines and only a few mostly inefficient procedures are described [28-32]. This prompted us to investigate a more practical access to halogenated 6H-1,2-oxazines. Gratifyingly, the desired 4-bromo-substituted 6H-1,2-oxazines 2a–2c could be prepared in a one-pot procedure by bromine addition to precursors 1a–1c [14] and HBr elimination by treatment with triethylamine (Scheme 1). The 4-bromo-6H-1,2-oxazines were obtained in reasonable to good yields. The bromination of 3-phenyl-substituted 6H-1,2-oxazine 1a often resulted in a mixture of several brominated products which are easily separable by chromatography. Depending on the reaction scale and the amount of bromine used (1.5 to 3 equiv) by-products such as 3a, 4 and 5 could be isolated in varying yields. The unexpected formation of 4,5-dibromo-6H-1,2-oxazine 3a can obviously be rationalized by addition of bromine to 2a and elimination of HBr during the bromination reaction of 1a.
![[1860-5397-5-44-i1]](/bjoc/content/inline/1860-5397-5-44-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Brominations of 6H-1,2-oxazines. a) Br2, Et2O, −30 °C, 2 h. b) Et3N, −30 °C to r.t., overnight.
Scheme 1: Brominations of 6H-1,2-oxazines. a) Br2, Et2O, −30 °C, 2 h. b) Et3N, −30 °C to r.t., overnight.
The literature describes just one related 4-chloro-substituted 6H-1,2-oxazine which was prepared by a hetero-Diels–Alder cycloaddition–elimination sequence of 2-chloro-1-nitroso-1-phenyl-ethene and 1-bromo-2-ethoxyethene in low yield (22%) [32]. As demonstrated in Scheme 2, a more efficient approach consists in chlorination of 6H-1,2-oxazines 1a,b by addition of chlorine and subsequent base-induced dehydrochlorination. The expected 4-chloro-6H-1,2-oxazines 6a,b were obtained in good yields. In analogy to the aforementioned bromination, the chlorination of 3-phenyl-6H-1,2-oxazine 1a also led to dihalogenation furnishing 4,5-dichloro-substituted compound 7a as a by-product in 13% yield.
![[1860-5397-5-44-i2]](/bjoc/content/inline/1860-5397-5-44-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Chlorinations of 6H-1,2-oxazines. a) Cl2, Et2O, −30 °C. b) Et3N, −30 °C to r.t.
Scheme 2: Chlorinations of 6H-1,2-oxazines. a) Cl2, Et2O, −30 °C. b) Et3N, −30 °C to r.t.
With the 4-halogenated 6H-1,2-oxazines 2 and 6 in hand, palladium-catalyzed cross couplings offer an efficient and useful approach for the synthesis of novel functionalized 6H-1,2-oxazines. The Suzuki-coupling of the 4-bromo-substituted heterocycles 2a,b with phenylboronic acid in the presence of Pd(PPh3)4 and sodium carbonate at 80 °C in toluene gave the expected 4-phenyl-substituted 6H-1,2-oxazines 8a or 8b in 82 and 77% yield (Scheme 3).
![[1860-5397-5-44-i3]](/bjoc/content/inline/1860-5397-5-44-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Suzuki-couplings of 4-bromo-6H-1,2-oxazines. a) ArB(OH)2, Pd(PPh3)4, Na2CO3, toluene, 80 °C, 3 h.
Scheme 3: Suzuki-couplings of 4-bromo-6H-1,2-oxazines. a) ArB(OH)2, Pd(PPh3)4, Na2CO3, toluene, 80 °C, 3 h.
4-Bromo-6H-1,2-oxazine 2a also serves as suitable model substrate for Sonogashira-reactions (Scheme 4). When the coupling reaction of 2a with various terminal alkynes, such as phenylacetylene, trimethylsilylethyne and 1-hexyne, was performed under typical conditions [PdCl2(PPh3)2, CuI, Et3N, toluene], the expected 4-alkynyl-substituted heterocycles 9a–9c were isolated in good yields. In contrast, when the same reaction conditions were applied to the coupling of 2a and methyl propargyl ether, product 9d was obtained only in very low yield. In addition, Sonogashira coupling of 2a and methyl propargyl ether performed by an alternative protocol (Pd(OAc)2, CuI, PPh3, NHiPr2 in DMF) afforded the expected product 9d and a byproduct bearing a 4-enyne moiety at 4-position. This indicates an addition of a second alkyne molecule to the primary product 9. Similar results were observed for the Sonogashira reaction of 2a with propargylic alcohol [33].
![[1860-5397-5-44-i4]](/bjoc/content/inline/1860-5397-5-44-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Sonogashira-couplings of 4-bromo-6H-1,2-oxazines. a) PdCl2(PPh3)2, CuI, Et3N, toluene, r.t., 6–20 h.
Scheme 4: Sonogashira-couplings of 4-bromo-6H-1,2-oxazines. a) PdCl2(PPh3)2, CuI, Et3N, toluene, r.t., 6–20 h....
After successful simple cross couplings of mono-halogenated 2, the 4,5-dibromo-3-phenyl-6H-1,2-oxazine 3a seemed to be an attractive candidate for a twofold Sonogashira reaction (Scheme 5). Treatment of 3a with an excess of phenylacetylene under conditions as described in Scheme 4 provided 5-bromo-4-alkynyl-substituted 6H-1,2-oxazine 10a as single product in 65% yield. When the Sonogashira coupling was performed with trimethylsilylethyne under the same reaction conditions an inseparable 85:15-mixture of mono-alkynylated product 10b and bis-alkynylated compound 11b was obtained in reasonable yield. These reactions certainly deserve further optimization, however, they already show the potential of compounds such as 3a to serve as precursors for two subsequent coupling reactions.
![[1860-5397-5-44-i5]](/bjoc/content/inline/1860-5397-5-44-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Sonogashira-couplings of 4,5-dibromo-6H-1,2-oxazines. a) PdCl2(PPh3)2, CuI, Et3N, toluene, r.t., 4 h to overnight.
Scheme 5: Sonogashira-couplings of 4,5-dibromo-6H-1,2-oxazines. a) PdCl2(PPh3)2, CuI, Et3N, toluene, r.t., 4 ...
Conclusion and Perspective
In conclusion, we have successfully demonstrated that a series of 4-aryl- and 4-alkynyl-substituted 6H-1,2-oxazines 8, 9, and 10 are easily accessible in short reaction sequences starting from precursors 1. These 6H-1,2-oxazines should allow access to many interesting five- and six-membered heterocycles. As illustrated in Scheme 6, the 4-hex-1-ynyl-3-phenyl-6H-1,2-oxazine 9c can be converted into the trisubstituted pyridine derivative 12 by treatment of 9c with boron trifluoride etherate in the presence of an excess of 1-hexyne via an azapyrylium intermediate [34,35]. Additional investigations are required to optimize the preparation diynes of type 11. Conversion of the new functionalized 6H-1,2-oxazines to highly substituted pyridine derivatives will also be reported in due course.
![[1860-5397-5-44-i6]](/bjoc/content/inline/1860-5397-5-44-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Preparation of trisubstituted pyridine derivatives: a) BF3·OEt2, CH2Cl2, −78 °C to r.t., overnight.
Scheme 6: Preparation of trisubstituted pyridine derivatives: a) BF3·OEt2, CH2Cl2, −78 °C to r.t., overnight.
Experimental
Bromination of 6H-1,2-oxazine 1a, typical procedure
6H-1,2-Oxazine 1a (5.35 g, 26.3 mmol) was dissolved in diethyl ether (200 mL) and treated with bromine (2.75 mL, 53.7 mmol) at −30 °C under argon atmosphere. After 2 h Et3N (54.0 mL, 390 mmol) was added. The reaction mixture was warmed to r.t. overnight and quenched with water (100 mL). The aqueous phase was extracted with CH2Cl2 (2 × 50 mL) and the combined organic phases were dried with Na2SO4. Purification of the crude product by column chromatography (SiO2, hexane:EtOAc 8:1, then 4:1) gave the 4-bromo-substituted 6H-1,2-oxazine 2a (5.11 g, 69%), the 4,5-dibromo-substituted by-product 3a (0.821 g, 9%), and starting material 1a (0.335 g, 6%).
4-Bromo-6-ethoxy-3-phenyl-6H-1,2-oxazine (2a): yellow–brown oil. 1H NMR (CDCl3, 300 MHz): δ = 1.23 (t, J = 7.1 Hz, 3 H, CH3), AB part of ABX3 system (δA = 3.68, δB = 3.95, JAX = JBX = 7.1 Hz, JAB = 9.8 Hz, 2 H, OCH2), 5.58 (d, J = 5.2 Hz, 1 H, 6-H), 6.70 (d, J = 5.2 Hz, 1 H, 5-H), 7.35–7.50, 7.50–7.60 (2 m, 3 H, 2 H, Ph) ppm. 13C NMR (CDCl3, 75.5 MHz): δ = 14.8 (q, CH3), 64.3 (t, OCH2), 94.7 (d, C-6), 112.9 (s, C-4), 127.8, 128.0, 128.8, 129.7, 133.1 (4 d, s, Ph, C-5), 156.2 (s, C-3) ppm. For the complete characterization, see ref. [31].
4,5-Dibromo-6-ethoxy-3-phenyl-6H-1,2-oxazine (3a): brown oil. 1H NMR (CDCl3, 300 MHz): δ = 1.25 (t, J = 7.1 Hz, 3 H, CH3), AB part of ABX3 system (δA = 3.76, δB = 3.97, JAX = JBX = 7.1 Hz, JAB = 9.7 Hz, 2 H, OCH2), 5.71 (s, 1 H, 6-H), 7.38–7.48, 7.49–7.56 (2 m, 3 H, 2 H, Ph) ppm. 13C NMR (CDCl3, 75.5 MHz): δ = 14.7 (q, CH3), 65.0 (t, OCH2), 99.8 (d, C-6), 114.4, 124.8 (2 s, C-4, C-5), 128.1, 128.9, 129.9, 133.3 (3 d, s, Ph), 155.9 (s, C-3) ppm. IR (neat): 3065–2900 (=C–H, C–H), 1630 (C=N), 1600 (C=C) cm−1. HRMS (80 eV, 40 °C) m/z calcd for C12H1179Br2NO2: 358.9157; found: 358.9160.
Chlorination of 6H-1,2-oxazine 1b, typical procedure
Chlorine gas was passed into diethyl ether (28 mL) at −30 °C until the solution became dark yellow. Then, 6H-1,2-oxazine 1b (0.200 g, 1.00 mmol) was added and the reaction mixture was monitored by TLC; upon complete consumption, triethylamine (2.00 mL, 27.8 mmol) was added at −30 °C and the mixture was slowly warmed to r.t. After addition of brine, the phases were separated, the aqueous phase was extracted with CH2Cl2 (2 × 20 mL) and the combined organic phases were dried with Na2SO4. Column chromatography (SiO2, hexane, hexane:EtOAc 9:1, then 4:1) afforded the 4-chloro-substituted product 6b (0.182 g, 78%) as pale–yellow oil.
Ethyl 4-chloro-6-ethoxy-6H-1,2-oxazine-3-carboxylate (6b): 1H NMR (CDCl3, 300 MHz): δ = 1.22 (t, J = 7.1 Hz, 3 H, CH3), 1.40 (t, J = 7.2 Hz, 3 H, CH3), AB part of ABX3 system (δA = 3.68, δB = 3.95, JAX = JBX = 7.1 Hz, JAB = 9.6 Hz, 2 H, OCH2), 4.40 (q, J = 7.2 Hz, 2 H, OCH2), 5.72 (d, J = 5.0 Hz, 1 H, 6-H), 6.34 (d, J = 5.0 Hz, 1 H, 5-H) ppm. 13C NMR (CDCl3, 75.5 MHz): δ = 13.9, 14.7 (2 q, CH3), 62.5, 64.6 (2 t, OCH2), 95.3 (d, C-6), 121.1 (s, C-4), 122.7 (d, C-5), 148.5 (s, C-3), 160.3 (s, C=O) ppm. IR (neat): 3105–2975 (=C–H, C–H), 1745 (C=O), 1615 (C=N) cm−1. C9H12ClNO4 (233.7): calcd. C, 46.27; H, 5.18; N, 5.99; found: C, 46.35; H, 5.16; N, 6.08.
Suzuki-coupling of 4-bromo-substituted 6H-1,2-oxazine 2a, typical procedure
6H-1,2-Oxazine 2a (0.0935 g, 0.33 mmol), phenylboronic acid (0.122 g, 1.00 mmol) and Pd(PPh3)4 (0.016 g, 0.0138 mmol) were dissolved in a mixture of toluene/MeOH (3 mL/0.75 mL) in a heat-gun-dried and argon-flushed flask. A 2M Na2CO3 solution (1.5 mL) was finally added and the reaction mixture was heated for 15 h at 80 °C. Then, the reaction mixture was cooled to r.t. and washed with 2M Na2CO3 (with 1% NH3) solution. After separation of the phases, the aqueous phase was extracted with CH2Cl2 (3 × 5 mL) and the combined organic phases were dried with Na2SO4. The crude product was purified by column chromatography (SiO2, hexane:EtOAc 9:1, then 4:1) to afford the Suzuki product 8a (0.076 g, 82%) as a pale–yellow solid, mp 68–70 °C.
6-Ethoxy-3,4-diphenyl-6H-1,2-oxazine (8a): 1H NMR (CDCl3, 300 MHz): δ = 1.25 (t, J = 7.1 Hz, 3 H, CH3), AB part of ABX3 system (δA = 3.75, δB = 4.01, JAX = JBX = 7.1 Hz, JAB = 9.8 Hz, 2 H, OCH2), 5.73 (d, J = 4.9 Hz, 1 H, 6-H), 6.37 (d, J = 4.9 Hz, 1 H, 5-H), 7.05–7.10, 7.15–7.27, 7.30–7.35 (3 m, 4 H, 4 H, 2 H, Ph) ppm. 13C NMR (CDCl3, 75.5 MHz): δ = 15.0 (q, CH3), 64.2 (t, OCH2), 93.0 (d, C-6), 124.0 (d, C-5), 128.0, 128.2, 128.4, 128.7, 129.0, 130.2, 133.9, 136.5 (5 d, 3 s, Ph, C-4), 157.7 (s, C-3) ppm. IR (KBr): 3040–2930 (=C–H, C–H), 1620 (C=N), 1600 (C=C) cm−1. C18H17NO2 (279.3): calcd. C, 77.39; H, 6.13; N, 5.01; found: C, 77.82; H, 6.37; N, 5.08.
Sonogashira-coupling of 4-bromo-substituted 6H-1,2-oxazine 2a, typical procedure
6H-1,2-Oxazine 2a (0.850 g, 3.19 mmol), trimethylsilylethyne (0.87 mL, 6.17 mmol), PdCl2(PPh3)2 (0.114 g, 0.16 mmol), CuI (0.019 g, 0.10 mmol) and Et3N (1.3 mL) were dissolved in toluene (15 mL) in a heat-gun-dried and argon-flushed flask and the reaction mixture was stirred at r.t. for 20 h. The reaction mixture was quenched with water (5 mL). The aqueous phase was extracted with CH2Cl2 (3 × 10 mL) and the combined organic phases were dried with Na2SO4. Purification of the crude product by column chromatography (SiO2, hexane: EtOAc 20:1, then 4:1) afforded the 4-alkynyl-substituted 6H-1,2-oxazine 9b (0.711 g, 74%) as a colorless oil.
6-Ethoxy-3-phenyl-4-(trimethylsilylethynyl)-6H-1,2-oxazine (9b): 1H NMR (CDCl3, 250 MHz): δ = 0.71 (s, 9 H, SiMe3), 1.21 (t, J = 7.1 Hz, 3 H, CH3), AB part of ABX3 system (δA = 3.68, δB = 3.96, JAX = JBX = 7.1 Hz, JAB = 9.7 Hz, 2 H, OCH2), 5.62 (d, J = 5.1 Hz, 1 H, 6-H), 5.69 (d, J = 5.1 Hz, 1 H, 5-H), 7.34–7.44, 7.67–7.73 (2 m, 3 H, 2 H, Ph) ppm. 13C NMR (CDCl3, 125.8 MHz): δ = −0.7 (q, SiMe3), 14.9 (q, CH3), 64.2 (t, OCH2), 92.0 (d, C-6), 99.5, 101.9 (2 s, C≡C), 114.0 (s, C-4), 127.7, 128.7, 129.5, 130.1, 132.9 (4 d, s, Ph, C-5), 155.5 (s, C-3) ppm. IR (neat): 3085–2900 (=C–H, C–H), 2160 (C≡C), 1620 (C=C), 1580 (C=N) cm−1. C17H21NO2Si (299.5): calcd. C, 68.19; H, 7.07; N, 4.68; found: C, 68.17; H, 7.08; N, 4.74.
Acknowledgments
Generous support by the Deutsche Forschungsgemeinschaft (Graduiertenkolleg at the Technische Universität Dresden “Struktur–Eigenschafts-Beziehungen bei Heterocyclen”), the Fonds der Chemischen Industrie and the Bayer–Schering Pharma AG is most gratefully acknowledged. We also thank Luise Schefzig and Ute Hain for their experimental assistance.
References
-
Gilchrist, T. L. Chem. Soc. Rev. 1983, 12, 53–73. doi:10.1039/cs9831200053
Return to citation in text: [1] -
Tsoungas, P. G. Heterocycles 2002, 57, 915–953. doi:10.3987/REV-02-548
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. In Science of Synthesis Houben-Weyl Methods of Molecular Transformations; Trost, B. M.; Molander, G. A., Eds.; Thieme: Stuttgart, Germany, 2006; Vol. 33, pp 371–389.
Return to citation in text: [1] -
Lyapkalo, I. M.; Ioffe, S. L. Russ. Chem. Rev. (engl. Transl.) 1998, 67, 467–485.
Return to citation in text: [1] -
Tishkov, A. A.; Reissig, H.-U.; Ioffe, S. L. Synlett 2002, 863–866. doi:10.1055/s-2002-31908
Return to citation in text: [1] -
Young, I. S.; Kerr, M. A. Angew. Chem. 2003, 115, 3131–3134.
Angew. Chem. Int. Ed. 2003, 42, 3023–3026. doi:10.1002/anie.200351573
Return to citation in text: [1] -
Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fisera, L.; Hlobilova, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627
Return to citation in text: [1] -
Cardona, F.; Goti, A. Angew. Chem. 2005, 117, 8042–8045.
Angew. Chem. Int. Ed. 2005, 44, 7832–7835. doi:10.1002/anie.200502640
Return to citation in text: [1] -
Sibi, M. P.; Ma, Z.; Jasperse, C. P. J. Am. Chem. Soc. 2005, 127, 5764–5765. doi:10.1021/ja0421497
Return to citation in text: [1] -
Kumarn, S.; Shaw, D. M.; Ley, S. V. Chem. Commun. 2006, 3211–3213. doi:10.1039/b606338a
Return to citation in text: [1] -
Lu, C.-D.; Zakarian, A. Angew. Chem. 2008, 120, 6935–6937.
Angew. Chem. Int. Ed. 2008, 47, 6829–6831. doi:10.1002/anie.200801652
Return to citation in text: [1] -
Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h
Return to citation in text: [1] -
Sukhorukov, A. Yu.; Lesiv, A. V.; Khomutova, Y. A.; Ioffe, S. L. Synthesis 2009, 741–754. doi:10.1055/s-0028-1083360
Return to citation in text: [1] -
Homann, K.; Angermann, J.; Collas, M.; Zimmer, R.; Reissig, H.-U. J. Prakt. Chem. 1998, 340, 649–655. doi:10.1002/prac.19983400709
Return to citation in text: [1] [2] -
Zimmer, R.; Orschel, B.; Scherer, S.; Reissig, H.-U. Synthesis 2002, 1553–1563. doi:10.1055/s-2002-33328
Return to citation in text: [1] [2] -
Zimmer, R.; Reissig, H.-U. Angew. Chem. 1988, 100, 1576–1577.
Angew. Chem., Int. Ed. Engl. 1988, 27, 1518–1519. doi:10.1002/anie.198815181
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U. Liebigs Ann. Chem. 1991, 553–562. doi:10.1002/jlac.1991199101101
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U. J. Org. Chem. 1992, 57, 339–347. doi:10.1021/jo00027a058
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U.; Lindner, H. J. Liebigs Ann. Chem. 1992, 621–624. doi:10.1002/jlac.1992199201106
Return to citation in text: [1] -
Zimmer, R.; Hoffmann, M.; Reissig, H.-U. Chem. Ber. 1992, 125, 2243–2248. doi:10.1002/cber.19921251012
Return to citation in text: [1] [2] -
Zimmer, R.; Homann, K.; Reissig, H.-U. Liebigs Ann. Chem. 1993, 1155–1157. doi:10.1002/jlac.1993199301186
Return to citation in text: [1] -
Zimmer, R.; Collas, M.; Czerwonka, R.; Hain, U.; Reissig, H.-U. Synthesis 2008, 237–244. doi:10.1055/s-2007-990946
Return to citation in text: [1] -
Buchholz, M.; Reissig, H.-U. Eur. J. Org. Chem. 2003, 3524–3533. doi:10.1002/ejoc.200300234
Return to citation in text: [1] -
Reissig, H.-U.; Homann, K.; Hiller, F.; Zimmer, R. Synthesis 2007, 2681–2689. doi:10.1055/s-2007-983802
Return to citation in text: [1] -
Zimmer, R.; Homann, K.; Angermann, J.; Reissig, H.-U. Synthesis 1999, 1223–1235. doi:10.1055/s-1999-3524
Return to citation in text: [1] -
Buchholz, M.; Reissig, H.-U. Synthesis 2002, 1412–1422. doi:10.1055/s-2002-33107
Return to citation in text: [1] -
Schmidt, E.; Reissig, H.-U.; Zimmer, R. Synthesis 2006, 2074–2084. doi:10.1055/s-2006-942403
Return to citation in text: [1] -
Birkofer, L.; Feldmann, H. Liebigs Ann. Chem. 1964, 677, 150–153. doi:10.1002/jlac.19646770121
Return to citation in text: [1] -
Abramovitch, R. A.; Shinkai, I.; Cue, B. W.; Ragan, F. A.; Atwood, J. C. J. Heterocycl. Chem. 1976, 13, 415–417. doi:10.1002/jhet.5570130247
Return to citation in text: [1] -
Baird, M. S.; Li, X.; Al-Dulayymi, J. R.; Kurdjukov, A. I.; Pavlov, V. A. J. Chem. Soc., Perkin Trans. 1 1993, 2507–2508. doi:10.1039/P19930002507
Return to citation in text: [1] -
Paulini, K.; Reissig, H.-U. Chem. Ber. 1994, 127, 685–689. doi:10.1002/cber.19941270418
Return to citation in text: [1] [2] -
Zimmer, R.; Angermann, J.; Hain, U.; Hiller, F.; Reissig, H.-U. Synthesis 1997, 1467–1474. doi:10.1055/s-1997-1373
Return to citation in text: [1] [2] -
Duhs, M.-A. Bachelor Thesis, Freie Universität Berlin, 2006.
Return to citation in text: [1] -
Zimmer, R.; Reissig, H.-U.; Homann, K. J. Prakt. Chem. 1995, 337, 521–528. doi:10.1002/prac.199533701112
Return to citation in text: [1] -
Homann, K.; Zimmer, R.; Reissig, H.-U. Heterocycles 1995, 40, 531–537. doi:10.3987/COM-94-S77
Return to citation in text: [1]
| 1. | Gilchrist, T. L. Chem. Soc. Rev. 1983, 12, 53–73. doi:10.1039/cs9831200053 |
| 2. | Tsoungas, P. G. Heterocycles 2002, 57, 915–953. doi:10.3987/REV-02-548 |
| 3. | Reissig, H.-U.; Zimmer, R. In Science of Synthesis Houben-Weyl Methods of Molecular Transformations; Trost, B. M.; Molander, G. A., Eds.; Thieme: Stuttgart, Germany, 2006; Vol. 33, pp 371–389. |
| 4. | Lyapkalo, I. M.; Ioffe, S. L. Russ. Chem. Rev. (engl. Transl.) 1998, 67, 467–485. |
| 5. | Tishkov, A. A.; Reissig, H.-U.; Ioffe, S. L. Synlett 2002, 863–866. doi:10.1055/s-2002-31908 |
| 6. |
Young, I. S.; Kerr, M. A. Angew. Chem. 2003, 115, 3131–3134.
Angew. Chem. Int. Ed. 2003, 42, 3023–3026. doi:10.1002/anie.200351573 |
| 7. | Helms, M.; Schade, W.; Pulz, R.; Watanabe, T.; Al-Harrasi, A.; Fisera, L.; Hlobilova, I.; Zahn, G.; Reissig, H.-U. Eur. J. Org. Chem. 2005, 1003–1019. doi:10.1002/ejoc.200400627 |
| 8. |
Cardona, F.; Goti, A. Angew. Chem. 2005, 117, 8042–8045.
Angew. Chem. Int. Ed. 2005, 44, 7832–7835. doi:10.1002/anie.200502640 |
| 9. | Sibi, M. P.; Ma, Z.; Jasperse, C. P. J. Am. Chem. Soc. 2005, 127, 5764–5765. doi:10.1021/ja0421497 |
| 10. | Kumarn, S.; Shaw, D. M.; Ley, S. V. Chem. Commun. 2006, 3211–3213. doi:10.1039/b606338a |
| 11. |
Lu, C.-D.; Zakarian, A. Angew. Chem. 2008, 120, 6935–6937.
Angew. Chem. Int. Ed. 2008, 47, 6829–6831. doi:10.1002/anie.200801652 |
| 12. | Brasholz, M.; Reissig, H.-U.; Zimmer, R. Acc. Chem. Res. 2009, 42, 45–56. doi:10.1021/ar800011h |
| 13. | Sukhorukov, A. Yu.; Lesiv, A. V.; Khomutova, Y. A.; Ioffe, S. L. Synthesis 2009, 741–754. doi:10.1055/s-0028-1083360 |
| 20. | Zimmer, R.; Hoffmann, M.; Reissig, H.-U. Chem. Ber. 1992, 125, 2243–2248. doi:10.1002/cber.19921251012 |
| 31. | Paulini, K.; Reissig, H.-U. Chem. Ber. 1994, 127, 685–689. doi:10.1002/cber.19941270418 |
| 20. | Zimmer, R.; Hoffmann, M.; Reissig, H.-U. Chem. Ber. 1992, 125, 2243–2248. doi:10.1002/cber.19921251012 |
| 19. | Zimmer, R.; Reissig, H.-U.; Lindner, H. J. Liebigs Ann. Chem. 1992, 621–624. doi:10.1002/jlac.1992199201106 |
| 14. | Homann, K.; Angermann, J.; Collas, M.; Zimmer, R.; Reissig, H.-U. J. Prakt. Chem. 1998, 340, 649–655. doi:10.1002/prac.19983400709 |
| 15. | Zimmer, R.; Orschel, B.; Scherer, S.; Reissig, H.-U. Synthesis 2002, 1553–1563. doi:10.1055/s-2002-33328 |
| 16. |
Zimmer, R.; Reissig, H.-U. Angew. Chem. 1988, 100, 1576–1577.
Angew. Chem., Int. Ed. Engl. 1988, 27, 1518–1519. doi:10.1002/anie.198815181 |
| 17. | Zimmer, R.; Reissig, H.-U. Liebigs Ann. Chem. 1991, 553–562. doi:10.1002/jlac.1991199101101 |
| 18. | Zimmer, R.; Reissig, H.-U. J. Org. Chem. 1992, 57, 339–347. doi:10.1021/jo00027a058 |
| 34. | Zimmer, R.; Reissig, H.-U.; Homann, K. J. Prakt. Chem. 1995, 337, 521–528. doi:10.1002/prac.199533701112 |
| 35. | Homann, K.; Zimmer, R.; Reissig, H.-U. Heterocycles 1995, 40, 531–537. doi:10.3987/COM-94-S77 |
| 25. | Zimmer, R.; Homann, K.; Angermann, J.; Reissig, H.-U. Synthesis 1999, 1223–1235. doi:10.1055/s-1999-3524 |
| 26. | Buchholz, M.; Reissig, H.-U. Synthesis 2002, 1412–1422. doi:10.1055/s-2002-33107 |
| 27. | Schmidt, E.; Reissig, H.-U.; Zimmer, R. Synthesis 2006, 2074–2084. doi:10.1055/s-2006-942403 |
| 14. | Homann, K.; Angermann, J.; Collas, M.; Zimmer, R.; Reissig, H.-U. J. Prakt. Chem. 1998, 340, 649–655. doi:10.1002/prac.19983400709 |
| 15. | Zimmer, R.; Orschel, B.; Scherer, S.; Reissig, H.-U. Synthesis 2002, 1553–1563. doi:10.1055/s-2002-33328 |
| 23. | Buchholz, M.; Reissig, H.-U. Eur. J. Org. Chem. 2003, 3524–3533. doi:10.1002/ejoc.200300234 |
| 24. | Reissig, H.-U.; Homann, K.; Hiller, F.; Zimmer, R. Synthesis 2007, 2681–2689. doi:10.1055/s-2007-983802 |
| 32. | Zimmer, R.; Angermann, J.; Hain, U.; Hiller, F.; Reissig, H.-U. Synthesis 1997, 1467–1474. doi:10.1055/s-1997-1373 |
| 22. | Zimmer, R.; Collas, M.; Czerwonka, R.; Hain, U.; Reissig, H.-U. Synthesis 2008, 237–244. doi:10.1055/s-2007-990946 |
| 21. | Zimmer, R.; Homann, K.; Reissig, H.-U. Liebigs Ann. Chem. 1993, 1155–1157. doi:10.1002/jlac.1993199301186 |
| 28. | Birkofer, L.; Feldmann, H. Liebigs Ann. Chem. 1964, 677, 150–153. doi:10.1002/jlac.19646770121 |
| 29. | Abramovitch, R. A.; Shinkai, I.; Cue, B. W.; Ragan, F. A.; Atwood, J. C. J. Heterocycl. Chem. 1976, 13, 415–417. doi:10.1002/jhet.5570130247 |
| 30. | Baird, M. S.; Li, X.; Al-Dulayymi, J. R.; Kurdjukov, A. I.; Pavlov, V. A. J. Chem. Soc., Perkin Trans. 1 1993, 2507–2508. doi:10.1039/P19930002507 |
| 31. | Paulini, K.; Reissig, H.-U. Chem. Ber. 1994, 127, 685–689. doi:10.1002/cber.19941270418 |
| 32. | Zimmer, R.; Angermann, J.; Hain, U.; Hiller, F.; Reissig, H.-U. Synthesis 1997, 1467–1474. doi:10.1055/s-1997-1373 |
© 2009 Zimmer et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)